

Abstract
Although elevated levels of homocysteine (Hcy), known as hyperhomocysteinemia (HHcy), is associated with inflammatory bowel disease, the mechanism of Hcy action is unclear. In the present study we tested the hypothesis that HHcy activates matrix metalloproteinase-9 (MMP-9), which in turn enhances permeability of human intestinal microvascular endothelial cell (HIMEC) layer by decreasing expression of endothelial junction proteins and increasing caveolae formation. HIMECs were grown in Transwells and treated with 500 μM Hcy in the presence or absence of MMP-9 activity inhibitor. Hcy-induced permeability to FITC-conjugated bovine serum albumin (FITC-BSA) was assessed by measuring fluorescence intensity of solutes in the Transwells’ lower chambers. The cell-cell interaction and cell barrier function was estimated by measuring trans-endothelial electrical impedance. Confocal microscopy and flow cytometry were used to study cell junction protein expressions. Hcy-induced changes in transcellular transport of HIMECs were estimated by observing formation of functional caveolae defined as caveolae labeled by cholera toxin and antibody against caveolin-1 and one that have taken up FITC-BSA. Hcy instigated HIMEC monolayer permeability through activation of MMP-9. The increased paracellular permeability was associated with degradation of vascular endothelial cadherin and zona occludin-1 and transcellular permeability through increased caveolae formation in HIMECs. Elevation of Hcy content increases permeability of HIMEC layer affecting both paracellular and transcellular transport pathways, and this increased permeability was alleviated by inhibition of MMP-9 activity. These findings contribute to clarification of mechanisms of inflammatory bowel disease development.
Keywords: Inflammatory bowel disease, microvascular remodeling, paracellular transport, transcellular transport
INTRODUCTION
Hyperhomocysteinemia (HHcy) has been shown to be associated with inflammatory bowel disease (IBD) (Mahmood et al., 2005), predisposing a patient to the high risk of mesenteric arteriolar/venular thromboembolic disorders. IBD has also been related to mutations of methylenetetrahydrofolate reductase (MTHFR), a critical enzyme in the metabolism of folate and methionine, both of which are important factors in DNA methylation and synthesis (Stocco et al., 2006). Two genotypes of the MTHFR gene are prevalent in humans: 677T and 677C. The 677T genotype resulting in an A222V amino acid change generates a thermolabile protein with reduced catalytic activity. The 677T genotype is associated with a variety of disease outcomes, including an increased risk of vascular disease. Although a mutated MTHFR genotype was associated with increased toxicity of methotrexate treatment, the mutated genotype was not associated with an increased risk of toxicity during thiopurine treatment in IBD (Stocco et al., 2006). Microvascular permeability is the hallmark of IBD (Cromer et al., 2011). IBD patients have increased permeability in the colonic microvasculature leading to the leakage of plasma proteins and other macromolecules (Szepes et al., 1997). During the perpetuation of IBD condition, microvascular injury is ubiquitously present. Intestinal microvessels showed marked endothelial dysfunction (Hatoum et al., 2003). Microvascular endothelial cell (EC) layer comprises of closely apposed ECs that form a semi-permeable barrier between blood and tissue for the transport of proteins, electrolytes, and lipids. Therefore, microvascular barrier dysfunction plays a critical role in the initiation and progression of inflammation, posttraumatic complications, sepsis, ischemia-reperfusion injury, atherosclerosis, diabetes, and IBD. Thus, changes induced in endothelium leading to microvascular permeability play a fundamental role during IBD. However, the associated mechanisms of these changes remain obscure.
Homocysteine (Hcy) is a non-protein amino acid that is generated as a breakdown product of S-adenosyl methionine, the universal cellular one-carbon unit donor. Its concentration in serum is increased in response to inflammation (Wilcken & Wilcken, 1976) and oxidative stress, infection, and injury (Gao and Xue, 2003). It has a marked effect on both smooth muscle and endothelial cytoskeleton and intercellular junctions (Starkebaum and Harlan, 1986). Hcy is an agent that arrests the growth of EC and promotes smooth muscle cell proliferation (Tsai et al., 1994).
ECs form a homeostatic barrier that regulates vascular permeability to plasma components by two different pathways. The one is the paracellular pathway, first reported by Majno and Palade in 1961, in which transported material passes across endothelial tight junctions (Majno and Palade, 1961). The other is the transcellular pathway, in which transported material is taken up at the apical surface of a cell, transported across the cell, and released at the basal surface of the cell, or vice-versa (Vasile et al., 1983). Previously it was thought that transcellular and paracellular routes were two distinct and independent pathways. However, with recent advancement in the field, it is apparent that the two pathways are dynamically interacting parallel pathways (Van et al., 2007).
The endothelial junction plays a crucial role in maintaining tissue integrity and also vascular permeability, leukocyte extravasation, and angiogenesis (Wallez and Huber, 2008). Vascular endothelial cadherin (VE-cadherin), exclusively expressed in ECs, is characterized as an adherence junction protein. VE-cadherin is involved in a marked reorganization of actin cytoskeleton (Mehta and Malik, 2006). The cytoplasmic membrane surface sealing proteins such as Zona Occludins (ZO-1, ZO-2, and ZO-3) are scaffold proteins of the tight junction (Mehta and Malik, 2006). ZO-1 serves as an important linker between the tight junction proteins (TJPs) and the actin cytoskeleton and is thought to be a functionally critical component of cell tight junctions (Fanning et al., 1998). The decreased expression of Occludin (a TJP) and ZO-1 (a tight junction-associate protein) in EC junctions results in formation of gaps between the cells and in increase in paracellular permeability (Patibandla et al., 2009; Tada et al., 2010).
The transcellular pathway is mainly regulated by small flask shaped organelles called caveolae (Mehta and Malik, 2006). Caveolae are small invaginations of the cell surface and thought to play a critical role in cell surface signaling, endocytosis, and intracellular cholesterol transport (Rothberg et al., 1992; Silva et al., 1998). Caveolin-1 (Cav-1), a scaffolding protein constitutes a critical component of caveolae membrane and also functions as a “master regulator” of signaling molecules in caveolae (Minshall et al., 2003). It has been shown that Cav-1 co-localizes with MMP’s on cellular surface which facilitate directed proteolysis essential for early migratory and invasive processes (Phillips and Birnby, 2004).
Alteration of microvascular permeability is an index of early tissue injury that occurs in result of contraction of stimulated ECs. Acute inflammatory reaction accompanied by increased permeability of microvessels to macromolecules such as albumin, results in tissue edema (Persson, 1986). Albumin is the most abundant plasma protein that maintains the colloidal osmotic pressure and protects endothelial barrier integrity by its interaction with glycocalyx (Malik et al., 1989). Thus, changes in albumin transport through the EC layer may reflect significant damage to tissue and induce inflammatory responses. During a systemic inflammatory response, the increased vascular permeability is believed to occur in all organ beds including intestinal vessels (Dong et al., 2009).
Active MMP-9 is a key pathogenic factor that causes barrier disruption during colitis (Castaneda et al., 2005). We recently demonstrated that Hcy induced MMP-9 activation in the mesenteric arcade, a part of gut circulation (Munjal et al., 2011). Therefore, the following study was designed to show that acute treatment with Hcy enhances paracellular transport through changes in human intestinal microvascular endothelial cell (HIMEC) tight junction integrity and increases transcellular transport through an enhanced formation of caveolae, the both pathways being inhibited by blocking of MMP-9 activity.
MATERIALS AND METHODS
Antibodies and reagents
Inhibitor of MMP-9 activity (MMP-9 inhibitor I; catalogue # 444278) was purchased from EMD Biosciences, Inc. (La Jolla, CA). Antibodies against VE-cadherin and ZO-1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against Cav-1 was purchased from Novus (Littleton, CO). Fluorescein isothiocyanate-conjugated bovine serum albumin (FITC-BSA), cholera toxin (CTX), Alexa Fluor-594 Phalloidin, Alexa Fluor dye-conjugated secondary antibodies were from Invitrogen (Carlsbad, CA).
Human intestinal microvascular endothelial cell culture
Although the endothelium may play similar role in vascular contraction and relaxation, for some responses the organ specific mechanism may be unique to the specific endothelium (for example skeletal muscle versus cerebral microvascular permeability). Since we were addressing problems related to IBD, we tested effects of Hcy on intestinal ECs. HIMECs were generous gift from Professor Claudio Fiocchi (Department of Gastroenterology and Hepatology, Cleveland Clinic Foundation, Cleveland, OH). HIMECs were plated onto fibronectin-coated tissue culture dishes and grown in MCDB 131 medium (Sigma Chemical, St. Louis, MO) supplemented with 20% fetal bovine serum (FBS) and EC growth factor (ECGF; Boehringer Mannheim, Indianapolis, IN).
Gelatin zymography
Gelatin zymography was done according to the previously published protocol (Moshal et al., 2006). Briefly, 24 h before initiation of assay, complete medium was replaced with serum-free medium. At the time of sample preparation, media was aspirated, and samples were prepared using equal volumes of supernatants and a non-reducing sample buffer (0.5 M). Tris/HCl, pH 6.8, 10 % (w/v) SDS, 10 % (v/v) glycerol and 0.02 % (w/v) bromophenol blue. Samples were loaded at the same concentration on 10% SDS-PAGE gel containing 1% gelatin. The gels were washed with 2.5% Triton X-100 after electrophoresis and were incubated overnight in the developing buffer at 37oC to induce gelatin lysis. The resultant gelatinize activity was qualitatively determined by Coommassie counterstaining on Gel-Doc system (Bio-Rad).
Another method, in-tissue zymography, defines activities of all MMPs and mainly that of MMP-2 and MMP-9 (Mook et al., 2003). To determine role of MMP-9 activity in Hcy-induced effects with this method in the present study, use of specific inhibitor of MMP-9 is still necessary followed with analyses of differences between the treatment groups (Hcy-treated and Hcy-treated+MMP-9 activity inhibitor). Since we found that Hcy activates manly MMP-9 (Lominadze et al., 2006), to determine role of MMP-9 activity in Hcy-induced effects in the present study, specific inhibitor of MMP-9 was used. The specificity of this inhibitor was assured by the manufacturer and confirmed by our experiments (see Fig. 1, inset). Therefore, the effects presented in this study that were caused by the MMP-9 activity inhibitor were most likely to be a result of inhibition of MMP-9 activity.
Figure 1. Hcy-induced albumin leakage through the HIMEC monolayer.
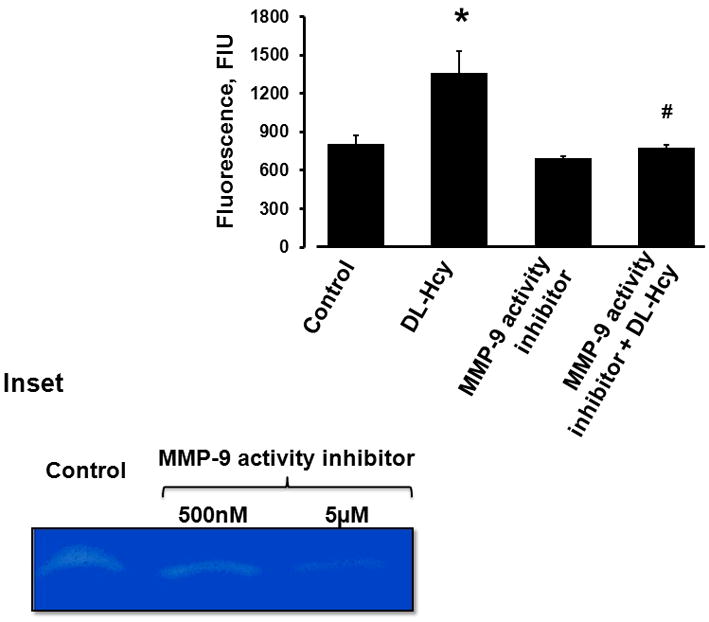
Fluorescence intensity values in samples from lower chambers of Transwells are presented. The cells were incubated for 1 hour with FITC-BSA in the presence of 500 μM Hcy alone (DL-Hcy), 500 μM Hcy and 5 μM of MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy), 5 μM MMP-9 activity inhibitor alone, and medium alone (control).
* P < 0.05 vs control, # P < 0.05 vs Hcy. n=4 for all groups.
Inset: In-gel gelatin zymography shows suppression in MMP-9 activity by MMP-9 activity inhibitor, which was more potent at 5 μM concentration than at the 500 nM concentration.
FITC-BSA permeability assay
Albumin leakage through HIMEC monolayer was assessed according to the method described previously (Tyagi et al., 2007). Transwell permeable supports (Corning, Corning, NJ) with polycarbonate membranes (Nuclepore Track-Etched membrane; 6.5 mm in diameter, 0.4-μm pore size, and pore density of 108/cm2) were coated with fibronectin for 1 h. The membranes were seeded with HIMECs and the cells were grown to confluence. To confirm the cell confluence and the presence of an intact monolayer on the membranes, cells in a separate test well (without membrane) with the same diameter were monitored by a light microscope. The permeability assay was done after confirming that the cells in the test wells and Transwells’ membranes were fully confluent and formed an intact monolayer. The surface levels of solutions in the luminal compartment (200 μL) and abluminal (lower chamber of a Transwell) compartment (650 μL) were the same.
For the permeability assay, cells were serum starved in plain medium and incubated with Hcy (500 μM) for 1 h. To determine a possible role of MMP-9 in Hcy-induced HIMEC permeability, cells were preconditioned with the specific inhibitor of MMP-9 activity (5 μM) followed by Hcy (500 μM). The dose of MMP-9 inhibitor was selected based on its functional activity determined during our preliminary experiments (see Figure 1, inset). Cells incubated with medium alone were used as a control group. Alexa Fluor 488-conjugated BSA (BSA-488, 3 mg/mL) was added to each of the wells described above at the time of Hcy addition.
The cells were incubated in humidified conditions at 37°C. After incubation, media samples were collected from lower and upper chambers. Extent of leaked BSA through a membrane was assessed by measuring fluorescence intensity in the samples with a microplate reader (SpectraMax M2; Molecular Devices, Sunnyvale, CA; excitation at 494 nm and emission at 520 nm). Experiments were performed five times in duplicate for each treatment. Results are expressed as fluorescence intensity units (FIU).
Transendothelial electrical impedance (TEEI) and capacitance (TEEC) measurements
The electrical cell-substrate impedance sensing system (ECIS; Applied Biophysics, Troy, NY) is designed to monitor the cellular behavior, their attachment to each other and adhesion to matrix, wound healing (cell migration), and cellular responses to drugs (Giaever and Keese, 1986). HIMECs were seeded in gold-covered ECIS microplate arrays (8W10E+) treated with electrode stabilizing solution followed by coating with fibronectin. Cells were allowed to grow for 12 hours. TEEI and TEEC of the cell layer were measured every 80 sec. The following groups were used: cells treated with Hcy, with Hcy and MMP-9 activity inhibitor, and MMP-9 activity inhibitor alone. Cells treated with medium alone were used as a control group. Average values for TEEI (collected at frequency of 8000 Hz) and TEEC (collected at frequency of 64000 Hz) for the last 30 min of observation before treatments were taken as baseline for each experimental group. TEEI and TEEC values for each experimental group were collected and plotted as relative (to baseline values) TEEI or TEEC (respectively) vs. time.
Filamentous actin (F-actin) formation
Formation of F-actin in cultured HIMEC cells was studied according to the method described earlier (Tyagi et al., 2008). HIMECs were grown until confluence in 8-well cover-glass plates coated with fibronectin. The cells were washed with phosphate buffered saline (PBS) and incubated with medium containing one of the following: Hcy (500 μM), preconditioned with MMP-9 activity inhibitor, followed by Hcy treatment (500 μM), at 37°C for 24hrs. Cells incubated with medium alone were used as a control. Medium was aspirated, and the cells were incubated with Alexa Fluor-594 Phalloidin for 30 min and fixed with paraformaldehyde (3.7 % in PBS) for 15 min at 4°C in dark.
Immunohistochemical analysis
For immunohistochemical staining, HIMECs were fixed with 4% formaldehyde for 15 minutes. After blocking with 2% BSA and permeabilization with and 0.05% Triton X-100, the samples were incubated with primary antibodies against ZO-1 and VE-cadherin for 1 hour at 4°C. After washing with PBS, the cells were incubated with fluorescein-conjugated respective secondary antibodies and counterstained with DAPI. Changes in expression of ZO-1 or VE-cadherin were observed with a laser-canning confocal microscope (Olympus FluoView FV1000; Tokyo, Japan).
Flow cytometric analysis of VE-cadherin expression in HIMECs
HIMECs were grown to subcon uence in six well plates and then treated with Hcy in the presence or absence MMP-9 activity inhibitor for 24 hours. After treatment cells were washed twice with PBS and harvested after incubation with 5 mmol/L ethylenediaminetetraacetic acid in Henck’s balanced salt solution for 5 to 10 minutes at 37°C. A single cell suspension of HIMECs was incubated with antibody against VE-cadherin followed by incubation with FITC-labeled secondary antibody for 30 minutes on ice. Cells were washed twice in PBS and fixed in 1% formaldehyde prepared in PBS. Changes in VE-cadherin cell surface expression were assessed by measuring fluorescence intensity of 10,000 cells using Accuri’s C6 Flow Cytometer (Accuri Cytometers, Ann Harbors, MI).
Formation of functional caveolae
In addition to Hcy-induced paracellular permeability, we sought to determine whether Hcy affects transcellular permeability; Hcy-induced formation of caveolae and uptake of FITC-BSA by caveolae was studied in cultured HIMECs, which were grouped as vehicle-treated (Figure 6A), Hcy (500 μM)-treated (Figure 6B), and MMP-9 activity inhibitor-preconditioned cells treated with (Figure 6D) or without Hcy (Figure 6C). Cells were incubated with FITC-BSA (50 μM) for 24 hours. To remove free FITC-BSA cells were washed four times with PBS, fixed, and were stained with Alexa Fluor-594-labeled CTX (1:1000 dilution) and Alexa Fluor-647-labeled antibody against Cav-1 (1:500 dilution). Changes in caveolae formation and up take of FITC-BSA by caveolae were observed with the Olympus laser-scanning confocal microscope (objective x100). CTX was used to label lipid vesicles and anti-Cav-1 antibody to label Cav-1 protein. Cav-1 is a main component of caveolae, the type of lipid vesicle, which can be present in the cellular membrane and its cytosol. Therefore, in the present study, it can be defined as co-localization of CTX and Cav-1. Co-localization of CTX, Cav-1, and FITC-BSA can indicate formation of functional caveolae that have taken up FITC-BSA.
Figure 6. Hcy-induced formation of functional caveolae in HIMECs.
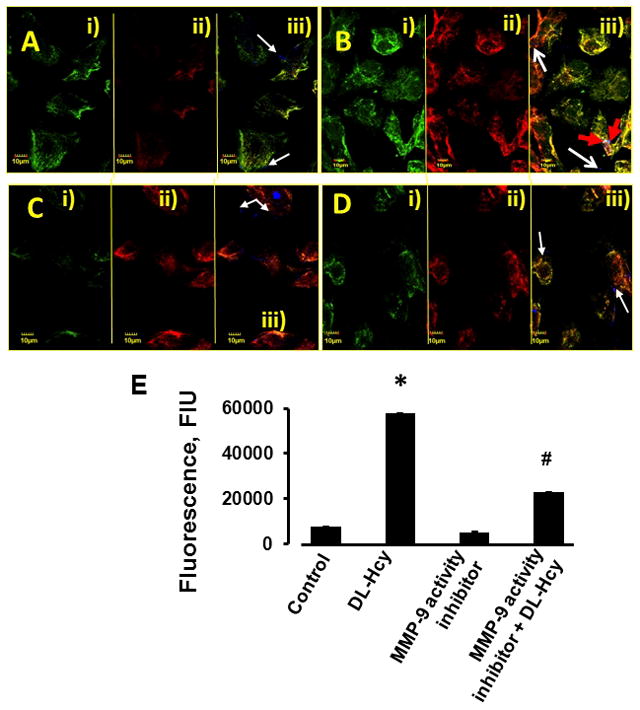
Confluent HIMECs were treated with: A) Medium alone (control); B) 500 μM Hcy (DL-Hcy); C) 5 μM of MMP-9 activity inhibitor alone; (MMP-9 activity inhibitor); and D) 500 μM Hcy in the presence of 5 μM of MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy). i) Cholera toxin (CTX)-labeled cell lipid vesicles (green); ii) Cell caveolin-1 (Cav-1) was labeled with antibody against Cav-1 (red), iii) Co-localization of CTX (green) and anti-Cav-1 antibody (red) defines caveolae (yellow). White arrows indicate free, fluorescently-labeled (blue color) bovine serum albumin (BSA) located in cell cytosol (iii), Red arrows indicate fluorescently-labeled BSA that was taken up by caveolae, which defines functional caveolae (cyan color) (B-iii).
E) Comparison of fluorescence intensity changes of CTX and Cav-1 as an indication of caveolae (shown in yellow - a result of co-localization of green and red) in HIMECs is presented by the histogram.
* P < 0.05 vs. control; # P < 0.05 vs. DL-Hcy. n=6 for all groups.
The confocal image quantification was carried out using Image-Pro Plus software and results were shown as fluorescence intensities of Cav-1 and CTX, Cav-1/CTX co-localization indicating caveolae (yellow), and expressed in fluorescence intensity units (FIU) (Figure 6E). Virtual colors were given for each dye using Olympus Flouview software; BSA is shown as blue dots in panel (iii), Cav-1 shown in red (ii), and CTX-B in green (i). Images showed that Hcy induced significant increase in Cav-1 expression (D ii); colocalization of Cav-1 and CTX, a marker of lipid vesicles (yellow) indicates caveolae formation, red arrows show BSA trapped in functional caveolae (cyan color) and untrapped BSA (blue dots) are shown with white arrows (D iii).
Statistical analysis
All data are presented as mean ± SEM. The experimental groups were compared by one-way analysis of variance (ANOVA). If ANOVA indicated a significant difference (P < 0.05), Tukey’s multiple comparison test was used to compare group means. Differences were considered significant if P < 0.05.
RESULTS
Hcy increases permeability of HIMEC layer through MMP-9 activation
We found that the content of FITC-BSA that leaked into the lower chambers of Hcy-treated Transwells in 1 hour was significantly greater (almost 60%) than that in control group (Figure 1). The treatment with MMP-9 activity inhibitor ameliorated Hcy-induced leakage defined by changes in cell layer permeability, which was 40% less than in the group treated by Hcy alone (Figure 1).
In-gel gelatin zymography indicated that higher dose (5 μM) of MMP-9 activity inhibitor almost completely blocked activation of MMP-9 in HIMECs (Figure 1, inset). Therefore, this dose of MMP-9 activity inhibitor was used in the present study.
MMP-9 activity inhibitor mitigated Hcy-mediated paracellular permeability
TEEI was significantly declined over the time when HIMECs were treated with Hcy alone compared to control group (Figure 2A). This decline was significantly abrogated when Hcy-treated HIMECs were preconditioned with specific inhibitor of MMP-9 activity (Figure 2A). Treatment of HIMECs with MMP-9 activity inhibitor alone did not change impedance values from that in control group (Figure 2A).
Figure 2. Hcy-induced changes in HIMEC layer integrity and attachment to the matrix.
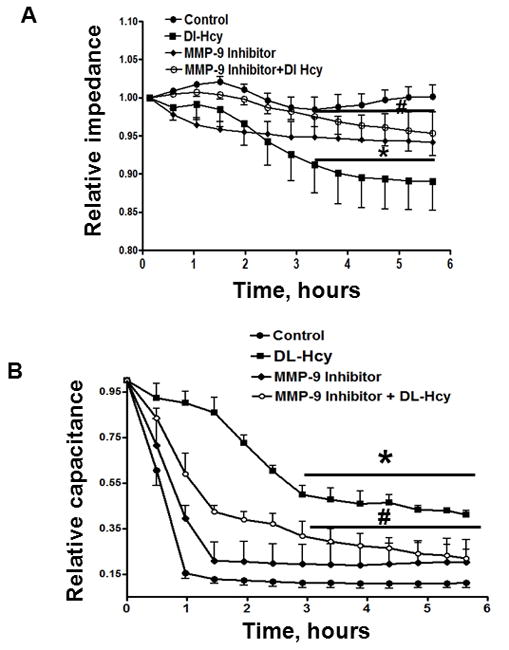
Confluent cells were treated with 500 μM Hcy alone (DL-Hcy), 500 μM Hcy and 5 μM of MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy), 5 μM MMP-9 activity inhibitor alone, and medium alone (control). A: Changes in TEEI (collected at 8000 Hz) are presented; B: Changes in TEEC (collected at 64000 Hz) are presented.
Relative TEEI (a measure of the cell layer integrity) was defined as ratio of the TEEI at the indicated time to the average TEEI determined during 30 min prior to experiment. Relative TEECI (a measure of cell attachment to the fibronectin matrix) was defined as ratio of the TEECI at the indicated time to the average TEEC determined during 30 min prior to experiment.
* P < 0.05 vs. control, # P < 0.05 vs. Hcy. n=8 for all experiments.
Over the period of 6 hours the TEEC sharply decreased in control group, vehicle-treated cells, and in cells treated with MMP-9 activity inhibitor alone indicating a tight attachment of cells to the matrix (Figure 2B). However, TEEC was higher in cells treated with Hcy indicating their weaker attachment to fibronectin matrix (Figure 2B). Pretreatment of the cells with MMP-9 activity inhibitor significantly improved the cell attachment to matrix (Figure 2B).
MMP-9 activity inhibitor mitigated Hcy-induced F-actin formation
Hcy caused a significant increase in F-actin formation in HIMEC (Figure 3). F-actin formation was almost 7 times greater than that in the vehicle treated cells (Figure 3). Cells pretreated with MMP-9 activity inhibitor and then treated with Hcy had a significantly lesser F-actin formation than the cells treated with Hcy alone (Figure 3). There was no significant differences in F-actin formation between the MMP-9 activity inhibitor pretreated cells and the cells treated with vehicle alone (Figure 3).
Figure 3. Hcy-induced formation of filamentous actin (F-actin) in HIMECs.
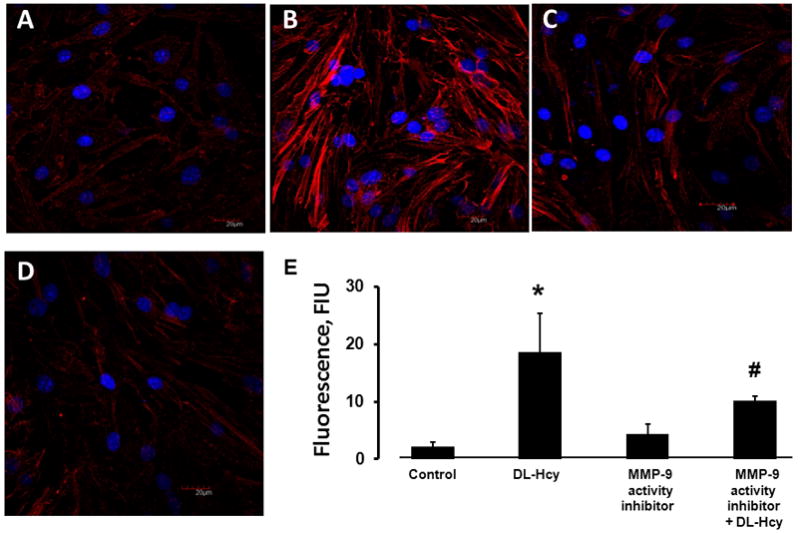
Confluent HIMECs were treated with: A) Medium alone (control); B) 500 μM Hcy (DL-Hcy); C) 5 μM of MMP-9 activity inhibitor alone; (MMP-9 activity inhibitor); and D) 500 μM Hcy in the presence of 5 μM of MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy). E) Comparison of fluorescence intensity changes of Alexa Fluor-594 Phalloidin (a measure of F-actin formation) in HIMECs is presented by the histogram.
* P < 0.05 vs. control, # P < 0.05 vs. DL-Hcy. n=4 for all groups.
Hcy-induced disruption of ZO-1 was MMP-9 activity dependent
Hcy caused a significant decrease in ZO-1 expression in HIMECs compared to that in vehicle treated control cells (Figure 4). Presence of MMP-9 activity inhibitor significantly improved Hcy-decreased ZO-1 expression (Figure 4). No difference in ZO-1 expression was found between the cells treated with MMP-9 activity inhibitor alone and the cells treated with vehicle alone (Figure 4).
Figure 4. Hcy-induced changes in expression of zona occludin (ZO-1) in HIMECs.
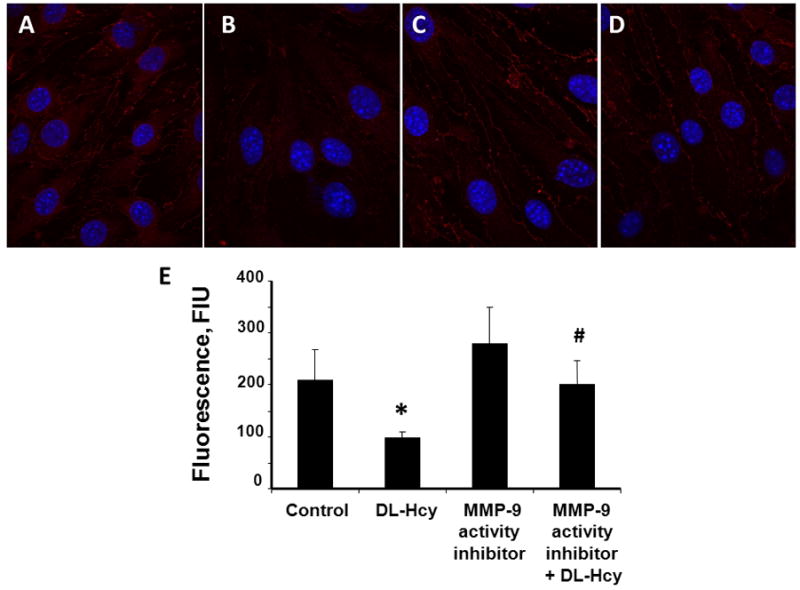
Confocal images show a visible decrease of ZO-1 expression in HIMECs after treatment with 500 μM Hcy (DL-Hcy) (B), which was significantly enhanced in the presence of 5 μM MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy) (D). No difference in ZO-1 expression was found between cells treated with medium alone (control) (A) and 5 μM of MMP-9 activity inhibitor alone (MMP-9 activity inhibitor) (C). E) Comparison of fluorescence intensity changes (a measure of ZO-1 expression) in HIMECs is presented by the histogram.
* P < 0.05 vs. control, # P < 0.05 vs. DL-Hcy. n=6 for all groups.
Hcy-induced VE-cadherin disruption was MMP-9 dependent
Hcy caused a significant decrease in VE-cadherin expression in HIMECs than in vehicle-treated cells (Figure 5). Presence of MMP-9 activity inhibitor with cells treated with Hcy had improved VE-cadherin expression around the cellular boundary, compared to the cells treated with Hcy alone (Figure 5). There was no significant difference in VE-cadherin expression between the cells treated with MMP-9 activity inhibitor alone and the vehicle-treated control cells (Figure 5). Surface expression of VE-cadherin on HIMECs detected by flow cytometry (Figure 5E) confirmed the results obtained by immunehistochemical analysis (Figure 5, A–D).
Figure 5. Hcy-induced changes in expression of vascular endothelial cadherin (VE-cadherin) in HIMECs.
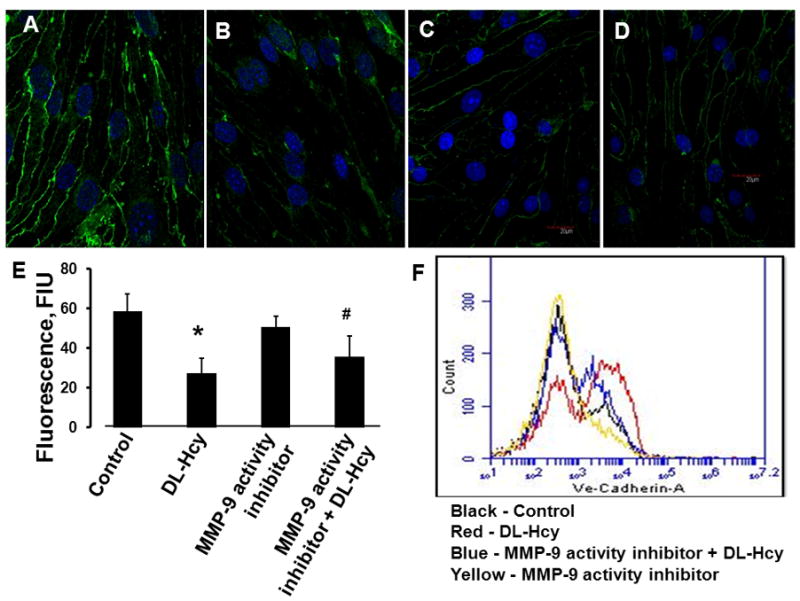
Confocal images show a visible decrease of VE-cadherin expression in HIMECs after treatment with 500 μM Hcy (DL-Hcy) (B), which was enhanced in the presence of 5 μM MMP-9 activity inhibitor (MMP-9 activity inhibitor + DL-Hcy) (D). No difference in VE-cadherin expression was found between cells treated with medium alone (control) (A) and 5 μM of MMP-9 activity inhibitor alone (MMP-9 activity inhibitor) (C). E) Comparison of fluorescence intensity changes (a measure of VE-cadherin expression) in HIMECs is presented by the histogram.
* P < 0.05 vs. control; # P < 0.05 vs. DL-Hcy. n=6 for all groups.
F) Changes in cell surface expression of VE-cadherin on HIMECs measured by flow cytometer confirm results obtained by immunohistochemical analysis and detected by confocal microscopy (see A, B, C, D, and E). Cell number is plotted on the Y axis and fluorescence intensity is plotted on the X axis (log scale). n=6 for all groups.
Hcy-induced increased transcellular permeability through caveolae formation was MMP-9 dependent
Changes in formation of functional caveolae induced by Hcy are presented in Figure 6. Hcy increased caveolae formation (Figure 6, B) compared to that in control cells (Figure 6, A). Although cell staining with CTX (Figure 6, Bi) and anti-Cav-1 antibody (Figure 6, Bii) were increased in Hcy-treated cell these formations cannot be positively considered as caveolae. Formation of caveolae was defined by co-localization of CTX (green) and anti-Cav-1 antibody (red) staining (Figure 6, Aiii, Biii, Ciii, and Diii). Hcy increased formation of caveolae (Figure 6, Biii) in comparison to that in control cells (Figure 6, Aiii). Hcy-induced increased formation of functional caveolae was detected as increased co-localization of CTX, anti-Cav-1 antibody, and FITC-BSA (blue) indicated by red arrows in Figure 6, Biii compared to that in control group (Figure 6, Aiii).Treatment of cells with MMP-9 activity inhibitor ameliorated effect of Hcy in formation of functional caveolae (Figure 6). MMP-9 activity inhibitor alone did not have effect on formation of functional caveolae in HIMECs (Figure 6).
DISCUSSION
In the present study we addressed the molecular mechanism that may be involved in Hcy-mediated increased paracellular and transcellular transport pathways. Since we found earlier that Hcy induces greater activations of MMP-9 than that of MMP-2 in brain vascular tissue (Lominadze et al., 2006), the main objective of the present study was to evaluate if Hcy-mediated MMP-9 activation induces EC permeability affecting either paracellular or transcellular pathways. It is well documented that Hcy causes MMP activation that is further known to cause increased microvascular permeability (Bonoiu et al., 2009; Lominadze et al., 2006). In our recent report we showed that Hcy induced gut vascular (mesenteric artery) remodeling by activating MMP-9 (Munjal et al., 2011). However, mechanism of its action on intestinal microvascular ECs has not been yet established. Here, we attempted to separate paracellular and transcellular transport pathways involved in Hcy-induced increased permeability of HIMEC.
As a normal level of Hcy in serum is below 13 μM, the ranges of HHcy have been referred to as: moderate (16 to 30 μM), intermediate (31 to 100 μM) or severe (> 100 μM) (Ji and Kaplowitz, 2004). The Hcy concentration (500 μM) used in the present study would correspond to conditions of severe HHcy. Previous reports from our lab showed that Hcy (50 μM) increased albumin leakage through brain EC monolayer over the period of 12 hour (Tyagi et al., 2007). In the present study, we showed that Hcy (500 μM) induced albumin leakage in an hour in HIMEC. Therefore, the underlying principle behind the efficacy of lower dose of Hcy is due to prolonged exposure and heterogeneity of response of ECs from different organs.
Activation of MMP-9 has been implicated in numerous pathological conditions such IBD (Garg et al., 2009), heart failure (Givvimani et al., 2011), and stroke (Rosell et al., 2006). Furthermore, MMP-9 inhibition has a therapeutic effect in treating stroke-induced vascular damage, brain edema and blood brain barrier disruption (Barr et al., 2010). In-addition, previous reports suggested MMP-9 activity inhibition may be beneficial in the treatment of colitis (Garg et al., 2009).
MMP-9, activated by extracellular growth factors, induced down-regulation of VE-cadherin expression in ovarian cancer cell line (Cowden Dahl et al., 2008). Incubation of isolated microvessels with purified MMP-9 caused degradation of a tight junction protein, occludin, and inhibition of MMP-9 activity prevented occludin protein loss in microvessels (Liu et al., 2009). Furthermore, inhibition of MMP activity by a metalloproteinase activity inhibitor prevented oxidative stress-induced TJP degradation and reduced the intercellular gap formation in brain ECs (Lischper et al., 2010). The results from the current study showed that Hcy-induced activation of MMP-9 caused disruption of tight junction-associated protein ZO-1 and this alteration in ZO-1 expression was mitigated by the presence of MMP-9 activity inhibitor. Effect of Hcy-activated MMP-9 on cell-cell interaction was confirmed by changes in TEEI of HIMECs induced by Hcy. Thus, these data suggest that Hcy affects paracellular pathway by disruption of TJPs mediated by MMP-9 activation.
MMP-9 activation inhibited cell attachment and wound healing in the intestinal epithelial cell line suggesting crucial role of MMP-9 in development of colitis by modulating cell-matrix interaction (Castaneda et al., 2005). These results were consistent with our present finding which shows that Hcy mediated MMP-9 activation caused weaker attachment defined by increase in TEEC of HIMEC layer.
Our study on HIMEC layer integrity (TEEI) and attachment (TEEC) to subendothelial matrix showed that Hcy effect and the effect of MMP-9 activity inhibition become significant after 3.5 hours of treatment (data were averaged from 8 experiments). However, differences in TEEC were more pronounced than differences in TEEI. These results suggest that Hcy affects the cellular attachment to subendothelial matrix (in our case fibronectin) more than it affects the cell-cell interaction. This may occur because of Hcy-activated MMP-9 affects the cell-matrix interaction first and then targets cell-cell junctions. To address this hypothesis further studies are necessary. These studies are in progress.
Cytoskeleton remodeling as a result of stress or other physiochemical factors is a key process required by many cell types for coupling to signaling pathways involved in retaining the cellular viability. It is known that F-actin is formed in ECs in-vitro and in-situ but its formation increases in response to shear stress. Actin disassembly from the cytoskeleton contributes to loss of EC barrier function (Boardman et al., 2004). Actin remodeling by HHcy, as we observed in the current study in the HIMECs suggests the involvement of the actin reorganization in increasing permeability of the cellular monolayer. This, in-turn, may cause stiffening of the cells and opening of inter-endothelial junctions that can contribute to abnormalities in the gut microvasculature.
Protein transport by caveolae has been reported to play a significant role in maintaining endothelial barrier properties and normal oncotic pressure gradient across the vessel wall (Mehta and Malik, 2006). Caveolae, first found in the ECs, are cholesterol- and sphingolipid-rich smooth invaginations of the plasma membrane involved in non-clathrin dependent endocytosis (Smith et al., 1973). Cav-1, a hetromeric oligomeric protein, is the defining protein constituent of caveolae (Lisanti et al., 1993). Previous reports demonstrated marked increase in the number of caveolae that in turn contributed to trans-endothelial albumin permeability (Tiruppathi et al., 2008) and decreased expression of eNOS as a result of lipopolysaccharides (LPS) exposure (Kamoun et al., 2006). The increased caveolae formation was evident by both the number of plasmalemmal-associated vesicles and free cytosolic vesicles. Interestingly, it was shown that interaction of eNOS with Cav-1 scaffolding domain appears to result in inhibition of NOS activity (Feron et al., 1998). A direct relationship has been observed between the expression of Cav-1 in ECs and the inhibition of NO release (Fulton et al., 2001). We have also shown that Hcy decreased eNOS expression in gut microvasculature (Munjal et al., 2011). Previous reports showed that Hcy induced albumin leakage from brain pial vessels through formation of endothelial gaps (paracellular pathway) mediated by MMP-9 activation (Lominadze et al., 2006). However, at that time we did investigate other possible routes of albumin transport that might be disrupted by the elevated Hcy (Lominadze et al., 2006). Our current findings demonstrate that Hcy increased transcellular permeability indicated by an increased formation of functional caveolae.
Furthermore to distinguish between membrane associated caveolae and cytosolic caveolae, HIMECs were treated with CTX (binds to lipid rafts) and labeled with antibody against Cav-1, to identify Cav-1 which mediates the internalization of sphingolipids and sphingolipid-binding toxins, such as CTX (Singh et al., 2003). Previous reports showed genetic deletion or pharmacologic inhibition of endothelial Cav-1 functions resulted in attenuation of IBD condition (Chidlow et al., 2009). However, these reports were unable to establish precise pathophysiologic mechanisms underlying the role of endothelial Cav-1 during experimental colitis. In the present study, we have demonstrated that Hcy leads to upregulation of Cav-1 and Cav-1/CTX staining representing functional caveolar rafts with increased BSA uptake in HIMECs compared to that in vehicle-treated control cells. This Hcy-induced increase in functional caveolae was abrogated in cells preconditioned with MMP-9 activity inhibitor, suggesting the role of MMP-9 activation in Hcy-induced EC layer permeability. It has been also demonstrated that phosphorylation of Cav-1 plays a crucial role in oxidative stress-induced pulmonary vascular hyperpermeability via transcellular and paracellular pathways (Sun et al., 2009). The microvasculature contributes to chronic inflammatory process in the gut through altered leukocyte recruitment, impaired perfusion, and angiogenesis further leading to tissue remodeling (Cromer et al., 2011).
During inflammation, enhanced accumulation of proteins in subendothelial matrix will increase edema formation in result of enhanced water transport from microvessels. Caveolae is the primary transcellular carrier for plasma components and particularly large proteins. To demonstrate the role of Hcy in transcellular transport mechanism, we showed here that there are also changes in caveolae formation following an increase in Hcy levels and the resultant activation of MMP-9. Effect of MMP-9 activity on caveolae transport has been shown previously (Phillips and Birnby, 2004).
In conclusion, Hcy had plethora of functional implications in terms of the paracellular and transcellular transport. It enhanced paracellular transport through changes in inter-endothelial tight junction integrity and increased trans-endothelial transport mediated by caveolae formation in HIMECs. These effects of Hcy were inhibited by the specific inhibitor of MMP-9 activity.
Limitation
Hcy is associated with a number of diseases with vascular involvement and Hcy levels can be reduced by B-vitamin supplementation in vivo without any clinical benefit for the patient (Ebbing et al., 2008). Thus, it seems that the precise role of Hcy in vivo is controversial. However, these studies did not measure the tissue levels of Hcy. Previously, we demonstrated robust increase in the tissue levels of Hcy in coronary artery disease (Tyagi et al., 1998).
Although it is possible to use siRNA against MMP-9 to mitigate MMP-9-mediated permeability, we showed that addition of specific inhibitor of MMP-9 attenuates Hcy-induced permeability of ECs. We previously showed that expression and/or activity of MMP-9 and MMP-2 were upregulated following the Hcy treatment. Importantly, the activity of MMP-9 was much greater than that of MMP-2 (Lominadze et al., 2006). The general inhibitor of MMPs, GM6001 mitigated the Hcy-induced permeability (Lominadze et al., 2006). These results suggested the prevailing role of MMP-9 over MMP-2 or other MMPs. However, specific role of MMP-9 was not clearly defined. Therefore, in the present study, to target exclusively MMP-9 we used its specific inhibitor.
Thus, we were able to demonstrate that Hcy-induced paracellular and transcellular permeability involved activation of MMP-9. However, we were unable to answer the question which pathway was affected the most by Hcy treatment. We also did not address the question related to Hcy-associated cross talk between the two paracellular and transcellular pathways.
Figure 7. Schematic representation of Hcy-induced mechanism of increased endothelial cell layer permeability.
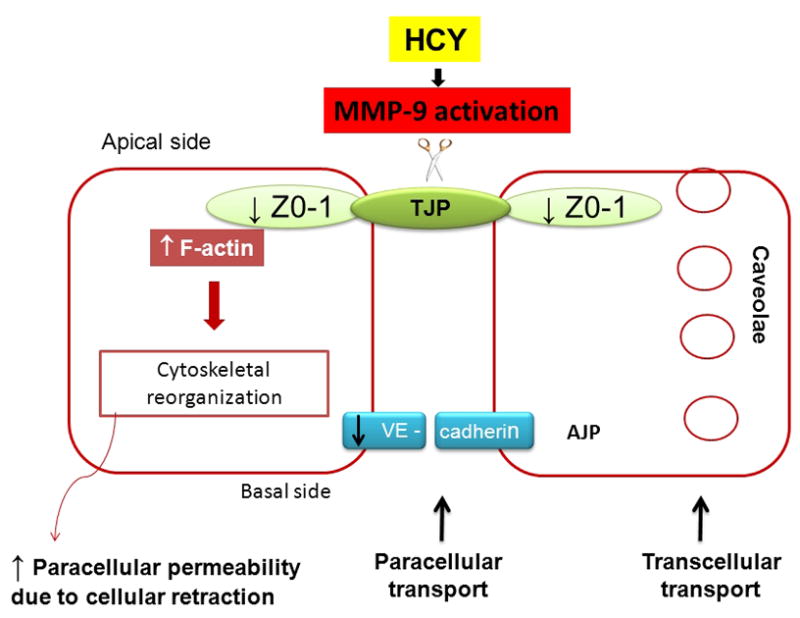
Hcy, through activation of MMP-9, affects transcellular transport by increasing the caveolae formation and paracellular transport by disrupting tight junction-associated protein ZO-1, adherence junction protein, VE-cadherin, and increases formation of F-actin that further causes cellular retraction.
Acknowledgments
Financial Support:
Contract grant sponsor: NIH; Contract grant number: HL-71010 (to SCT).
Contract grant sponsor: NIH; Contract grant number: HL-74185 (to SCT).
Contract grant sponsor: NIH; Contract grant number: NS-51568 (to SCT).
Contract grant sponsor: NIH; Contract grant number: HL-80394 (to DL).
Reference List
- Barr TL, Latour LL, Lee KY, Schaewe TJ, Luby M, Chang GS, El-Zammar Z, Alam S, Hallenbeck JM, Kidwell CS, Warach S. Blood-brain barrier disruption in humans is independently associated with increased matrix metalloproteinase-9. Stroke. 2010;41(3):e123–e128. doi: 10.1161/STROKEAHA.109.570515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman KC, Aryal AM, Miller WM, Waters CM. Actin re-distribution in response to hydrogen peroxide in airway epithelial cells. J Cell Physiol. 2004;199(1):57–66. doi: 10.1002/jcp.10451. [DOI] [PubMed] [Google Scholar]
- Bonoiu A, Mahajan SD, Ye L, Kumar R, Ding H, Yong KT, Roy I, Aalinkeel R, Nair B, Reynolds JL, Sykes DE, Imperiale MA, Bergey EJ, Schwartz SA, Prasad PN. MMP-9 gene silencing by a quantum dot-siRNA nanoplex delivery to maintain the integrity of the blood brain barrier. Brain Res. 2009;1282:142–155. doi: 10.1016/j.brainres.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, Rojas M, Wang L, Oprea G, Garg P, Gewirtz AT, Roman J, Merlin D, Sitaraman SV. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129(6):1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Greer JJ, Anthoni C, Bernatchez P, Fernandez-Hernando C, Bruce M, Abdelbaqi M, Shukla D, Granger DN, Sessa WC, Kevil CG. Endothelial caveolin-1 regulates pathologic angiogenesis in a mouse model of colitis. Gastroenterology. 2009;136(2):575–584. doi: 10.1053/j.gastro.2008.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowden Dahl KD, Symowicz J, Ning Y, Gutierrez E, Fishman DA, Adley BP, Stack MS, Hudson LG. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008;68(12):4606–4613. doi: 10.1158/0008-5472.CAN-07-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer WE, Mathis JM, Granger DN, Chaitanya GV, Alexander JS. Role of the endothelium in inflammatory bowel diseases. World J Gastroenterol. 2011;17(5):578–593. doi: 10.3748/wjg.v17.i5.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong GH, Wang CT, Li Y, Xu B, Qian JJ, Wu HW, Jing H. Cardiopulmonary bypass induced microcirculatory injury of the small bowel in rats. World J Gastroenterol. 2009;15(25):3166–3172. doi: 10.3748/wjg.15.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbing M, Bleie Ø, Ueland PM, Nordrehaug JE, Nilsen DW, Vollset SE, Refsum H, Ringdal Pedersen EK, Nygård O. Mortality and Cardiovascular Events in Patients Treated With Homocysteine-Lowering B Vitamins After Coronary Angiography. JAMA: The Journal of the American Medical Association. 2008;300(7):795–804. doi: 10.1001/jama.300.7.795. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. JBiolChem. 1998;273(45):29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- Feron O, Saldana F, Michel JB, Michel T. The endothelial nitric-oxide synthase-caveolin regulatory cycle. JBiolChem. 1998;273(6):3125–3128. doi: 10.1074/jbc.273.6.3125. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299(3):818–824. [PubMed] [Google Scholar]
- Gao J, Xue A. Study on the oxidative injury of ECV304 cell induced by homocysteine. Wei Sheng Yan Jiu. 2003;32(1):20–21. [PubMed] [Google Scholar]
- Garg P, Vijay-Kumar M, Wang L, Gewirtz AT, Merlin D, Sitaraman SV. Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G175–184. doi: 10.1152/ajpgi.90454.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever I, Keese CR. Use of electric fields to monitor the dynamical aspect of cell behavior in tissue culture. IEEE Trans Biomed Eng. 1986;33(2):242–247. doi: 10.1109/TBME.1986.325896. [DOI] [PubMed] [Google Scholar]
- Givvimani S, Munjal C, Gargoum R, Sen U, Tyagi N, Vacek JC, Tyagi SC. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J Appl Physiol. 2011 doi: 10.1152/japplphysiol.01064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum OA, Binion DG, Otterson MF, Gutterman DD. Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology. 2003;125(1):58–69. doi: 10.1016/s0016-5085(03)00699-1. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World Journal of Gastroenterology. 2004;10(12):1699–1708. doi: 10.3748/wjg.v10.i12.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun WS, Karaa A, Kresge N, Merkel SM, Korneszczuk K, Clemens MG. LPS inhibits endothelin-1-induced endothelial NOS activation in hepatic sinusoidal cells through a negative feedback involving caveolin-1. Hepatology. 2006;43(1):182–190. doi: 10.1002/hep.20940. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Tang ZL, Sargiacomo M. Caveolin forms a hetero-oligomeric protein complex that interacts with an apical GPI-linked protein: implications for the biogenesis of caveolae. J Cell Biol. 1993;123(3):595–604. doi: 10.1083/jcb.123.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010;1326:114–127. doi: 10.1016/j.brainres.2010.02.054. [DOI] [PubMed] [Google Scholar]
- Liu W, Hendren J, Qin XJ, Shen J, Liu KJ. Normobaric hyperoxia attenuates early blood-brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. JNeurochem. 2009;108(3):811–820. doi: 10.1111/j.1471-4159.2008.05821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. AmJPhysiol Heart CircPhysiol. 2006;290(3):H1206–H1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood A, Needham J, Prosser J, Mainwaring J, Trebble T, Mahy G, Ramage J. Prevalence of hyperhomocysteinaemia, activated protein C resistance and prothrombin gene mutation in inflammatory bowel disease. EurJGastroenterolHepatol. 2005;17(7):739–744. doi: 10.1097/00042737-200507000-00008. [DOI] [PubMed] [Google Scholar]
- Majno G, Palade GE. Studies on inflammation. 1. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AB, Lynch JJ, Cooper JA. Endothelial barrier function. JInvest Dermatol. 1989;93(2 Suppl):62S–67S. doi: 10.1111/1523-1747.ep12581072. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. AmJPhysiol Lung Cell MolPhysiol. 2003;285(6):L1179–L1183. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- Mook ORF, Overbeek CV, Ackema EG, Maldegem FV, Frederiks WM. In Situ Localization of Gelatinolytic Activity in the Extracellular Matrix of Metastases of Colon Cancer in Rat Liver Using Quenched Fluorogenic DQ-gelatin. Journal of Histochemistry & Cytochemistry. 2003;51(6):821–829. doi: 10.1177/002215540305100613. [DOI] [PubMed] [Google Scholar]
- Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N, Zhang H, Tyagi SC. Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. AmJPhysiol Heart CircPhysiol. 2006;291(6):H2825–H2835. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- Munjal C, Givvimani S, Qipshidze N, Tyagi N, Falcone JC, Tyagi SC. Mesenteric vascular remodeling in hyperhomocysteinemia. Molecular and Cellular Biochemistry. 2011;348(1–2):99–108. doi: 10.1007/s11010-010-0643-y. [DOI] [PubMed] [Google Scholar]
- Patibandla PK, Tyagi N, Dean WL, Tyagi SC, Roberts AM, Lominadze D. Fibrinogen induces alterations of endothelial cell tight junction proteins. Journal of Cellular Physiology. 2009;221(1):195–203. doi: 10.1002/jcp.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson CC. The role of microvascular permeability in the pathogenesis of asthma. EurJRespirDisSuppl. 1986;144:190–216. [PubMed] [Google Scholar]
- Phillips PG, Birnby LM. Nitric oxide modulates caveolin-1 and matrix metalloproteinase-9 expression and distribution at the endothelial cell/tumor cell interface. AmJPhysiol Lung Cell MolPhysiol. 2004;286(5):L1055–L1065. doi: 10.1152/ajplung.00262.2003. [DOI] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabín J, Fernández-Cadenas I, Ribó M, Molina CA, Lo EH, Montaner J. Increased Brain Expression of Matrix Metalloproteinase-9 After Ischemic and Hemorrhagic Human Stroke. Stroke. 2006;37(6):1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68(4):673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Silva W, Maldonado H, Chompre G, Mayol N. Caveolae a new subcellular transport organelle. BolAsocMedPR. 1998;90(1–3):30–33. [PubMed] [Google Scholar]
- Singh RD, Puri V, Valiyaveettil JT, Marks DL, Bittman R, Pagano RE. Selective caveolin-1-dependent endocytosis of glycosphingolipids. MolBiolCell. 2003;14(8):3254–3265. doi: 10.1091/mbc.E02-12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U, Ryan JW, Smith DS. Freeze-etch studies of the plasma membrane of pulmonary endothelial cells. J Cell Biol. 1973;56(2):492–499. doi: 10.1083/jcb.56.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest. 1986;77(4):1370–1376. doi: 10.1172/JCI112442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocco G, Martelossi S, Sartor F, Toffoli G, Lionetti P, Barabino A, Fontana M, Decorti G, Bartoli F, Giraldi T, Ventura A. Prevalence of methylenetetrahydrofolate reductase polymorphisms in young patients with inflammatory bowel disease. DigDisSci. 2006;51(3):474–479. doi: 10.1007/s10620-006-3158-3. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. CircRes. 2009;105(7):676–685. 615. doi: 10.1161/CIRCRESAHA.109.201673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szepes Z, Kiss J, Molnar T, Lamarque D, Jancso G, Laszlo F. Capsaicin-sensitive mechanisms in the modulation of rat colonic vascular permeability under physiological and pathological conditions. JPhysiol Paris. 1997;91(3–5):123–126. doi: 10.1016/s0928-4257(97)89475-2. [DOI] [PubMed] [Google Scholar]
- Tada Y, Yagi K, Kitazato KT, Tamura T, Kinouchi T, Shimada K, Matsushita N, Nakajima N, Satomi J, Kageji T, Nagahiro S. Reduction of endothelial tight junction proteins is related to cerebral aneurysm formation in rats. JHypertens. 2010;28(9):1883–1891. doi: 10.1097/HJH;0b013e32833c2273. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Shimizu J, Miyawaki-Shimizu K, Vogel SM, Bair AM, Minshall RD, Predescu D, Malik AB. Role of NF-kappaB-dependent caveolin-1 expression in the mechanism of increased endothelial permeability induced by lipopolysaccharide. JBiolChem. 2008;283(7):4210–4218. doi: 10.1074/jbc.M703153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Perrella MA, Yoshizumi M, Hsieh CM, Haber E, Schlegel R, Lee ME. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proceedings of the National Academy of Sciences. 1994;91(14):6369–6373. doi: 10.1073/pnas.91.14.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Moshal KS, Tyagi SC, Lominadze D. gamma-Aminbuturic acid A receptor mitigates homocysteine-induced endothelial cell permeability. Endothelium. 2007;14(6):315–323. doi: 10.1080/10623320701746164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem. 2008;307(1–2):13–22. doi: 10.1007/s11010-007-9579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi SC, Smiley LM, Mujumdar VS. Reduction-oxidation (Redox) and vascular tissue level of homocyst(e)ine in human coronary atherosclerotic lesions and role in extracellular matrix remodeling and vascular tone. Molecular and Cellular Biochemistry. 1998;181(1):107–116. doi: 10.1023/a:1006882014593. [DOI] [PubMed] [Google Scholar]
- Van DW, Kreindler JL, Malik AB, Margulies S, Lewis SA, Kim KJ. Interrelations/cross talk between transcellular transport function and paracellular tight junctional properties in lung epithelial and endothelial barriers. AmJPhysiol Lung Cell MolPhysiol. 2007;293(3):L520–L524. doi: 10.1152/ajplung.00218.2007. [DOI] [PubMed] [Google Scholar]
- Vasile E, Simionescu M, Simionescu N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. JCell Biol. 1983;96(6):1677–1689. doi: 10.1083/jcb.96.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. BiochimBiophysActa. 2008;1778(3):794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]