

Abstract
Hypothalamic-pituitary-adrenal (HPA) axis responses following naloxone administration have been assumed to provide a measure of opioid receptor activity. Employing positron emission tomography (PET) using the mu opioid receptor (MOR) selective ligand [11C] Carfentanil (CFN), we demonstrated that cortisol responses to naloxone administration were negatively correlated with MOR availability (Wand et al, 2011). In this study we examined whether naloxone-induced cortisol and ACTH responses in 15 healthy control and 20 recently detoxified alcohol dependent subjects correlated with delta opioid receptor (DOR) availability in 15 brain regions using the DOR-selective ligand [11C] methyl-naltrindole (MeNTL) and PET imaging. The day after the scan, cortisol responses to cumulative doses of naloxone were determined. Peak cortisol and adrenocorticotropin (ACTH) levels and area under the cortisol and ACTH curve did not differ by group. There were negative relationships between cortisol AUC to naloxone and [11C] MeNTL BPND in the ventral striatum, anterior cingulate, fusiform cortices, temporal cortex, putamen and a trend in the hypothalamus of healthy control subjects. However, in alcohol dependent subjects, cortisol responses did not correlate with [11C]MeNTL BPND in any brain region. Plasma ACTH levels did not correlate with [11C]MeNTL BPND in either group. The study demonstrates that naloxone provides information about individual differences in DOR availability in several mesolimbic structures. The data also show that the HPA axis is intimately connected with mesolimbic stress pathways through opioidergic neurotransmission in healthy subjects but this relationship is disrupted during early abstinence in alcohol dependent subjects.
Keywords: mu opioid receptors, naloxone, PET imaging, HPA axis, cortisol
Introduction
Central nervous system (CNS) opioid pathways are important modulators in the adaptation to stress as well as to alcohol. Strong evidence indicates that endogenous opioids attenuate or terminate the emotional consequences of the stress response as part of the allostatic reaction (Drolet et al, 2001). There is also strong evidence that CNS opioid systems play a significant role in alcohol dependence (Oswald and Wand, 2004). The mesocorticolimbic system mediates the rewarding effects of most drugs of abuse including alcohol, and both dopamine and opioid neurotransmission play a crucial role in this reward pathway. Within this key region of the brain, the reinforcing effects of alcohol are modulated by release of opioid peptides and release of dopamine.
Investigators have administered opioid receptor antagonists to interrogate the HPA axis in part to elucidate the pathophysiologic role of opioid systems in regulating stress and contributing to substance abuse disorders. The non-selective opioid receptor antagonist, naloxone, activates the HPA axis by blocking opioid inhibitory tone directed at the hypothalamic and possibly mesolimbic regulators of ACTH secretion. ACTH, in turn, stimulates cortisol release. Studies using opioid receptor antagonists have assumed to identify differences in opioid activity due to nicotine and alcohol dependence, family history of alcoholism, gender, neuroticism and genetic variations in the MOR (Oswald and Wand, 2004; Roche et al, 2010; Adinoff et al, 2005; Wand et al, 1998; Wand et al, 1999; Mangold et al, 2000; Wand et al 2001; Wand et al, 2002; Uhart et al, 2006; Mangold and Wand, 2006).
Employing the MOR selective PET ligand, [11C]CFN to measure [11C]CFN binding potential (BPND), in healthy subjects, we recently demonstrated that cortisol responses to a naloxone challenge were negatively correlated with MOR availability in the mesolimbic region (Wand et al, 2011). This finding demonstrated that naloxone is not merely a non-specific pharmacologic activator of the HPA axis but it also provides information about individual differences in MOR availability in important brain regions involved in responding to stress. The data also suggested that in addition to the hypothalamus, the ventral striatum, caudate and putamen may contribute endorphinergic inhibitory tone directed to the hypothalamic regulators of ACTH, and that this inhibition could be relieved by blocking MORs.
Similar to MORs, the DOR system is also important in stress regulation and is involved in maintaining alcohol consumption (Mendez and Morales-Mulia, 2008). The majority of stress-related brain nuclei receives enkephalinergic innervation, or contains enkephalin perikarya (Drolet et al, 2001). In addition to MORs, DORs also place inhibitory tone on the hypothalamic regulators of HPA axis (Drolet et al, 2001). Indeed enkephalinergic neurons are present not only in the paraventricular (PVN) nucleus of the hypothalamus and median eminence contiguous to CRF neurons, but also in other stress-related regions including the anterior cingulated cortex, infra-limbic cortex, central and medial amygdale and the bed nucleus of the stria terminalis (Fallon and Leslie, 1986; Guthrie and Basbaum, 1984; Harlan et al, 1987; Hurd, 1996; Khachaturian et al, 1983; Petrusz et al, 1985; Watson et al, 1982). The relationship between naloxone-induced cortisol and DOR availability has never been studied.
Individuals at increased risk for alcohol dependence (eg., non-alcohol dependent offspring of alcohol dependent parents) have altered cortisol responses to naloxone and naltrexone compared to individuals at low risk for alcohol dependence, suggesting differences in opioids receptor activity (Wand, 2008) Moreover, opioid receptor antagonists have been an important therapeutic modality for the treatment of alcohol dependence (Kranzler and Edenberg, 2010). Employing PET imaging with [11C]CFN and [11C]MeNTL, our group reported significantly greater MOR availability in mesolimbic regions in alcohol dependent subjects when compared to controls; there was a similar trend for higher DOR availability in many of these brain regions in alcohol dependent subjects, although differences did not achieve statistical significance (Weerts et al, 2011). In laboratory animals, antagonists that are selective for the MOR or DOR decrease alcohol consumption in laboratory animals (Krishnan-Sarin et al, 1985; Froehlich et al, 1991; Franck et al, 1998).
The current study examined the utility of using a naloxone challenge procedure to characterize the relationship between naloxone-induced cortisol and DOR availability in healthy control subjects and to also detect defects in these systems in alcohol dependent subjects. Cortisol responses were assessed using a technique which allows administration of 5 incremental doses of naloxone in a single session (Mangold et al, 2000). This cumulative naloxone dosing procedure is believed to activate the HPA axis by blocking MORs at lower doses and DORs at higher doses (Traynor et al, 1990). Based on our recent findings that naloxone-induced cortisol is negatively correlated with MOR availability, we hypothesized a negative correlation between naloxone-induced cortisol and DOR availability in stress-related brain regions in healthy control subjects. Since chronic alcohol exposure is known to disrupt opioid and stress systems, we hypothesized that the relationship between cortisol and DOR availability would be disrupted in alcohol dependent subjects.
Methods
Subjects
Current heavy, alcohol dependent drinkers and healthy control subjects between 21 and 58 years of age were recruited by advertisement and provided informed consent, in the sober state, using JHU Institutional Review Board approved procedures. A Masters-level research assistant utilized a battery of standardized diagnostic and psychological instruments to interview potential subjects as described previously (Weerts et al, 2008). Inclusion criteria for alcohol dependent subjects included meeting DSM-IV criteria for alcohol dependence based on the Semi-Structured Assessment of the Genetics of Alcoholism (Bucholz et al, 1994) and active drinking at NIAAA-defined hazardous levels as determined by completion of a 90-day Time Line Follow Back (Sobel and Sobel, 1992). Aged matched healthy control subjects did not drink above the NIAAA recommended guidelines (less than 8 drinks/week for women and less than 15 drinks/week for men) and did not meet current or lifetime DSM-IV criteria for either alcohol abuse or dependence. For both groups, exclusionary criteria included: 1) current or lifetime DSM-IV diagnostic criteria for any other Axis I disorder, including other drug abuse/dependence (except nicotine), 2) positive urine drug toxicology at screening or hospital admission, 3) other ongoing health problems or 4) subject report of maternal drinking during pregnancy. In addition, alcohol dependent subjects were excluded if they reported alcohol-related seizures or the need for medication during previous detoxifications. Basic demographic characteristics for alcohol dependent and healthy control subjects are shown in Table 1. The Alcohol Dependence Scale (ADS) (Skinner and Allen, 1982) was administered to characterize alcohol use and associated problems. Prior to the CRU admission, subjects underwent magnetic resonance imaging (MRI) to rule out persons with clinically significant brain abnormalities, and allow within-subject anatomical localization of brain regions (Meltzer et al, 1990).
Table 1.
Demographics for alcohol dependent (AD) and healthy control (HC) subjects. Data shown are group means (SD) or number (n) of subjects as indicated.
| HC (n=15) | AD (n=20) | P | |
|---|---|---|---|
| Mean Years of Age (SD) | 46.7 (9.7) | 44.3 (7.4) | 0.263 |
| Gender (n) | 0.344 | ||
| Male | 9 | 15 | |
| Female | 6 | 5 | |
| Race (n) | |||
| Caucasian | 9 | 12 | 1.000 |
| Black | 6 | 8 | |
| Smoking Status (n) | |||
| Non-smokers | 13 | 4 | <.001 |
| Alcohol-related measures: mean (SD) | |||
| Age (Yrs) met criteria for Alcohol | N/A | 28 (6.1) | |
| Years of Dependent Alcohol Drinking | N/A | 16.4 (9.1) | |
| Alcohol Dependence Scale (ADS) | 0.1 (0.4) | 20.3 (7.3) | <.001 |
| Number of Drinks per drinking day | 1.5 (0.9) | 12.9 (7.1) | <.001 |
| Number of Drinking Days per Week | 0.8 (1.3) | 5.5 (1.2) | <.001 |
| Psychological Assessments on | <.001 | ||
| Beck Depression Inventory (BDI-II) | 1.3 (1.4) | 11.8 (8.9) | <.001 |
| Beck Anxiety Inventory (BAI) | 1.3 (2.5) | 9.9 (8.2) | <.001 |
| Brief Symptom Inventory (BSI) | 0.1 (0.1) | 0.5 (0.6) | 0.003 |
Inpatient Procedures following admission to Clinical Research Unit (CRU)
Healthy control subjects were admitted to the clinical research unit and completed an inpatient protocol that included PET imaging and the naloxone challenge. Alcohol dependent subjects completed the study under an inpatient protocol that included hospital admission and medically supervised alcohol withdrawal prior to PET imaging on day 5 of supervised abstinence. Alcohol dependent subjects remained on the clinical research unit for subsequent naltrexone treatment and PET imaging to determine naltrexone blockade of MORs and DORs (Weerts et al, 2008). Both groups underwent the naloxone challenge the day after the PET scans.
The CRU nursing staff completed the Clinical Institute Withdrawal Assessment-Alcohol Revised (CIWA-Ar) (Sullivan et al, 1989) with alcohol dependent participants 3 times each day for the first 5 days. No subject required withdrawal medication based on CIWA scores, vital signs and physician assessment. During the CRU stay and all study procedures, cigarette smoking was prohibited. Nicotine dependent smokers determined by a FNDT score of 3 or higher, received a transdermal nicotine patch (21 mg nicotine) at hospital admission, each morning while on the CRU, and three hours prior to the PET scan. This standardized approach was used to mitigate potential impact of nicotine withdrawal on scan outcome.
PET procedures
PET scans were acquired in 3D mode on a GE Advance PET scanner (GE Medical Systems, Milwaukee, WI) as previously described (Weerts et al, 2011). In brief, subjects underwent two PET scans in a fixed order on the same day; the [11C]MeNTL, a specific DOR antagonist (33,34) and [11C]CFN, a specific mu opioid agonist (Titeler et al, 1989; Frost et al, 1990)) scans were conducted at 8:30 and 10:45 am, respectively. This study only presents results from the [11C] MeNTL scan.
Twenty five frames with variable time period (6 × 30 sec, 5 × 60 sec, 5 × 120 sec, 9 × 480 sec) were acquired during a 90-min period for each study. Arterial blood samples were collected every 5 seconds initially, and then increasing intervals throughout out the [11C] MeNTL scans (e.g.,1-min, 2-min, 5-min, 10-min and 15-min) and counted for the radioactivity. Blood samples taken at 0, 5, 15, 30, 60 and 90 min were analyzed with HPLC for radioactive metabolites (Hilton et al, 2000). PET images were reconstructed employing the back projection algorithm using the software provided by the manufacturer (Kinahan et al, 1989).
Volumes of Interest (VOI) Analyses
Fifteen VOIs were selected to include brain regions that had moderate to high [11C] MeNTL BPND and included regions that are involved in responding to stressful stimuli and/or are altered in alcohol dependence (table 4). The VOIs were manually defined as previously described (Weerts et al, 2011).
Table 4.
Correlation of cortisol AUC (0 minute to 240 minute) response to naloxone and [11C]MeNTL BPND adjusted by sex and smoking. The adaptive step-up Bonferroni method was used to adjust P values for multiple comparisons.
| Region Region |
HC | AD | ||||
|---|---|---|---|---|---|---|
| Partial correlation coefficient | Adjusted P | Unadjusted P | Partial correlation coefficient | Adjusted P | Unadjusted P | |
| Fusiform | −0.826 | 0.0015 | 0.0005 | −0.115 | 0.974 | 0.651 |
| Ventral Striatum* | −0.839 | 0.0019 | 0.0006 | 0.288 | 0.974 | 0.246 |
| Cingulate | −0.661 | 0.0418 | 0.0139 | −0.05 | 0.974 | 0.843 |
| Temporal | −0.633 | 0.0604 | 0.0201 | 0.017 | 0.974 | 0.948 |
| Putamen | −0.619 | 0.0727 | 0.0242 | 0.098 | 0.974 | 0.699 |
| Hypothalamus | −0.547 | 0.1593 | 0.0531 | 0.027 | 0.974 | 0.916 |
| Thalamus | −0.527 | 0.1936 | 0.0645 | 0.246 | 0.974 | 0.326 |
| Caudate | −0.495 | 0.2574 | 0.0858 | −0.188 | 0.974 | 0.455 |
| Insula | −0.486 | 0.2764 | 0.0921 | 0.045 | 0.974 | 0.86 |
| Frontal Cortex | −0.455 | 0.3537 | 0.1179 | 0.008 | 0.974 | 0.974 |
| Hippocampus | −0.374 | 0.6241 | 0.208 | 0.247 | 0.974 | 0.324 |
| Globus Pallidus | −0.246 | 0.6892 | 0.4174 | −0.119 | 0.974 | 0.638 |
| Parietal | −0.198 | 0.6892 | 0.5161 | 0.009 | 0.974 | 0.971 |
| Amygdala | −0.185 | 0.6892 | 0.5442 | −0.243 | 0.974 | 0.33 |
| DLPFC** | −0.123 | 0.6892 | 0.6892 | −0.059 | 0.974 | 0.815 |
, one outlier removed;
DLPFC, dorsal lateral prefrontal cortex
Pharmacokinetic modeling
The primary outcome variable of interest for DOR was binding potential (BPND) (Innis et al, 2007) of [11C] MeNTL. We analyzed data using plasma reference graphical analysis (PRGA) (Logan et al., 1990) to obtain distribution volume (VT); [11C]MeNTL BPND was obtained as target-reference VT ratio less 1 using cerebellum as the reference region. Plasma Logan plots for [11C] MeNTL approached asymptotes in all regions examined by 20 min in all subjects. Therefore t* was set at 20 min (Weerts et al, 2011).
Naloxone cumulative dosing procedure
Generating a dose response curve to naloxone in a single session was carried out based on our previously published procedures (Wand et al, 2011; Mangold et al, 2000). The session was conducted the afternoon following the PET scan. Following a calorie controlled lunch, participants had an intravenous catheter inserted into a forearm vein 60 min before placebo administration. Baseline blood samples were obtained −30 min, −15 min and immediately prior to placebo administration. At time 0, placebo (0.9% saline) was administered as a bolus. Sequential doses of naloxone (25, 50, 100 and, 250 ug/kg) dissolved in 0.9% saline were administered every 30 min thereafter. Blood samples were obtained 15 and 30 min after placebo and each naloxone dose, and then every 30 min through 240 min. Plasma cortisol (Diagnostic Products Corporation, Inc.; Los Angeles, CA). and ACTH concentrations (DiaSorin immunoradiometric assay) were assayed by radioimmunoassay. Intra-assay and inter-assay coefficients of variance were less than 8% for cortisol and 6% and 9%, respectively, for plasma ACTH.
Statistical Plan
Chi-square tests, t-tests or the non-parametric equivalent were performed to compare demographics, smoking status, alcohol-related-measures and psychological assessment between the two groups.
The hormone level at baseline and following each naloxone dose was plotted for each group. The hormone level for each dose was calculated as the average of the two measures 15 and 30 min after the dose was given. To compare hormone levels following various naloxone doses to the placebo level, a linear mixed model with random intercept was constructed for each group. An unstructured covariance matrix was used to obtain robust standard errors.
We calculated the hormone area under the curve (AUC) measures to estimate the total amount of response. AUC was calculated as the sum of the trapezoids between every pair of consecutive time points subtracting baseline hormone level. Calculated AUC value had units of μg/dl • hour for cortisol and pg/mL • hour for ACTH. The hormone response AUC for the time period from time 0 through time 240 minutes was calculated as the main outcome. For each subject, the hormone peak response and time to peak was also calculated. Then linear models were constructed to compare AUC, peak, and time to peak between the two groups. Smoking status and sex were included as covariates. Similar linear models were constructed to compare [11C] MeNTL level between AD and HC group on indicated regions. The obtained p values across the regions were adjusted using the adaptive step-up Bonferroni method for multiple comparisons (Hochberg and Benjamin, 1990).
To model the correlation between hormone response and [11C] MeNTL BPND, multi-linear models were constructed with BPND as the dependent variables and cortisol or ACTH response AUC as the independent variable. Although the covariates of sex and smoking did not have significant effects on any dependent measures, both covariates have been shown to modulate opioid activity(Micevych et al, 1997; al’Absi M et al, 2008; Berrendero et al, 2010). Therefore we included sex and smoking status (smoker/non-smoker) as covariates. Partial residual plots with the component line were plotted for both groups in selected regions to show the relationship between hormone AUC and [11C] MeNTL BPND given that the covariates were also in the model. Again the adaptive step-up Bonferroni method was used for multiple comparison correction. All the analyses were carried out using SAS 9.2, and a two-sided p-value of <0.05 was considered statistically significant.
Results
Baseline cortisol levels (mean of −30, −15 and 0 time points) did not differ by group. Compared to the effect of placebo, cumulative naloxone dose administration induced statistically significant cortisol responses in both healthy and alcohol dependent subjects (Figure 1). Magnitude of peak cortisol responses did not differ by group nor did area under the cortisol curve (AUC) (Table 2). In contrast, time to peak response did differ by group. The majority of the healthy control subjects mounted a peak cortisol response between 75–90 min corresponding to the 50 ug/kg dose whereas the majority of the alcohol dependent subjects mounted a peak cortisol response between 105–120 min corresponding to the 100ug dose (Table 2; p=.05).
Figure 1.

Cortisol responses to five graded doses of naloxone. Data points are mean (SEM). BASE=baseline; PBO=placebo. * denotes time points in alcohol dependent subjects that were significantly different from mean of placebo time points with p<.05, adjusting for sex and smoking status. + denotes time points in healthy control subjects that were significantly different from mean of placebo time points with p<.05, adjusting for sex and smoking status.
Table 2.
Comparisons of mean cortisol variables between healthy control (HC) alcohol dependent (AD), adjusted for smoking status and sex.
| Variable | Adjusted mean (SE) | P | |
|---|---|---|---|
| HC | AD | ||
| Peak (ug/dL) | 19.4 (1.4) | 20.2 (1.2) | 0.712 |
| Response AUC (ug/dL·h) | 17.2 (3.4) | 16.7 (2.8) | 0.923 |
| Time at peak (min) | 87.5 (10.2) | 118.1 (8.5) | 0.050 |
We measured [11C] MeNTL BPND in fifteen brain regions in healthy control and alcohol dependent subjects a day before the naloxone session was completed. We previously reported mean [11C] MeNTL BPND on 8 regions in healthy control and alcohol dependent subjects: cingulated cortex, amygdala, insula, ventral striatum, putamen, caudate, globus pallidus, and thalamus (Weerts et al, 2011). We found no statistical differences in [11C] MeNTL BPND between healthy control and alcohol dependent subjects, although there were trends for alcohol dependent subjects to have higher [11C] MeNTL BPND in the insula, putamen, caudate and globus pallidus. In table 3 we compare [11C] MeNTL BPND in 7 additional brain regions. Again there were no group differences in [11C] MeNTL BPND except for a trend in the temporal cortex.
Table 3.
Mean [11C]MeNTLBPND healthy control (HC) and alcohol dependent (AD) subjects. Data shown are group means and SEM adjusted for sex and smoking for each VOI.
| VOI | HC (N = 15) | AD (N = 20) | Adjusted P | Unadjusted P |
|---|---|---|---|---|
| Fusiform | 0.548 (0.037) | 0.601 (0.03) | 0.696 | 0.330 |
| Temporal | 0.864 (0.05) | 0.993 (0.042) | 0.266 | 0.089 |
| Hypothalamus | 0.082 (0.062) | −0.002 (0.051) | 0.696 | 0.357 |
| Frontal Cortex | 0.801 (0.044) | 0.89 (0.037) | 0.526 | 0.175 |
| Hippocampus | 0.455 (0.047) | 0.528 (0.039) | 0.696 | 0.292 |
| Parietal | 0.849 (0.053) | 0.925 (0.044) | 0.696 | 0.333 |
| DLPFC** | 0.727 (0.059) | 0.761 (0.049) | 0.696 | 0.696 |
Adjusted for multiple comparison using the adaptive step-up Bonferroni method for multiple comparisons (Hochberg and Benjamini, 1990).
dorsal lateral prefrontal cortex
We then examined the correlation between [11C] MeNTL BPND and naloxone-induced cortisol response in all 15 brain regions. There were no significant correlations between baseline cortisol concentration and [11C] MeNTL BPND in healthy subjects or alcohol dependent subjects. However there were significant negative correlations between area under the cortisol response curve and [11C] MeNTL BPND in the fusiform gyrus, ventral striatum and cingulate cortex in healthy subjects (Table 4). The data, without adjustment for multiple comparisons, also includes significant correlations with the temporal cortex and putamen as well as a trend in the hypothalamus (p=.0531). In contrast, there was no significant correlations between naloxone-induced cortisol responses and [11C] MeNTL BPND in alcohol dependent subjects in any of the 15 brain region (Table 4).
As an example of these relationships, the partial residual plots show the association between [11C] MeNTL BPND in the ventral striatum, fusiform gyrus and cingulate cortex with cortisol responses to naloxone in both groups (Figure 2A–F). Figure 3 shows brain images of mean [11C] MeNTL BPND in the ventral striatum for the control and alcohol dependent subjects in the lowest cortisol response tertile compared to subjects in the highest cortisol response tertile.
Figure 2.


Partial residual plot of [11C]MeNTL BPND and cortisol response AUC (0–240 min) adjusted for sex and smoking. Statistics are displayed in Table 4. A and B, ventral striatum, C and D, fusiform; E and F, cingulate
Figure 3.
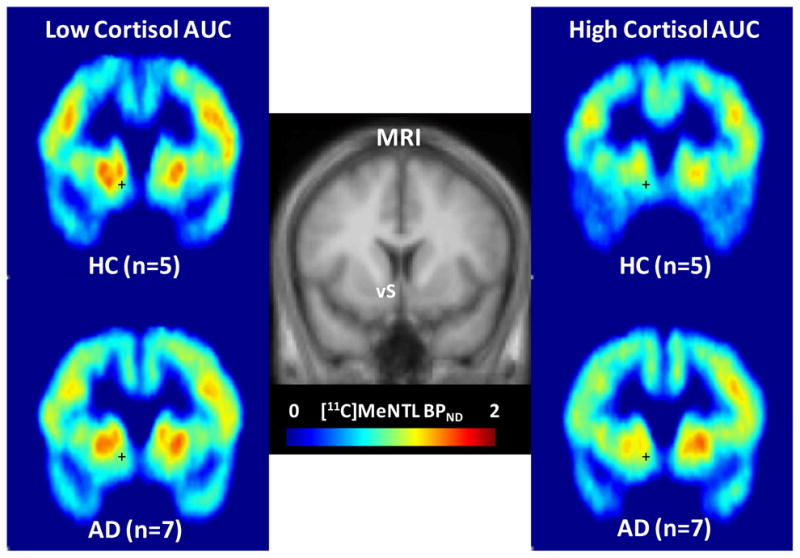
Coronal view images of [11C]MeNTL BPND, averaged across the subjects in the lowest tertile of cortisol responses (left) compared to the subjects in the highest tertile of cortisol responses (right). A standard MRI to which BPND images were spatially normalized is displayed in the middle panel to indicate image locations around the anterior-posterior center of ventral striatum (vS). Colored legend depicts [11C]MeNTL BPND from 0 (light blue) to 2.0 (red).
The magnitude of peak ACTH responses did not differ by group nor did area under the ACTH curve or time to peak (data not shown). There were no significant correlations between ACTH AUC and [11C] MeNTL BPND in any VOI in either group (data not shown).
Discussion
In the current study we compared two CNS measures in healthy control and alcohol dependent subjects. Cortisol response to opioid receptor blockade was interrogated by administrating 5 incremental doses of naloxone. Naloxone has been shown to dose-dependently block mu, delta-1 and delta-2 opioid receptors (Hirose et al, 2005; Lewandowitsch and Irvine, 2003). The alcohol dependent subjects mounted a cortisol response that was minimally different compared to the healthy control subjects. The peak and the AUC cortisol response did not differ between groups; however time to peak response differed significantly. The physiological significance of this group difference in time to peak is unclear. It could reflect differential sensitivity to naloxone dosing between alcohol dependent and control subjects or may indicate a more global injury of the HPA axis in alcohol dependent subjects. If the latter is true it implies that the alcohol dependent group may have an impaired response to both pharmacologic and physiologic activation. Mounting a delayed response to stress could jeopardize health of the organism.
Second, we examined whether the naloxone-induced cortisol response correlated with DOR availability. To do this we first measured [11C] MeNTL BPND in 15 brain regions in the healthy control and alcohol dependent subjects. We previously published these data in 8 of the 15 regions (Weerts et al, 2011) finding no significant group differences in [11C] MeNTL BPND. However there were trends in many brain regions for alcohol dependent subjects to have higher MeNTL than control subjects, although differences did not achieve statistical significance due to limited sample sizes (Weerts et al, 2011). Interestingly, DOR availability in the caudate was correlated with recent drinking (average drinks per drinking day) in alcohol dependent subjects. In the present study we showed [11C] MeNTL BPND measured in 7 additional brain regions that had not been previously reported. Again we found no group differences in [11C] MeNTL BPND except for a trend in the temporal cortex. With these measurements in hand we went on to correlate [11C] MeNTL BPND and cortisol AUC. In the healthy control subjects, there were significant negative relationships between cortisol response to naloxone and [11C] MeNTL BPND in several mesolimbic structures. Subjects who mounted a higher cortisol response to naloxone had lower [11C] MeNTL BPND compared to subjects with a higher cortisol response. Lower [11C] MeNTL BPND indicates either increased enkephalin peptide occupancy or decreased DOR number/affinity compared to a state of higher [11C] MeNTL BPND; the scanning technique cannot distinguish among these possibilities. In contrast, in alcohol dependent subjects, cortisol responses did not correlate with [11C] MeNTL BPND in any brain region. Apparently factors associated with alcohol dependence and/or withdrawal disrupted the relationship identified in the healthy control participants.
The primary regulators of ACTH secretion, and thus cortisol, are CRF and AVP neurons located in the parvocellular region of the paraventricular nucleus. Enkephalins and other opioid peptides modify the synthesis and secretion of hypothalamic releasing factors like CRF (Boosook and Hyman 1995; Szekely, 1990). Furthermore, endorphinergic and enkephalinergic innervation places inhibitory tone on these neurons; opioid receptor antagonists can release this inhibition resulting in stimulation of ACTH and cortisol (Schluger et al, 1998). In the healthy control group we identified negative correlations between [11C] MeNTL BPND and cortisol responses to naloxone in the fusiform gyrus, ventral striatum and the cingulate cortex; these relationships remained significant after statistically adjusting for analyses in the 15 brain regions. The three regions have high [11C] MeNTL BPND and are enriched in enkephalin perikarya (Drolet et al, 2001; Narita et al, 2006). DORs in the ventral striatum and cingulate cortex have been shown to modulate anxiety-like behaviors in rodents (Narita et al, 2006; Hebb et al, 2004). When we did not statistically adjust for the analyses across the 15 brain regions, additional correlations with several other brain regions including the hypothalamus were significant. The weaker correlation for the hypothalamus may relate to this region being difficult to study with PET.
There are several possible mechanisms to explain the correlation between naloxone-induced activation of the HPA axis and [11C] MeNTL BPND in the identified brain regions in healthy controls. The mechanisms are not mutually exclusive. Although causality cannot be proven by these data, the findings suggest that the three regions are under enkephalinergic inhibitory tone and release of that inhibition by naloxone results in communications with the PVN to activate the HPA axis. In this scenario, cumulative naloxone dosing activated the HPA axis proportionately to DOR availability. This assumes that these brain regions communicate to PVN neurons though the enkephalinergic system. In fact preclinical studies have shown that mesolimbic brain regions that participate in responding to stressful stimuli also communicate with PVN neurons to stimulate CRF and AVP release or are involved in glucocorticoid negative feedback (Jankord and Herman, 2008). It is thought that the release of enkephalin peptides from several mesolimbic structures attenuates the impact of the stress response on emotional and affective states helping the organism cope with the stressful event (Drolet et al, 2001). Supporting this are studies showing higher anxiety levels in mice with a targeted disruption of the DOR gene (Filliol et al, 2000)) or the proenkephalin gene (Konig et al, 1996). Also naloxone-induced activation of the HPA response is stronger in chronically stressed animals compared to unstressed control animals showing the impact of chronic stress on opioid systems (Janssens et al, 1995). It is interesting that [11C] MeNTL BPND measured in the fusiform gyrus correlated with cortisol responses. Similar to the amygdala, recent studies have shown that the fusiform gyrus is also involved in the perception of threatening facial stimuli (Radua et al, 2010).
It is noteworthy that our previous paper demonstrated negative correlations between MOR availability and cortisol responses to naloxone in the ventral striatum, putamen, caudate and hypothalamus of healthy control subjects using the mu selective ligand [11C]Carfentanil (CFN) (Wand et al, 2011). Taken together these observations suggest that certain mesolimbic regions that activate the hypothalamic regulators of ACTH are under enkephalinergic inhibitory tone (fusiform and cingulate), other regions are predominantly under endorphinergic inhibitory tone (caudate and putamen) and still other regions are under both forms of opioidergic inhibition (ventral striatum and hypothalamus).
There is ample evidence that genotype, early life traumas and current life stressors create individual differences in chronic cortisol exposure (cortisol burden) (Stephens and Wand, 2011). Although a less likely explanation for the relationship between [11C] MeNTL BPND and cortisol response to naloxone, it is possible that individual differences in cortisol burden modulate DOR availability. In this model, individuals with a phenotype of high chronic cortisol production have lower DOR availability compared to subjects with a low chronic cortisol exposure phenotype. The effects of chronic cortisol exposure on DOR expression would then account for the association of cortisol and [11C] MeNTL BPND. Indeed it has been shown that stress causes adaptive increases in pro-enkephalin gene expression in the paraventricular nucleus and other brain regions conducted using several different stress procedures including intraperitoneal injection of hypertonic saline (Young and Lightman, 1992; Ceccatelli and Orazzo, 1993; Watts, 1991). Cortisol-induced increases in synaptic concentrations of enkephalinergic peptides could reduce [11C] MeNTL BPND through competitive inhibition. This is precisely the relationship we see in our study. If we had observed a relationship between baseline cortisol levels and [11C] MeNTL BPND, this would have provided stronger support for this model. Whatever the mechanisms responsible for these interesting correlations, the data show that the naloxone challenge procedure as conducted in this study provide measurement related to DOR availability in healthy control subjects.
The findings in the ventral striatum are particularly interesting. In this region, the correlations between cortisol and [11C] MeNTL BPND in this study and [11C] CFN BPND in our previous work (Wand et al, 2011) are robust and particularly important because endogenous opioids modulate dopaminergic transmission in the nucleus accumbens, modulating reinforcement and reward (Wise, 2008). This brain region is central in understanding the neurobiology of alcohol and other drug dependencies (Barson et al, 2009; Barson, et al 2010). Moreover, naloxone has been shown to dose-dependently block mu, delta 1 and delta 2 opioid receptors attenuating the effects of DAMGO, DPDPE and [D-Ser(2)]Leu-enkephalin-Thr on dopamine release in the nucleus accumbens (Hirose et al, 2005).
A third finding of this paper is that the alcohol dependent subjects did not show correlations between cortisol responses to naloxone and [11C]MeNTL BPND in any brain region. This suggests disruption of signaling between enkephalinergic pathways and the paraventricular nucleus. This analysis reveals an abnormality in the relationship between DOR availability and the HPA stress response in alcohol dependent subjects that neither technique alone can identify. These data support a role of the DOR in alcohol dependence, particularly when coupled to our previous study showing that the FDA approved dose of naltrexone (50 mg) produced only partial inhibition (21%) of DORs in alcohol dependent subjects when the same dose of naltrexone produces near complete inhibition (95%) of MORs (Weerts et al, 2008). Moreover, there is evidence from preclinical research showing efficacy of DOR antagonists for relapse prevention (Marinelli et al., 2009; Nielsen et al., 2011). It remains uncertain whether the failure to see a correlation between cortisol and [11C] MeNTL BPND in alcohol dependent subjects reflects a trait or state phenomenon. Through a variety of possible mechanisms (e.g., chronic alcohol toxicity, acute alcohol withdrawal, genetic regulation), this relationship has become disrupted. The clinical implications of this disruption merit further investigation, particularly in terms of the possible contribution to alcohol relapse in early recovery.
There are several limitations to the study. First, the sample size is small as this was an exploratory study. Second, we studied the relationship between DOR availability and naloxone challenge only using a cumulative dosing procedure developed in our laboratory (Mangold et al, 2000). It remains to be seen whether this would be observed following a single dose of naloxone or using other HPA provocateurs. A third limitation is the potential confound of smoking which plagues most studies with alcohol dependent subjects. Over 80% of alcohol-dependent subjects report regular tobacco use and smoke at high rates (Dawson, 2000) when compared to social drinkers. It is somewhat reassuring that the covariate of smoking did not have a significant effect on any dependent measures. In addition all smokers had nicotine patches placed to control for smoking, and smoking was a covariate in the statistical model. Fourth, we did not identify any association between ACTH AUC and [11C] CFN BPND in any VOI. It is possible that cortisol negative feedback attenuates the ACTH response to naloxone, and therefore a full ACTH response cannot be realized. It is also possible that the short half-life of ACTH in plasma prohibited capturing the true area under the ACTH curve given our blood sampling frequency. Finally, because the same measure of naloxone response is correlated with both mu and delta receptor availability in the brain, this raises the issue of ligand specificity. In vitro affinity, Ki values of MeNTL were reported to be 0.02, 14, and 65 nM for δ, μ, and κ opioid receptors, respectively (Portoghese et al. 1990), while Ki values of CFN were 0.024, 3.28. and 43.1 nM for μ, δ, and κ opioid receptors, respectively (Cometta-Morini et al, 1992). This supports that [11C]MeNTI and [11C]CFN are specific radioligands of δ and μ opioid receptors (Madar et al. 1997). Accordingly the two radioligand displayed distinctively different regional neuroreceptor distribution patterns in the human brain (Madar et al. 1997; Weerts et al., 2011).
In summary the study demonstrates that naloxone provides information about individual differences in DOR availability in several mesolimbic structures. The data also show that the HPA axis is intimately connected with mesolimbic stress pathways through opioidergic neurotransmission in healthy subjects but this relationship is disrupted during early abstinence in alcohol dependent subjects. The mechanism and clinical significance behind this disruption needs further study.
Acknowledgments
The National Institute of Alcohol Abuse and Alcoholism (NIAAA) provided financial support for research related to the subject matter of this manuscript from the grants R37AA12303 (PI: G.S. Wand) and R01AA11855 (PI:M.E. McCaul). Dr. Wand is the recipient of a gift fund from the Kenneth Lattman Foundation. He is an investigator in a post marketing study for Eli Lilly & Company, entitled The Global Hypopituitary Control and Complications Study (HypoCCS). He is an investigator in a post marketing study for Ipsen entitled, Somatuline Depot (lanreotide) Injection for Acromegaly (SODA). Dr. Wong is a consultant for Amgen. Between 2009 and present, Dr. Wong has received funding from the following companies: Acadia, Amgen, Avid, Biotie, Bristol Myers Squibb, GE, Intracellular, J&J, Lilly, Luhdeck, Merk, Orexigen, Otuska, Roche, Sanofi-Aventis and Sepracor.
Footnotes
Drs. Kuwabara, Xu and Weerts have no financial disclosures.
References
- 1.al’Absi M, Wittmers LE, Hatsukami D, Westra R. Blunted opiate modulation of hypothalamic-pituitary-adrenocortical activity in men and women who smoke. Psychosom Med. 2008;70(8):928–35. doi: 10.1097/PSY.0b013e31818434ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adinoff B, Krebaum SR, Chandler PA, Ye W, Brown MB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal pathology in one-month abstinent alcohol-dependent men: Response to CRH and Naloxone. Alcohol: Clin Exp Res. 2005;29:528–537. doi: 10.1097/01.ALC.0000158939.25531.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barson JR, Carr AJ, Soun JE, Sobhani NC, Leibowitz SF, Hoebel BG. Opioids in the nucleus accumbens stimulate ethanol intake. Physiol Behav. 2009;98(4):453–9. doi: 10.1016/j.physbeh.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barson JR, Carr AJ, Soun JE, Sobhani NC, Rada P, Leibowitz SF, Hoebel BG. Opioids in the hypothalamic paraventricular nucleus stimulate ethanol intake. Alcohol: Clin Exp Res. 2010;34(2):214–222. doi: 10.1111/j.1530-0277.2009.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berrendero F, Robledo P, Trigo JM, Martín-García E, Maldonado R. Neurobiological mechanisms involved in nicotine dependence and reward: participation of the endogenous opioid system. Neurosci Biobehav Rev. 2010;35(2):220–31. doi: 10.1016/j.neubiorev.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borsook D, Hyman SE. Proenkephalin gene regulation in the neuroendocrine hypothalamus: A model of gene regulation in the CNS. Am J Physiol. 1995;269:E393–E408. doi: 10.1152/ajpendo.1995.269.3.E393. [DOI] [PubMed] [Google Scholar]
- 7.Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, et al. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol. 1994;55(2):149–58. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 8.Ceccatelli S, Orazzo C. Effect of different types of stressors on peptide messenger ribonucleic acids in the hypothalamic paraventricular nucleus. Acta Endocrinol. 1993;128:485–492. doi: 10.1530/acta.0.1280485. [DOI] [PubMed] [Google Scholar]
- 9.Cometta-Morini C, Maguire PA, Loew GH. Molecular determinants of mu receptor recognition for the fentanyl class of compounds. Mol Pharmacol. 1992;41(1):185–96. [PubMed] [Google Scholar]
- 10.Dawson DA. Drinking as a risk factor for sustained smoking. Drug Alcohol Depend. 2000;59(3):235–49. doi: 10.1016/s0376-8716(99)00130-1. [DOI] [PubMed] [Google Scholar]
- 11.Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, Trottier JF. Role of endogenous opioid system in the regulation of the stress response. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(4):729–41. doi: 10.1016/s0278-5846(01)00161-0. [DOI] [PubMed] [Google Scholar]
- 12.Fallon JH, Leslie FM. Distribution of dynorphin and enkephalin peptides in the rat brain. J Comp Neurol. 1986;249:293–336. doi: 10.1002/cne.902490302. [DOI] [PubMed] [Google Scholar]
- 13.Filliol D, Ghozland S, Chluba J, Martin M, Matthews HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- 14.Franck J, Lindholm S, Raaschou P. Modulation of volitional ethanol intake in the rat by central delta-opioid receptors. Alcohol Clin Exp Res. 1998;22(6):1185–9. [PubMed] [Google Scholar]
- 15.Froehlich JC, Zweifel M, Harts J, Lumeng L, Li TK. Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology. 1991;103(4):467–72. doi: 10.1007/BF02244246. [DOI] [PubMed] [Google Scholar]
- 16.Frost JJ, Mayberg HS, Sadzot B, Dannals RF, Lever JR, Ravert HT, et al. Comparison of [11C]diprenorphine and [11C]carfentanil binding to opiate receptors in humans by positron emission tomography. J Cereb Blood Flow Metab. 1990;10(4):484–92. doi: 10.1038/jcbfm.1990.90. [DOI] [PubMed] [Google Scholar]
- 17.Guthrie J, Basbaum A. Colocalization of immunoreactive proenkephalin and prodynorphin products in medullary neurons of the rat. Neuropeptides. 1984;4:437–445. doi: 10.1016/0143-4179(84)90087-8. [DOI] [PubMed] [Google Scholar]
- 18.Harlan RE, Shivers BD, Romano GJ, Howells RD, Pfaff DW. Localization of proenkephalin mRNA in the rat brain and spinal cord by in situ hybridization. J Comp Neurol. 1987;258:159–184. doi: 10.1002/cne.902580202. [DOI] [PubMed] [Google Scholar]
- 19.Hebb A, Zacharko RM, Gauthier M, Trudei F, LaForest S, Drolet G. Brief exposure to predator odor and resultant anxiety enhances mesocorticolimbic activity and enkephalin expression in CD-1 mice. Eur Neurosci. 2004;20:2415–18. doi: 10.1111/j.1460-9568.2004.03704.x. [DOI] [PubMed] [Google Scholar]
- 20.Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF. Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol. 2000;27(6):627–30. doi: 10.1016/s0969-8051(00)00125-6. [DOI] [PubMed] [Google Scholar]
- 21.Hirose N, Murakawa K, Takada K, Oi Y, Suzuki T, Nagase H, et al. Interactions among mu- and delta-opioid receptors, especially putative delta1- and delta2-opioid receptors, promote dopamine release in the nucleus accumbens. Neuroscience. 2005;135(1):213–25. doi: 10.1016/j.neuroscience.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 22.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–8. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 23.Hurd YL. Differential messenger RNA expression of prodynorphin and proenkephalin in the human brain. Neuroscience. 1996;72:767–783. doi: 10.1016/0306-4522(96)00002-4. [DOI] [PubMed] [Google Scholar]
- Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;148:64–73. doi: 10.1196/annals.1410.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 25.Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;148:64–73. doi: 10.1196/annals.1410.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khachaturian H, Lewis M, Watson SJ. Enkephalin systems in the diencephalon and brainstem of the rat. J Comp Neurol. 1983;220:310–320. doi: 10.1002/cne.902200305. [DOI] [PubMed] [Google Scholar]
- 27.Kinahan PE, Rogers JG. Analytic 3D image reconstruction using all detected events. Nuclear Science, IEEE Transactions on. 1989;36(1):964–968. [Google Scholar]
- 28.Konig M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A. Pain responses, anxiety and aggression in mice deficient in pre- proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- 29.Kranzler HR, Edenberg HJ. Pharmacogenetics of alcohol and alcohol dependence treatment. Curr Pharm Des. 2010;16(19):2141–8. doi: 10.2174/138161210791516387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnan-Sarin S, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben selectively attenuates alcohol intake in rats bred for alcohol preference. Pharmacol Biochem Behav. 1995;52(1):153–9. doi: 10.1016/0091-3057(95)00080-g. [DOI] [PubMed] [Google Scholar]
- 31.Lewanowitsch T, Irvine RJ. Naloxone and its quaternary derivative, naloxone methiodide, have differing affinities for mu, delta, and kappa opioid receptors in mouse brain homogenates. Brain Res. 2003;964(2):302–5. doi: 10.1016/s0006-8993(02)04117-3. [DOI] [PubMed] [Google Scholar]
- 32.Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10(5):740–7. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 33.Madar I, Lesser RP, Krauss G, Zubieta JK, Lever JR, Kinter CM, Ravert HT, Musachio JL, Mathews WB, Dannals RF, Frost JJ. Imaging of delta- and mu-opioid receptors in temporal lobe epilepsy by positron emission tomography. Ann Neurol 1997. 1997;41(3):358–67. doi: 10.1002/ana.410410311. [DOI] [PubMed] [Google Scholar]
- 34.Mangold D, Wand G. Cortisol and ACTH responses to naloxone in subjects with high and low neuroticism. Biol Psychi. 2006;60:850–855. doi: 10.1016/j.biopsych.2006.03.049. [DOI] [PubMed] [Google Scholar]
- 35.Mangold D, McCaul M, Ali M, Wand GS. Generating a dose response curve to naloxone in a single session. Biol Psychi. 2000;48:310–314. doi: 10.1016/s0006-3223(00)00885-4. [DOI] [PubMed] [Google Scholar]
- 36.Marinelli PW, Funk D, Harding S, Li Z, Juzytsch W, Lê AD. Roles of opioid receptor subtypes in mediating alcohol-seeking induced by discrete cues and context. Eur J Neurosci. 2009;(4):671–8. doi: 10.1111/j.1460-9568.2009.06851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meltzer CC, Bryan RN, Holcomb HH, Kimball AW, Mayberg HS, Sadzot B, et al. Anatomical localization for PET using MR imaging. J Comput Assist Tomogr. 1990;14(3):418–26. doi: 10.1097/00004728-199005000-00019. [DOI] [PubMed] [Google Scholar]
- 38.Méndez M, Morales-Mulia M. Role of mu and delta opioid receptors in alcohol drinking behaviour. Curr Drug Abuse Rev. 2008;1(2):239–52. doi: 10.2174/1874473710801020239. [DOI] [PubMed] [Google Scholar]
- 39.Micevych PE, Eckersell CB, Brecha N, Holland KL. Estrogen modulation of opioid and cholecystokinin systems in the limbic-hypothalamic circuit. Brain Res Bull. 1997;44(4):335–43. doi: 10.1016/s0361-9230(97)00212-8. [DOI] [PubMed] [Google Scholar]
- 40.Narita M, Kuzumaki N, Narita M, Kaneko C, Hareyama N, Miyatake M, et al. Chronic pain-induced emotional dysfunction is associated with astrogliosis due to cortical delta-opioid receptor dysfunction. J Neurochem. 2006;97(5):1369–78. doi: 10.1111/j.1471-4159.2006.03824.x. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen CK, Simms JA, Bito-Onon JJ, Li R, Ananthan S, Bartlett SE. The delta opioid receptor antagonist, SoRI-9409, decreases yohimbine stress-induced reinstatement of ethanol-seeking. Addict Biol. 2011 doi: 10.1111/j.1369-1600.2010.00295.x. Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oswald L, Wand G. Opioids and Alcohol. Physiol Behav. 2004;81(2):339–358. doi: 10.1016/j.physbeh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Petrusz P, Merchenthaler I, Maderdrut JL. Distribution of enkephalin-containing neurons in the central nervous system. In: Björklund A, Hökfelt T, editors. Handbook of Chemical Neuroanatomy. Elsevier Science; Amsterdam: 1985. pp. 273–334. [Google Scholar]
- 44.Portoghese PS, Sultana M, Takemori AE. Design of peptidomimetic delta opioid receptor antagonists using the message-address concept. J Med Chem. 1990;33(6):1714–20. doi: 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- 45.Radua J, Phillips M, Tamara R, Lawrence N, Marshall N, Kalidindi S, et al. Neural response to specific components of fearful faces in healthy and schizophrenic adults. NeuroImage. 2010;49 (1):939–946. doi: 10.1016/j.neuroimage.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 46.Roche DJ, Childs E, Epstein AM, King AC. Acute HPA axis response to naltrexone differs in female vs. male smokers. Psychoneuroendocrinology. 2010;35(4):596–606. doi: 10.1016/j.psyneuen.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schluger JH, Ho A, Borg L, Porter M, Maniar S, Gunduz M, Perret G, King A, Kreek MJ. Nalmefene causes greater hypothalamic-pituitary-adrenal axis activation than naloxone in normal volunteers: implications for the treatment of alcoholism. Alcohol Clin Exp Res. 1998:1430–6. doi: 10.1111/j.1530-0277.1998.tb03931.x. [DOI] [PubMed] [Google Scholar]
- 48.Skinner HA, Allen BA. Alcohol dependence syndrome: measurement and validation. J Abnorm Psychol. 1982;91(3):199–209. doi: 10.1037//0021-843x.91.3.199. [DOI] [PubMed] [Google Scholar]
- 49.Sobell LC, Sobell MB. Timeline followback: A technique for assessing self-reported alcohol consumption. In: Litten RZ, Allen J, editors. Measuring alcohol consumption: Psychosocial and biological methods. Humana Press; New Jersey: 1992. pp. 41–72. [Google Scholar]
- 50.Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar) Br J Addict. 1989;84(11):1353–7. doi: 10.1111/j.1360-0443.1989.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 51.Stephens M, Wand G. Stress and Alcohol use disorders: the role of glucocorticoids. National Institutes of Health’s Alcohol Health Research; 2011. In Press. [Google Scholar]
- 52.Szekely JL. Opioid peptides and stress. Crit Rev Neurobiol. 1990;6:1–12. [PubMed] [Google Scholar]
- 53.Titeler M, Lyon RA, Kuhar MJ, Frost JF, Dannals RF, Leonhardt S, et al. Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur J Pharmacol. 1989;167(2):221–8. doi: 10.1016/0014-2999(89)90582-7. [DOI] [PubMed] [Google Scholar]
- 54.Traynor JR, Hunter JC, Rodriguez RE, Hill RG, Hughes J. Delta-opioid receptor binding sites in rodent spinal cord. Br J Pharmacol. 1990;100(2):319–23. doi: 10.1111/j.1476-5381.1990.tb15802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uhart M, Chong R, Oswald L, Ping L, Wand GS. Gender differences in hypothalamic-pituitary-adrenal (HPa) axis reactivity. Psychoneuroendocrinol. 2006;31(5):642–52. 12. doi: 10.1016/j.psyneuen.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Wand GS. The influence of stress on the transition from drug use to addiction. National Institutes of Health’s Alcohol Health Res. 2008;31(2):119–36. [PMC free article] [PubMed] [Google Scholar]
- 57.Wand GS, Mangold D, Ali M. Adrenocorticotropin response to naloxone in sons of alcohol dependent men. J Clin Endocrinol Metab. 1999;84:64–68. doi: 10.1210/jcem.84.1.5373. [DOI] [PubMed] [Google Scholar]
- 58.Wand GS, Mangold D, El Diery S, McCaul M, Hoover D. Family history of alcoholism and hypothalamic opioidergic activity. Arch Gen Psychi. 1998;55:1114–1119. doi: 10.1001/archpsyc.55.12.1114. [DOI] [PubMed] [Google Scholar]
- 59.Wand GS, McCaul M, Gotjen D, Reynolds J, Lee S. Confirmation that offspring from families with alcohol dependent individuals have greater HPA axis activation-induced by naloxone compared to offspring without a family history of alcohol dependence. Alcohol: Clin Exp Res. 2001;25:1134–1139. [PubMed] [Google Scholar]
- 60.Wand GS, McCaul M, Gotjen D, Reynolds J, Lee S. The mu opioid receptor gene polymorphism A(118)G alters HPA axis activation induced by opioid receptor blockade. Neuropsychopharmacol. 2002;26:106–114. doi: 10.1016/S0893-133X(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 61.Wand G, Weerts E, Kuwabara H, Frost JJ, Xu X, McCaul ME. Naloxone-induced cortisol predicts mu opioid receptor binding potential in specific brain regions. Psychoneuroendocrinology. 2011 doi: 10.1016/j.psyneuen.2011.03.019. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Watts A. Ether anesthesia differentially affects the content of prepro-corticotropin-releasing hormone, prepro-neurotensin/neuromedin N and prepro-enkephalin mRNAs in the hypothalamic paraventricular nucleus of the rat. Brain Res. 1991;544:353–357. doi: 10.1016/0006-8993(91)90080-f. [DOI] [PubMed] [Google Scholar]
- 63.Watson SJ, Khachaturian H, Akil H, Coy DH, Goldstein A. Comparison of the distribution of dynorphin systems and enkephalin systems in brain. Science. 1982;218:1134–1136. doi: 10.1126/science.6128790. [DOI] [PubMed] [Google Scholar]
- 64.Weerts EM, Wand G, Kuwabara H, Munro CA, Dannals DF, Hilton J, Frost JJ, McCaul ME. PET imaging of mu- and delta-opioid receptor binding in alcohol dependent and healthy control subjects. Alcohol: Clin Exp Res. 2011 doi: 10.1111/j.1530-0277.2011.01565.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weerts E, Kim Y, Wand G, Dannals R, Lee J, Frost J, McCaul M. Differences in mu- and delta opioid receptor blockade measured by PET in Naltrexone-treated recently abstinent alcohol dependent subjects. Neuropsychopharmacology. 2008;33(3):653–65. doi: 10.1038/sj.npp.1301440. [DOI] [PubMed] [Google Scholar]
- 66.Wise RA. Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox Res. 2008;14(2–3):169–183. doi: 10.1007/BF03033808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Young E, Lightman M. Chronic stress elevates enkephalin expression in the rat paraventricular and supraoptic nuclei. Mol Brain Res. 1992;13:111–117. doi: 10.1016/0169-328x(92)90050-l. [DOI] [PubMed] [Google Scholar]