

Abstract
Cell-type-specific expression of optogenetic molecules allows temporally precise manipulation of targeted neuronal activity. Here we present a toolbox of 4 knock-in mouse lines engineered for strong, Cre-dependent expression of channelrhodopsins ChR2-tdTomato and ChR2-EYFP, halorhodopsin eNpHR3.0, and archaerhodopsin Arch-ER2. All 4 transgenes mediate Cre-dependent, robust activation or silencing of cortical pyramidal neurons in vitro and in vivo upon light stimulation, with ChR2-EYFP and Arch-ER2 demonstrating light sensitivity approaching that of in utero or virally transduced neurons. We further show specific photoactivation of parvalbumin-positive interneurons in behaving ChR2-EYFP reporter mice. The robust, consistent, and inducible nature of our ChR2 mice represents a significant advancement over previous lines, whereas the Arch-ER2 and eNpHR3.0 mice are the first demonstration of successful conditional transgenic optogenetic silencing. When combined with the hundreds of available Cre-driver lines, this optimized toolbox of reporter mice will enable widespread investigations of neural circuit function with unprecedented reliability and accuracy.
Introduction
A major challenge in neuroscience is to understand how brain functions are mediated by particular cell types within neural networks. Dissection of such complex networks requires the ability to manipulate the activities of specific cell types and to examine the resulting effects. One of the most exciting recent innovations in experimental neuroscience has been the development of light-activated channels or pumps, derived from microbial photosynthetic systems, to modulate neural activity, known as optogenetics. The best-known prototypes for the application of optical control in neurons include the neural-activating cation channel, channelrhodopsin-2 (ChR2) 1-3, and the neural-silencing chloride transporter halorhodopsin (NpHR) 4, 5 and proton pump archaerhodopsin-3 (Arch) 6. These and related optogenetic molecules 7 allow activation or silencing of neurons with unprecedented specificity and excellent temporal precision on a millisecond scale, and are in widespread use.
In mice, cell-type-specific genetic manipulation is most widely achieved through the Cre/lox recombinase system. Hundreds of Cre mouse lines, generated in individual labs and through large-scale efforts 8-10, have been established to direct specific gene expression or deletion in a wide range of cell types or populations throughout the nervous system. A major challenge to developing optogenetic tools is the need to express high levels of the opsins, due to the relatively small optical current mediated by each opsin molecule. For this reason, opsin genes have been most often introduced in vivo using recombinant viral vectors or by in utero electroporation (IUE). By delivering an adeno-associated virus (AAV) that expresses an opsin in a Cre-dependent manner, e.g. using floxed-stop or floxed-inverse (FLEX) cassettes 11, 12, it is possible to virally deliver an opsin to a brain region, where only Cre-positive cells will activate expression of the opsin. Although successful for many applications, these approaches possess intrinsic limitations. They can result in incomplete coverage of neurons within the region, which may limit experiments requiring complete labeling (e.g. neural silencing), and variable opsin expression levels across cells from the injection center out. The variability in the number and location of opsin-expressing cells between animals necessitates laborious validation for each animal, introducing variability in data interpretation. The brain targets of interest may be very small, very large, or hard for the virus to access for other reasons (e.g. a difficult location for injection, a particular cell type that cannot be infected by any serotype of virus, or during brain development).
A transgenic mouse approach can overcome many of these limitations. However, exogenous opsin gene expression in transgenics is typically regulated by a specific linked promoter, and so, is predetermined to occur in a particular and fixed cell population 13-19. A Cre-dependent system of opsin expression would exploit the abundant resource available Cre-driver lines constitute and would offer a powerful approach for controlling the activity of a wide range of cells. However, transgenic mice with robust Cre-dependent expression of opsins have been difficult to generate, as indicated by recent characterization of a ChR2 Cre-reporter 20. To date, no transgenic line with Cre-dependent expression of a silencing opsin has been described. Efforts to overcome the limitations of early versions of silencing opsins, such as protein aggregation and low conductance 21, has led to the development of newer optical silencing molecules, including eNpHR 22, eNpHR3.0 23, and various forms of Arch 6, 24, with great improvement in membrane expression and related increase in photoconductance, making reliable genetic silencing achievable now.
Here we report the creation of a toolbox of 4 new mouse lines with high-level and Cre-dependent expression of ChR2(H134R)-tdTomato, ChR2(H134R)-EYFP, Arch-EGFP-ER2, or eNpHR3.0-EYFP. Inducible expression of these opsins is driven by a unique expression cassette in a modified Rosa26 locus, which we recently showed capable of mediating efficient fluorescent labeling 9. Here we demonstrate for the first time that high-performance optogenetic manipulation in Cre-dependent transgenic mice is enabled for all the classes of opsin molecules using this optimized strategy.
For all 4 lines, we found that cortical pyramidal neurons were highly responsive to either light activation (ChR2s) or light inhibition (eNpHR3.0, Arch-ER2), in both in vitro brain slice preparations and in vivo brains of awake animals. In addition, we found that light activation of hippocampal and reticular parvalbumin (Pvalb)-positive interneurons, as well as cortical rhythms resulting from synchronized reticular Pvalb+ neuron activation, can be readily achieved in behaving ChR2-EYFP mice. These results demonstrate that selective optical activation and silencing can be applied to different cell types in different brain regions in these mice, using a variety of photostimulation paradigms. Thus, when combined with the plethora of publically available Cre-driver lines, these transgenics should greatly facilitate the study of various neuronal cell types including those inaccessible to previous approaches.
Results
Generation of mice conditionally expressing opsin genes
We previously demonstrated strong, ubiquitous, Cre-dependent expression of fluorescent markers from a modified Rosa26 locus by incorporating a CAG promoter and the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) 9. Using the same expression strategy, we created 4 new Cre-dependent mouse lines that express optogentic tools: the H134R mutant version of ChR2 25 fused to either tdTomato (Ai27; ChR2(H134R)-tdTomato) or EYFP (Ai32; ChR2(H134R)-EYFP); a modified version of Arch (Ai35; ss-Arch-EGFP-ER2, abbreviated as Arch-ER2) 6; or eNpHR3.0 (Ai39; eNpHR3.0-EYFP) 23 (Fig. 1a). ChR2(H134R) was chosen because it produces larger conductance changes compared to wild-type ChR2, likely due to its slower deactivation kinetics. Both Arch-ER2 and eNpHR3.0 have Kir2.1 ER-exporting signals which were shown to enhance proper expression of microbial opsin proteins to the cell membrane 6, 21-23.
Figure 1.
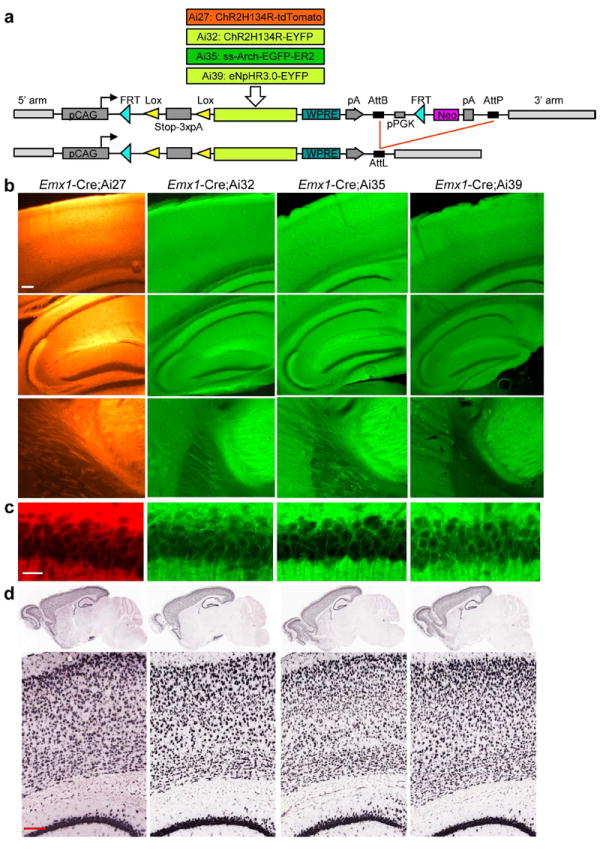
Generation and expression characterization of the Ai27, Ai32, Ai35 and Ai39 Cre-reporter lines. (a) Gene targeting vectors were designed to insert the Cre-dependent reporter cassettes into intron 2 of the Rosa26 locus. After obtaining germline-transmitted F1 mice, the PGK-neo selection cassette can be deleted by PhiC31-mediated recombination between the AttB and AttP sites, which combine into an AttL site, by breeding with a Rosa26-PhiC31 deleter line. (b) tdTomato, EYFP, and EGFP native fluorescence in Emx1-Cre;Ai27, Emx1-Cre;Ai32, Emx1-Cre;Ai35 and Emx1-Cre;Ai39 mice. Scale bar, 200 μm. (c) Confocal images of the CA1 pyramidal neurons in the same mice as in b, showing the cell membrane localization of tdTomato, EYFP and EGFP fluorescence. Scale bar, 20 μm. (d) Reporter gene mRNA expression in Emx1-Cre;Ai27, Emx1-Cre;Ai32, Emx1-Cre;Ai35 and Emx1-Cre;Ai39 mice (ages all ~P56), using in situ hybridization (Ai27, tdTomato riboprobe; Ai32, Ai35 and Ai39, EGFP/EYFP riboprobe). Scale bar, 200 μm.
To examine Cre-dependent expression of the optogenetic reporters, each line was bred with Emx1-Cre. Consistent with Emx1-Cre’s recombination pattern, all mice displayed strong native fluorescence throughout cortex and hippocampus (Fig. 1b). In each line, the fluorescent fusion proteins were primarily localized to the cell membrane with minimal accumulation in the cytoplasm, which was best seen in the cell body layer of all hippocampal subfields (CA1, CA3, and dentate gyrus) where the fluorescence was lower than in the dendritic layers and was ring-shaped (Fig. 1b-c). Axon fibers extending from the cortex and hippocampus, as well as their termination zones (e.g. thalamus), were also strongly labeled (Fig. 1b). Axon fiber fluorescence in Emx1-Cre;Ai39 mice appeared weaker than in other lines, indicative of possible lower protein expression. The mRNA expression of the opsin-fusion genes was mapped by in situ hybridization (ISH) on brain sections (Fig. 1d). All showed strong ISH signals in the cortex and hippocampus at the single-cell level, similar to expression seen in our Cre reporter mouse lines that express fluorescent proteins 9.
In the absence of Cre, no leaky expression at mRNA or fluorescence level was seen in Ai27 and Ai32 mice, although some leakage at mRNA level was seen in Ai35 and Ai39 mice (Supplementary Figs. 1a and 2a). In addition to Emx1-Cre, strong Cre-dependent expression of opsin-fusion genes was also seen throughout the brain in other Cre-driver crosses (e.g. Pvalb-IRES-Cre, Camk2a-CreERT2, Chat-IRES-Cre) (Supplementary Figs. 1b, 2b and 3), consistent with previous studies of our Cre-dependent fluorescent reporter mice 9. Plasma membrane targeting of all 4 transgenes was also seen in all cases with no detectable intracellular protein aggregates, as shown in the Pvalb+ interneurons in the cortex of the 4 types of Pvalb-IRES-Cre/reporter mice (Supplementary Fig. 1b). Long-term expression of these optogenetic transgenes in the various Cre-defined cell populations did not produce observable toxicity (Supplementary Fig. 4). The robust and widespread opsin-fusion expression observed in these multiple lines thus suggests that a variety of cell populations will be amenable to photo-manipulation by these Cre-dependent optogenetic tools.
Effective light-activation of cortical pyramidal neurons
We investigated the photoexcitability of cortical pyramidal neurons from Emx1-Cre;Ai27, Emx1-Cre;Ai32 mice (abbreviated as E-Ai27 and E-Ai32), and Ai32 alone mice not crossed to any Cre, by whole-cell recordings in the barrel cortex of acute brain slices with a previously established photostimulation paradigm 26, 27 (Fig. 2a), and compared them to neurons in which ChR2 was expressed by in utero electroporation (IUE). ChR2-expressing neurons showed normal resting membrane potentials (E-Ai27, -65.7 ± 1.4 mV, n = 14; E-Ai32, -65.4 ± 1.4 mV, n = 9; Ai32 (-Cre), -66.6 ± 2.2 mV, n = 9; IUE, -66.0 ± 2.4 mV, n = 6; mean ± s.e.m.). For each trial, a series of 1-ms pulses of constant-power blue light (473 nm) was applied through an air objective, sequentially on an 8 × 16 grid pattern overlaying cortical layers 1-5 (Fig. 2a). The grid covered the soma, dendrites, and parts of the axonal arbor of the recorded layer 2/3 pyramidal neuron (Fig. 2b). In voltage clamp mode, large photocurrents were evoked in E-Ai27 and E-Ai32 cells with modest laser powers (Fig. 2b). With high laser powers (1350-1700 μW), the large photocurrents (peak current for Ai27: 1.51 ± 0.24 nA, n = 10; Ai32: 2.01 ± 0.04 nA, n = 7) were often accompanied by spikes. Even under high power stimulation conditions, no detectable photocurrent was recorded from Ai32 (-Cre) control cells (Fig. 2b, n = 9). In current clamp mode, action potentials (APs) were evoked in 12 of 14 E-Ai27 cells (2 cells showed subthreshold or no response), in 9 of 9 E-Ai32 cells which is comparable to IUE cells 26, 28, and in 0 of 9 Ai32 (-Cre) cells (Fig. 2c-d).
Figure 2.
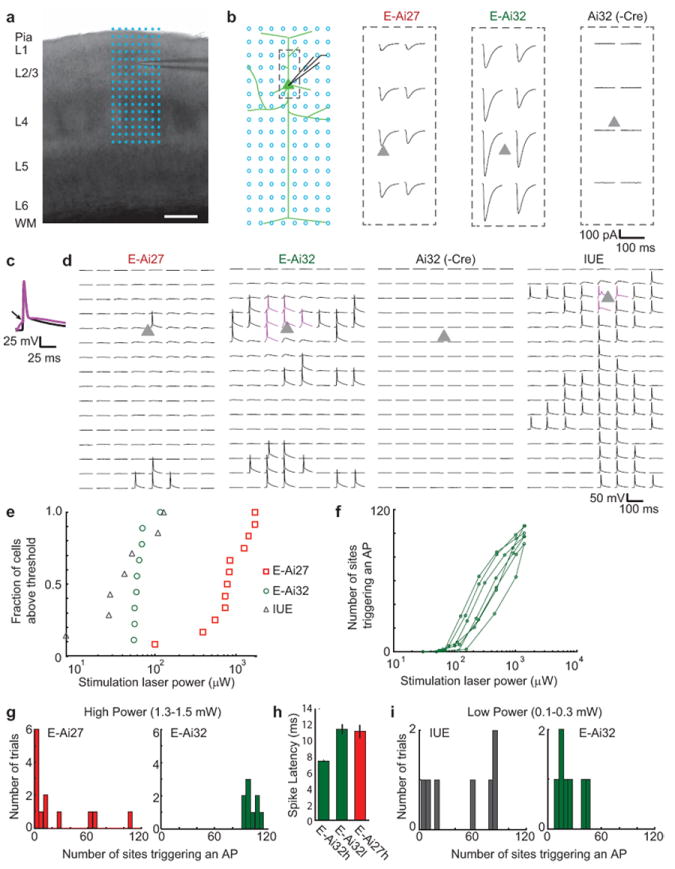
Photostimulation of pyramidal neurons in cortical slices of Emx1-Cre;Ai27 (abbreviated as E-Ai27), Emx1-Cre;Ai32 (E-Ai32), and Ai32 alone (-Cre) mice. (a) A barrel cortex slice with the 8 × 16 photostimulation grid overlaid (blue dots; spacing 50 μm). Scale bar, 200 μm. (b) Schematic of the photostimulation geometry and example traces. Whole-cell voltage-clamp traces from the dashed box area (left) are shown for E-Ai27 (with 840 μW light), E-Ai32 (70 μW) and Ai32 (1490 μW). Triangles, soma locations. (c) Waveforms of APs evoked by photoactivating the somata and dendrites (magenta) and axons (black). The arrow marks the inflection point. (d) Whole-cell current-clamp traces showing evoked APs in the 8 × 16 grid for a typical cell each of E-Ai27 (1700 μW), E-Ai32 (155 μW), Ai32 (1500 μW), and IUE (155 μW). (e) Minimum laser power required to evoke an AP from at least one stimulation site. (f) Number of photostimulation sites evoking an AP as a function of laser power in E-Ai32 neurons (n = 7). (g) Number of stimulation sites triggering APs using high laser powers. (h) Spike latencies of somatic/dendritic APs from the light onset. E-Ai32h: E-Ai32 cells (n = 9 cells, 143 APs) under high powers (≥1 mW). E-Ai32l: E-Ai32 cells (n = 9 cells, 28 APs) under low powers (≤100 μW). E-Ai27: E-Ai27 cells (n = 2 cells, 4 APs) under high powers. Spike latencies of axonal APs (not shown) varied greatly. (i) Number of stimulation sites triggering APs using low laser powers.
To compare the photosensitivity, each cell was stimulated with a series of varying laser powers to determine the threshold for evoking an AP (Fig. 2e). We found that E-Ai27 cells showed substantial heterogeneity in their thresholds for spiking, whereas E-Ai32 cells demonstrated consistently low thresholds (Fig. 2e-f). The average threshold powers were: E-Ai27, 919 ± 146 μW (ranging 550-1720 μW, n = 12); E-Ai32, 67 ± 6 μW (ranging 55-116 μW, n = 9); IUE, 56 ± 17 μW (ranging 8-128 μW, n = 7) (Fig. 2e). We further found that high power stimulation consistently evoked APs on more sites of the grid for E-Ai32 cells than E-Ai27 cells (Fig. 2g) and with shorter spike latencies (Fig. 2h). Notably, low laser powers evoked APs in E-Ai32 cells similar to IUE cells, but E-Ai32 cells showed less variability in the number of sites that could trigger an AP (Fig. 2i).
Photoexcitation of axons is desirable for stimulating long-range postsynaptic neurons. The subcellular distribution of ChR2 will also influence the spatial resolution of photostimulation in brain slices and in vivo 29. Antidromic APs triggered in axons can be distinguished from those triggered in the somata and dendrites, because somatic/dendritic APs are associated with a slow charging phase preceding the AP threshold whereas axon-initiated APs arrive in the soma without prior charging 30 (Fig. 2c). By analyzing the charging phase, each AP was categorized as originating from either somatic/dendritic or axonal stimulation (Fig. 2d). Under our photostimulation conditions both E-Ai27 and E-Ai32 cells were preferentially excited in axons, whereas E-Ai32 cells, similar to IUE cells, were also more readily excited in the somata and dendrites than E-Ai27 (Supplementary Fig. 5). In sum, our results demonstrate that whereas E-Ai27 cells are less sensitive to photostimulation, the light response properties of E-Ai32 cells are comparable in many aspects to those of IUE ChR2-expressing cells.
Effective light-silencing of cortical pyramidal neurons
To test light-evoked neuronal silencing, we performed whole-cell recordings from layer 2/3 or layer 5 cortical pyramidal neurons (Fig. 3a) in acute slice preparations taken from adult Emx1-Cre;Ai35 or Emx1-Cre;Ai39 mice (abbreviated as E-Ai35 or E-Ai39). In all pyramidal neurons tested (29/29), illumination with a 200-μm optical fiber coupled to a 593-nm yellow laser caused marked hyperpolarization from the resting membrane potential. The response was very similar to a hyperpolarizing current injection prior to the light pulse (Fig. 3b). Recorded neurons exhibited a variety of responses to hyperpolarizing currents injected under current clamp conditions. Some exhibited Ih currents and/or rebound firing of APs in response to a transient application of a hyperpolarizing current, while others did not. In each case, the effect of the input current could be effectively reproduced by light (Fig. 3b). Upon repeated cycles of light stimulation (23 mW mm-2), the extent of hyperpolarization diminished to about two-thirds of the initial value, then reached a plateau in E-Ai39 cells (Fig. 3c). The E-Ai39 cells could recover from such diminished response to >90% original level after ~5-min rest between the cycles. Unlike E-Ai39, the E-Ai35 neurons did not exhibit diminishing effects with repeated light stimulation (Fig. 3c).
Figure 3.
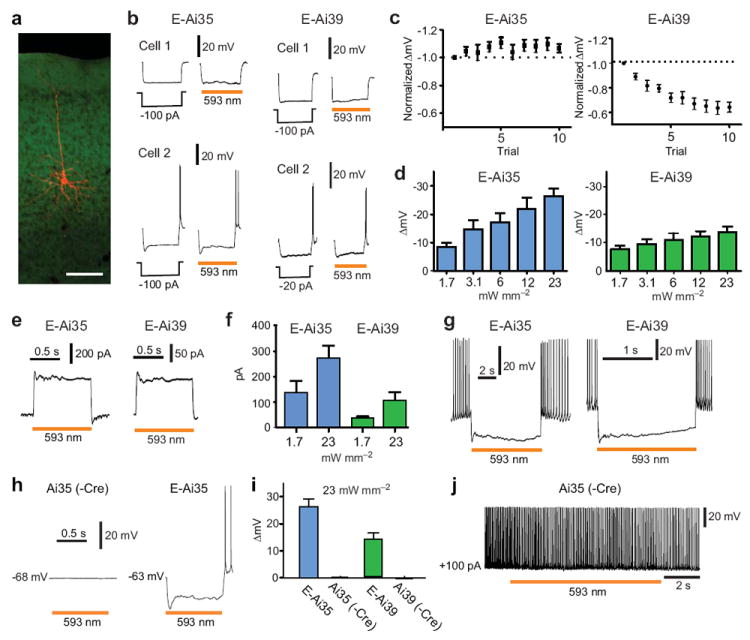
Effective silencing of cortical pyramidal neurons by Arch-ER2 in Emx1-Cre;Ai35 (E-Ai35) and eNpHR3.0 in Emx1-Cre;Ai39 (E-Ai39) mice. (a) Biocytin staining (red) of a cortical pyramidal neuron after recording. Scale bar, 100 μm. (b) Voltage responses of representative neurons to a 1-s negative current injection or a 1-s light pulse. Under both conditions, Cell 2 from E-Ai35 and E-Ai39 exhibits rebound firing at the end of the stimulus, whereas Cell 1 does not. (c) Voltage response of a neuron to 10 consecutive trials of laser stimulation (1-s pulse, 1-s interpulse interval). Values are normalized to the first trial; first trial ΔV was -27.05 ± 2.38 mV (n = 8, E-Ai35) or -19.43 ± 2.82 mV (n = 10, E-Ai39). (d) Average hyperpolarization from the resting membrane potential (mean of 10 stimulations as in c) evoked by different light intensities. (e) Representative photocurrent traces under voltage clamp (-70 mV) and 12 mW mm-2 illumination. (f) Mean photocurrents evoked by low and high light intensity. (g) Effective suppression of AP firing in E-Ai35 or E-Ai39 neurons evoked by positive current injection (+50-100 pA). (h) Voltage responses of Ai35 (-Cre) and E-Ai35 neurons under maximal light illumination (23 mW mm-2). (i) Comparison of light-induced hyperpolarization in Cre-positive (E-Ai35, n = 15; E-Ai39, n = 14) and Cre-negative (Ai35, n = 4; Ai39, n = 7) neurons. (j) Maximum light (23 mW mm-2) failed to slow or silence the firing of a Cre-negative Ai35 neuron evoked by positive current injection (+100 pA).
Both E-Ai35 and E-Ai39 cells showed larger hyperpolarization with increased light intensities (Fig. 3d). The mean maximal hyperpolarization (measured at 23 mW mm-2) was 26.39 ± 2.67 mV for E-Ai35 (n = 15) and 12.96 ± 1.39 mV for E-Ai39 (n = 14). Under voltage clamp conditions, light-induced currents were corresponding to light intensity and were highly reproducible from cell to cell (Fig. 3e-f). The photocurrents attained in E-Ai35 neurons at low and high light levels were 2-3 fold greater than those observed in E-Ai39 neurons (1.7 mW mm-2: p < 0.05, n = 5 for E-Ai35, n = 7 for E-Ai39; 23 mW mm-2: p < 0.01, n = 8 for E-Ai35, n = 9 for E-Ai39) (Fig. 3f). APs induced under current clamp by constant positive current injections were rapidly and reversibly inhibited by light illumination (23 mW mm-2) in all trials in both E-Ai35 and E-Ai39 neurons (Fig. 3g). We conclude that Ai35-expressed Arch-ER2 is somewhat more efficient at generating photocurrents than Ai39-expressed eNpHR3.0, but that both transgenically-expressed opsins are effective tools for the silencing of neural activity.
To ensure that no physiologically significant opsins are expressed from the un-induced reporter loci, we examined light-induced responses in cortical pyramidal neurons (n = 4) of Ai35 mice, as well as cortical (n = 4) and hippocampal CA1 (n = 3) pyramidal neurons of Ai39 mice, in the absence of any Cre allele. No light-induced changes were observed in the Ai35 (-Cre) cells at all, either at resting membrane potential (Fig. 3h-i) or when activated by a positive current injection (Fig. 3j), even with the highest intensity (23 mW mm-2). Similarly for Ai39 (-Cre) cells, light (593 nm, 23 mW mm-2; or 640 nm, 24 mW mm-2) caused no or negligible change in membrane potential (-0.02 ± 0.08 mV, n = 7, Fig. 3i). This is consistent with the leaky mRNA but no leaky protein expression in Ai39 (-Cre) mice (Supplementary Figs. 1a and 2).
Silencing by alternative light sources
In addition to the yellow light (593 nm) which is near the excitation maximum of the Arch and NpHR proteins 4, 6, 31, we also tested neural silencing by two alternative light sources, a 640-nm red laser and a white LED, that offer several practical advantages. Red lasers are less expensive and more easily modulated than the yellow lasers. Though somewhat red-shifted relative to the excitation peaks of Arch and NpHR, longer wavelengths are also less prone to attenuation by light scattering, so the off-peak excitation may be partly offset by improved tissue penetration. Red light also offers better spectral separation from the blue light used to activate ChR2, so they may be better suited for binary control of activity 4, 32. LEDs are inexpensive light sources and are potentially portable or implantable for experiments in moving animals.
These alternative light sources efficiently silenced both E-Ai35 and E-Ai39 cortical pyramidal neurons (Fig. 4). Both the LED and 640-nm red laser produced an intensity-dependent drop in membrane potential (Fig. 4b,e,g,i), and effectively silenced current-induced spiking (Fig. 4c,f,h,j). At similar powers, the 640-nm light response of E-Ai35 neurons was ~43% of that obtained with 593-nm light (Figs. 3d and 4e), while the 640-nm light response of E-Ai39 neurons was ~100% of that obtained with 593-nm light (Figs. 3d and 4i), consistent with the red-shifted spectrum reported for eNpHR3.0 23.
Figure 4.
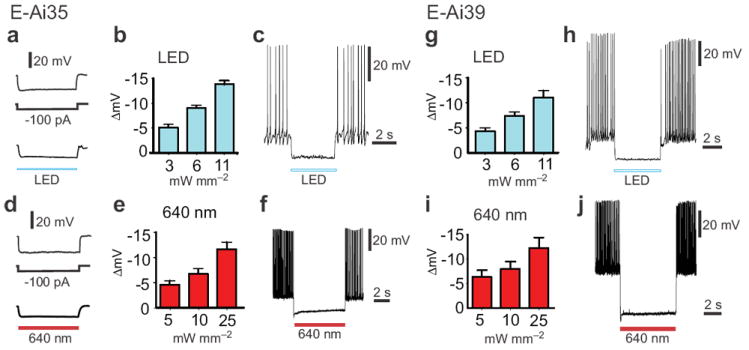
Alternative light sources for silencing of cortical pyramidal neurons in Emx1-Cre;Ai35 (E-Ai35) and Emx1-Cre;Ai39 (E-Ai39) mice. (a-c) Inhibition of E-Ai35 neurons by a white LED. (a) An example E-Ai35 neuron exhibited similar hyperpolarization response to negative current injection (-100 pA) and illumination by white light. (b) White light dose-response curve (n = 3). (c) An example of effective silencing by white light (11 mW mm-2) of APs evoked by a positive current injection (+100 pA). (d-f) Inhibition of E-Ai35 neurons by red laser light (640 nm). (d) An example E-Ai35 neuron exhibited similar hyperpolarization response to negative current injection (-100 pA) and illumination by red light. (e) Red light dose-response curve (n = 7). At the highest tested intensity (25 mW mm-2) the 640-nm laser illumination achieved 43% ± 5% of the hyperpolarization achieved with the 593-nm laser. (f) An example of silencing of current-evoked APs by red laser light. (g-h) Inhibition of E-Ai39 neurons by a white LED. (g) White light dose-response curve (n = 5). (h) An example of silencing of current-evoked APs by white light. (i-j) Inhibition of E-Ai39 neurons by red laser light (640 nm). (i) Red light dose-response curve (n = 7). At the highest tested intensity (25 mW mm-2) the 640-nm laser illumination achieved 100% ± 35% of the hyperpolarization achieved with the 593-nm laser. (j) An example of silencing of current-evoked APs by red laser light.
Light inhibition of hippocampal network activity
In order to test the effectiveness of Emx1-Cre;Ai35 mice as a model for inhibiting neural network activity, we tested light-mediated silencing of a well-described monosynaptic pathway from area CA3 to area CA1 of the hippocampus (Fig. 5a) 33. In this model, firing of hippocampal neurons was induced in brain slice preparations by elevated K+, and population activity was recorded from CA1 (Fig. 5b). Focused illumination of the CA3 region produced an immediate drop in CA1 activity (Fig. 5c). With sustained illumination, CA3 firing increased, but remained below baseline level. Partial release from silencing may occur over time because of compensatory effects in the network and/or because only a portion of the CA3 neurons with outputs to CA1 are inhibited by the focal illumination. Following light off, firing rates returned quickly to baseline, and there was no rebound of spiking activity.
Figure 5.
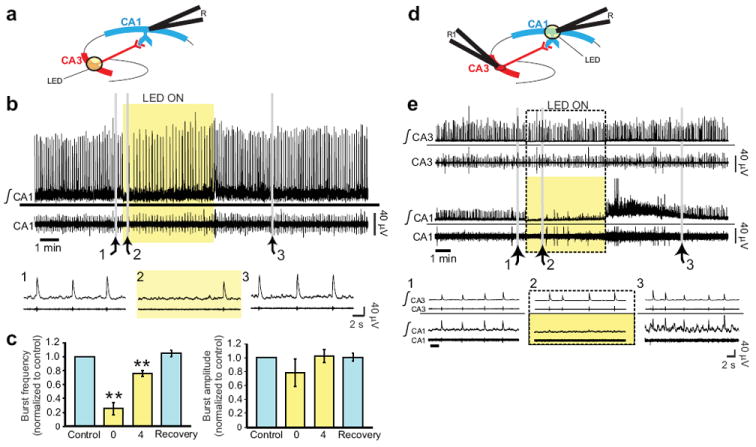
Effective silencing of induced population bursting in the hippocampal circuit in Emx1-Cre;Ai35 mice. (a) Schematic for the inhibition of presynaptic neurons in the hippocampal circuit. K+ (8 mM) induced population bursting in CA1 was recorded with an extracellular electrode (R). A white light source (LED, yellow circle) was positioned over CA3 to activate Arch-ER2 in presynaptic neurons. (b) Representative traces of the integrated (top) and raw (bottom) population bursting activity from CA1 before (expanded traces in inset 1), during (2), and following (3) the illumination of CA3. Raw population bursting activity is the direct measure of unit activity. Integrated population activity represents the change of unit activity (time constant = 200 ms). (c) Quantified response of population bursting from CA1 (n = 4 experiments) during 4 intervals: light-off control; the first (0) and final (4) minute of a 5-min exposure to white light; and 5 minutes after light exposure (recovery). Values were normalized to light-off control (** p < 0.01, repeated measures ANOVA; mean ± s.e.m.). (d) Schematic for the inhibition of postsynaptic neurons in the hippocampal circuit. Neurons were recorded simultaneously using dual extracellular electrodes (CA1: R, CA3: R1). A white LED was positioned over CA1 to activate Arch-ER2 in postsynaptic neurons. (e) Representative traces of the integrated (top) and raw (bottom) population bursting activity from CA3 and CA1 before (inset 1), during (2), and following (3) illumination to CA1. Light to CA1 led to suppressed bursting from that region, but bursting activity in CA3 was unaffected.
Dual recording from CA1 and CA3, combined with focal illumination of CA1, confirmed that these results were consistent with the silencing of a unidirectional synaptic input from CA3 to CA1 (Fig. 5d-e). First, illumination of CA1 had no effect on population activity in CA3. By contrast, direct illumination of CA1 caused immediate and nearly complete silencing of this region, and a large rebound increase in firing was observed upon recovery from direct illumination of CA1. In addition, the extent of CA1 inhibition was dependent on the area of illumination on CA3, under conditions of constant light intensity per unit area (Supplementary Fig. 6). We conclude that illumination of CA3 results in the selective inhibition of synaptic inputs to CA1, thus demonstrating the potential of Ai35 (and Ai39) mice for optogenetic studies of brain circuitry.
In vivo activation and silencing of cortical neurons
To evaluate the effectiveness of light modulation in vivo in these transgenic mice lines, we performed extracellular recordings in awake, head-fixed adult mice. We estimated that light intensity at the electrode recording site to be 1-5% of that at the optical fiber tip, which was 500-900 μm above the recording site, based on the Monte Carlo simulation of light attenuation in brain tissues 6.
In Emx1-Cre;Ai32 mice, we recorded a total of 16 units, including 7 single units and 9 multi units. Of the 7 single units, firing rates significantly increased in 6 units (Fig. 6a), and decreased in 1 unit, upon blue light (473 nm) illumination (0.8 mW out of a 100-μm optical fiber, corresponding to ~100 mW mm-2 at the fiber tip and ~2 mW mm-2 at the recording sites). The light induced spiking is fast, reliable and precise after each pulse illumination (Supplementary Fig. 7). Precise activation was observed even when the fiber tip was 1.5-2 mm away from the recording site, where light intensity is expected to drop to ~0.17% of that at the tip. Of the 9 multi units, firing rates significantly increased in 8 units, and decreased in 1 unit. Light illumination and recording was also performed in an Ai32 (-Cre) mouse as a negative control. Of the 3 cortical neurons recorded, none were modulated by light at the highest power. In Emx1-Cre;Ai27 mice, we recorded a total of 11 units, including 4 single units and 7 multi units. Of the 4 single units, firing rates significantly increased in only 1 unit, and decreased in the other 3, with the same blue light illumination (2.2 mW, ~280 mW mm-2 at the 100-μm fiber tip, and ~5 mW mm-2 at the recording sites). However, all 7 multi units increased their firing rates. Overall, the single and multi units with increased firing rates showed an enhancement of 350-1400% from the baseline for E-Ai32 (768 ± 94%, n = 14), and 150-350% for E-Ai27 (198 ± 25%, n = 8) (Fig. 6b). The magnitude of light-induced activation in E-Ai32 is significantly higher than that in E-Ai27, consistent with that observed in vitro (Fig. 2).
Figure 6.
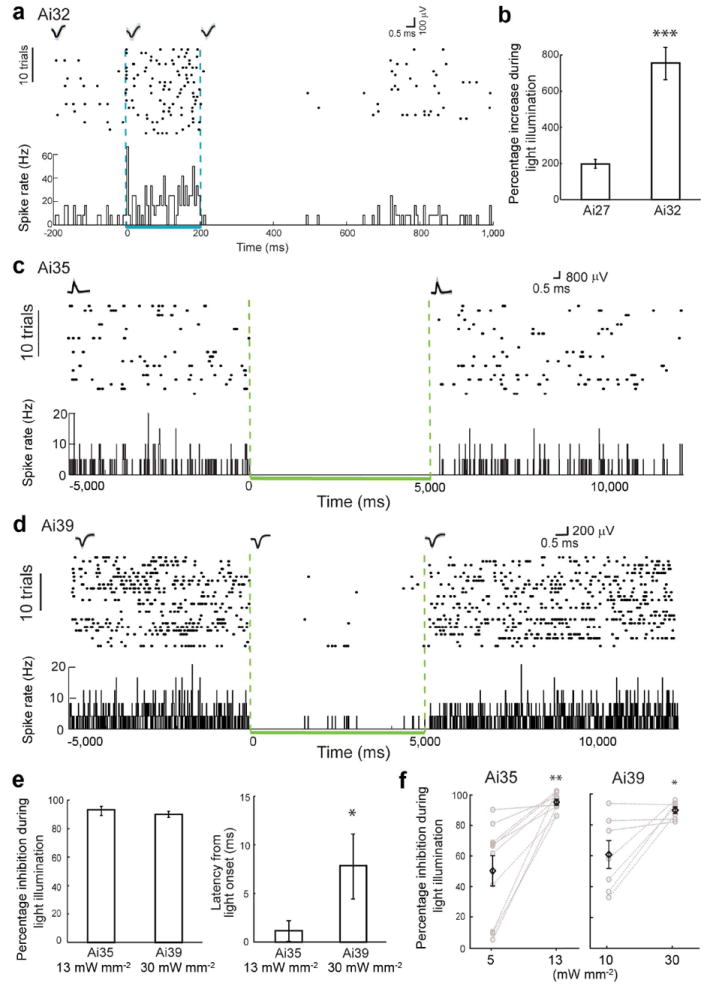
Optical activation or silencing of pyramidal neuron activities in the neocortex of awake Emx1-Cre;Ai27, Emx1-Cre;Ai32, Emx1-Cre;Ai35 and Camk2a-CreERT2;Ai39 mice (abbreviated as Ai27, Ai32, Ai35 or Ai39, respectively, here). (a) Neural activity and spike waveforms in a representative Ai32 neuron before, during and after 200-ms blue light illumination (3 mW mm-2). Top, spike raster plot; bottom, histogram of instantaneous firing rate averaged across trials (bin size, 5 ms). (b) Average changes in firing rates upon blue light illumination in Ai27 and Ai32 mice. (*** p < 0.001) (c) Neural activity and spike waveforms in a representative Ai35 neuron before, during and after 5-s green light illumination (13 mW mm-2). (d) Neural activity and spike waveforms in a representative Ai39 neuron before, during and after 5-s green light illumination (30 mW mm-2). For c and d, top, spike raster plot; bottom, histogram of instantaneous firing rate averaged across trials (bin size, 10 ms). (e) Average changes in firing rates (left) and latencies (right) observed in Ai35 and Ai39 single units during green light illumination at indicated light intensities. (* p < 0.05) (f) Green light illumination at higher intensity induced more powerful silencing in Ai35 and Ai39 mice. (* p < 0.05, ** p<0.01) All data points are mean ± s.e.m.
To evaluate the efficiency of optical silencing in vivo mediated by eNpHR3.0 and Arch-ER2, we used Emx1-Cre;Ai35, Camk2a-CreERT2;Ai35, and Camk2a-CreERT2;Ai39 mice. Following tamoxifen induction, Camk2a-CreERT2 drives eNpHR3.0 or Arch-ER2 expression in majority of cortical pyramidal neurons similar to Emx1-Cre (Supplementary Fig. 1) 9. In an Emx1-Cre;Ai35 mouse, we recorded a total of 10 single units in cell attached mode with glass electrodes. All single units significantly reduced their firing rates upon green light illumination (532 nm, 16 mW, ~470 mW mm-2 at the 200-μm fiber tip, and ~13 mW mm-2 at the recording sites) (Fig. 6c). The recorded neurons showed a reduction of 83-100% from the baseline firing rates (93 ± 2%, n = 10) (Fig. 6e, left panel). Light-induced silencing was instantaneous, with 0 ms latency for 9 of the 10 neurons recorded. The population latency was 1 ± 1 ms from light onset (Fig. 6e, right panel). Similar results of complete silencing of single unit activities were observed in a tamoxifen-induced Camk2a-ERT2;Ai35 mouse (93 ± 7% silencing at ~10 mW mm-2 at the recording sites, n = 2). Light illumination and recording was also performed in an Ai35 (-Cre) mouse as a negative control. Of the 8 cortical neurons recorded, none were modulated by light at the highest power used (~16 mW mm-2 at the recording sites).
In a tamoxifen-induced Camk2a-CreERT2;Ai39 mouse, we recorded a total of 13 single units. Of these, 7 significantly reduced their firing rates (Fig. 6d), 3 did not change, and 3 increased their firing rates upon green light illumination (11 mW, ~1400 mW mm-2 at the 100-μm fiber tip, and ~30 mW mm-2 at the recording sites). The single units with reduced firing rates showed a reduction of 83-97% from the baseline (90 ± 2%, n = 7), with 0-20 ms latency from light onset (8.6 ± 4.0 ms) (Fig. 6e). The magnitude of silencing in Ai39 mice with 30 mW mm-2 light is comparable to that observed in Ai35 mice with 10 mW mm-2 light (p = 0.4), but the latency of silencing in Ai39 mice is significantly longer than that in Ai35 mice (p < 0.05). The observed incomplete inhibition could result from the neurons that did not express Camk2a-CreERT2, and thus were disinhibited or activated through neural network mechanisms, consistent with previous observations using viral labeling method in vivo 24.
We then compared the responses of the same single units to different light intensities. These single units in both Emx1-Cre;Ai35 and Camk2a-CreERT2;Ai39 mice showed less inhibition at lower light intensities (Fig. 6f). In Ai35, firing rates were reduced by 49 ± 10% at 5 mW mm-2, and by 93 ± 2% at 13 mW mm-2 (n = 10, p = 0.001). In Ai39, firing rates were reduced by 62 ± 9% at 10 mW mm-2, and by 90 ± 2% at 30 mW mm-2 (n = 7, p = 0.02).
In vivo activation of Pvalb+ interneurons
To examine the photoexcitability in an inhibitory neuronal type, we performed extracellular recordings in awake behaving Pvalb-IRES-Cre;Ai32 mice using a custom-designed optoelectronic probe 34 containing 5-20 μm optical fibers that deliver locally focused light (~1 mW at the tip). For an independent assessment of the specificity of Pvalb+ neuron activation in these mice, two brain areas, the hippocampal CA1 region and the thalamus, were chosen because in these areas interneurons and principal cells can be reliably separated by physiological means 35-37. In the hippocampus, the neuron with the typical autocorrelogram (ACG) of fast firing perisomatic interneurons 38 was directly activated by both single pulses and the sinus pattern (Fig. 7a). In contrast, the bursting, putative pyramidal cell was silenced at the time of the firing epoch of the activated neuron. Using previously established waveform criteria, trough-to-peak time and the width of the spike of the wide-band (1 Hz – 5 kHz) unit at 20% of its peak amplitude 35, 39, we were able to segregate all recorded neurons into two major groups (Fig. 7b and Supplementary Fig. 8). All of the light activated neurons fell into the ‘narrow-spike category’. In another comparison, neurons were identified as excitatory or inhibitory neurons on the basis of putative monosynaptic excitation and inhibition estimated by their short-time cross-correlograms 35, 39, 40. Again, the two groups were segregated and all activated neurons fell into the physiologically identified interneuron cluster (Fig. 7b and Supplementary Fig. 8). These results show that ChR2-mediated activation was absent in CA1 pyramidal cells, and present in fast firing interneurons, corresponding to the perisomatic Pvalb+ interneurons.
Figure 7.
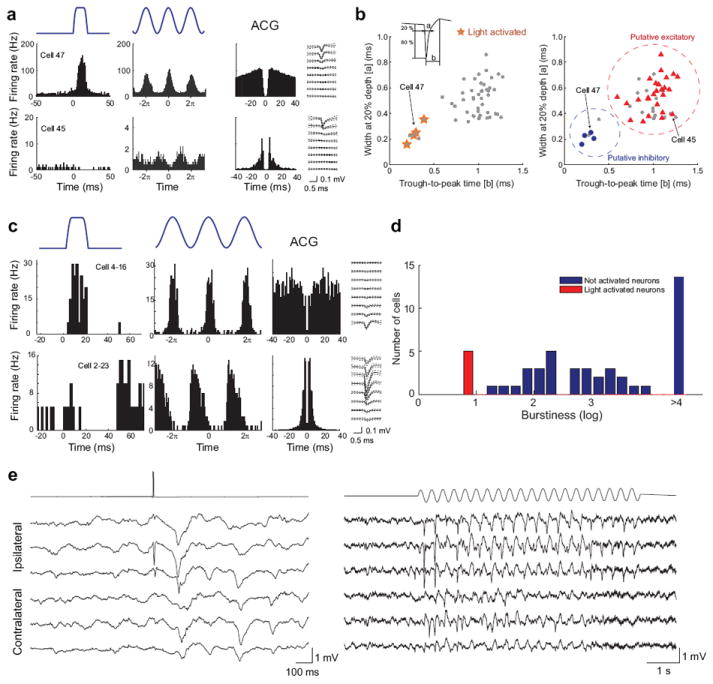
In vivo identification of light-activated neurons in the hippocampus and thalamus of Pvalb-IRES-Cre;Ai32 mice. (a) Excitation of ChR2-expressing neurons in the hippocampal CA1 region during waking state. Top row shows peristimulus histogram of a Pvalb+ neuron (Cell 47) transiently activated by single pulses or 8-Hz sinus light stimulation (1 mW). Also shown are the autocorrelogram (ACG) and waveform (± s.e.) of the neuron. Note typical ACG for a fast firing putative basket cell 35. Bottom row shows the same arrangement for a nearby pyramidal cell (Cell 45). Note ACG typical of bursting neurons. (b) Optogenetic (left) and physiological (right) classifications of neuron types in the hippocampus strongly overlap. Physiological segregation of simultaneously recorded neurons is based on two parameters – spike width and trough-to-peak time (inset). (c) Activation of ChR2-expressing neurons in the thalamus during anesthesia. Top row shows a reticular nucleus Pvalb+ neuron (Cell 4-16) in response to single pulses or 10-Hz sinus light stimulation. Bottom row shows a simultaneously recorded thalamocortical neuron (Cell 2-23) with typical bursting pattern in ACG 36, 37. (d) Distribution of burst index in the activated (putative reticular, red) and suppressed (putative thalamocortical, blue) neurons. Burst index is the ratio of spikes with short (<6 ms) inter-spike intervals relative to other spikes in the same session. (e) Light-evoked cortical patterns in response to reticular nucleus stimulation in the waking Pvalb-IRES-Cre;Ai32 mice. Single pulse (left) and sinus pattern (10 Hz, right) evoked activities are epidural recordings from the ipsilateral and contralateral parietal areas.
In the thalamus, the characteristic low-threshold spike burst patterns of thalamocortical neurons can be reliably separated from the fast firing inhibitory interneurons of the reticular nucleus 37. In the mouse reticular nucleus the majority of interneurons are PVALB-immunoreactive 41. Simultaneous recordings from reticular and the adjacent thalamocortical neurons and their local optogenetic activation generated two separate groups. Neurons that were activated by either single pulses or sinusoid pattern had ACGs characteristic of reticular neurons, whereas bursty neurons were suppressed by light stimulation (Fig. 7c and Supplementary Fig. 9). Light-activated and light-suppressed neurons had significantly different burst index magnitudes (Fig. 7d). Thus, these findings establish further support for the specificity of ChR2-mediated activation in reticular Pvalb+ interneurons. Although these physiological methods cannot distinguish between Pvalb+ and other classes of interneurons 42, the physiological results are consistent with the previously demonstrated recombination specificity of Pvalb-IRES-Cre to Pvalb+ interneurons in both hippocampus and thalamus 9.
Since reticular inhibitory neurons are critical for pacing thalamocortical rhythm 37, we examined their light activation on neocortical activity of waking mice. Single pulse stimulation (5 ms, 20 mW) evoked a primary response, likely through rebound spike activation of thalamocortical neurons 36, 37, followed by several cycles of activity typical of thalamocortical reverberation (Fig. 7e, left panel). Repetitive activation of reticular neurons at 10 Hz effectively entrained the thalamocortical circuit 36 (Fig. 7e, right panel). These findings demonstrate that reticular neurons can be recruited effectively by local light stimulation in Pvalb-IRES-Cre;Ai32 mice to induce physiologically relevant cortical patterns.
Discussion
We have established 4 new transgenic mouse lines with robust Cre-dependent expression of ChR2, Arch-ER2 or eNpHR3.0. Compared to a previously published R26∷ChR2-EGFP Cre-reporter mouse line 20, our Ai32 ChR2-EYFP-expressing line exhibits significantly greater light sensitivity. In the R26∷ChR2-EGFP mice, using very similar stimulation paradigms in cortical slices, very long and strong laser pulses (~20 ms, ~2 mW) were needed to induce single spiking in interneurons, and even so spiking could not be induced in pyramidal neurons. The necessity to use mice with homozygous floxed-ChR2 allele and to feed the mice with retinol was also reported. In contrast, the Ai32 mice possess excitability with 1-ms light pulses at intensities as low as 30 μW, much larger photocurrents, and much shorter latency to spiking, in pyramidal neurons. We further show that pyramidal neurons and interneurons are also readily excitable in vivo with low light in heterozygous animals. These greatly improved properties are likely to be proven critical in future in vitro and in vivo studies. Using such an efficient transgenic expression strategy is particularly essential for expressing the silencing opsins (Arch and eNpHR) to functional levels.
The transgenic targeting strategy used in the R26∷ChR2-EGFP line 20 is similar to our strategy described here. Both targeted the Rosa26 locus and used a strong and ubiquitous CAG promoter. But, two main differences may be responsible for the improved expression level in the new lines reported here. First, our expression cassettes included a WPRE sequence, which we previously showed to enhance protein expression of Rosa26-targeted transgenes 9. Second, while the R26∷ChR2-EGFP line incorporated a commonly-utilized floxed-stop cassette that contains the bacterial neomycin-resistant (neo) gene in front of the stop, we intentionally excluded the neo gene from our own floxed-stop cassette and instead placed it separately downstream from the transgene expression cassette. Although it is unclear whether the relocation of the neo gene is helpful to the significantly improved expression in our mice, it is well-known that integration of bacterial sequences into mammalian genomes could cause epigenetic modifications that affect expression of nearby genes.
Each of our optogenetic lines exhibited distinct photo-response properties that could be useful for different applications. Neurons from Ai32 appeared significantly more photosensitive than those from Ai27, both in vitro and in vivo. Although the basis for this difference between Ai27 and Ai32 neurons is unresolved, and no apparent difference in expression level or membrane localization was seen, we speculate that the bulkier tdTomato fluorescent tag in the Ai27 transgene may interfere with some channel properties. Although Ai32’s photosensitivity seems desirable for most experiments, Ai27’s red label and/or preferential axonal excitation may be advantageous in some applications 28, 30. Our in vitro and in vivo data on Ai35 and Ai39 neurons suggest that Arch-ER2 produces greater photocurrents and larger hyperpolarization than eNpHR3.0 under both green and yellow light. This could be due to the observed lower protein level for eNpHR3.0 even though the mice were generated using identical designs. However, due to its broad and red-shifted activation spectrum, eNpHR3.0 is equally effective as Arch-ER2 under red light, and may be preferable for dual-channel work together with blue light-responsive depolarizing opsins.
The kinetics of the in vivo light response of ChR2-expressing neurons in Ai27 and Ai32 mice were identical to those observed with virally expressed opsins, with a rapid increase in spiking at light onset, followed by a stable, lower steady-state firing level during light stimulation and often a period of suppression immediately following light termination 32. Photoinhibition of Arch-ER2 or eNpHR3.0-expressing neurons in Ai35 or Ai39 mice in vivo showed a near instantaneous reduction in firing rate, consistent with that observed with viral infection method 6. Many of the neurons recorded in Ai35 and Ai39 mice were nearly completely silenced. The homogenous and complete silencing in these mice presents a major advantage of the consistent expression achieved in these transgenic mice compared to viral infection method 6, 24.
An important question is the specificity of neuronal activity manipulation in the targeted populations. While this can be addressed by exhaustive anatomical multiple labeling methods, physiological verification of specificity is also useful. We have chosen two brain areas, the CA1 pyramidal layer and the thalamus because identification of principal cells and interneurons in these regions is possible by physiological means 35-40. While these methods cannot distinguish among the large family of interneurons 38, light-activated neurons displayed well-known features of short-duration spikes and high firing rates, typical of perisomatic Pvalb+ interneurons. Conversely, putative CA1 pyramidal neurons and thalamocortical cells with bursting properties and excitatory connections were never directly activated by light. Instead, the majority of them were suppressed by light stimulation. These excellent physiological-optogentic correlations in the intact brain support the cell type specificity of ChR2 activation. They also suggest that optogenetic activation of genetically labeled cell types, especially those difficult to distinguish by physiological or other means, will enable more refined in vivo identification and characterization of their functional properties 43.
Systematic expression characterization data from our previously generated fluorescent reporter lines (e.g. Ai14), crossed to dozens of different Cre-driver lines, have shown that Cre-dependent activation of transgene expression can be obtained in nearly all neuronal types 9. Thus, although data presented here are confined to the light responses of cortical/hippocampal pyramidal neurons and hippocampal/thalamic Pvalb+ interneurons, we believe it highly likely that these optogenetic tools will effectively modulate activity in a wide range of neurons. It should be noted that different types of neurons may have different excitability properties for both intrinsic and/or local circuitry reasons, and hence may require individual optimization of the conditions for activation and silencing.
Here we not only provide a novel set of transgenic tools with superior properties for both stimulating and silencing neuronal activity, we also demonstrate a transgenic expression strategy having several significant conceptual advantages. First, since the opsins are expressed as single copies in an identical manner from a consistent genomic environment, reliable comparisons of in vivo performance can be made between different opsin genes and different transgenic lines. Second, the Cre-dependent on-off switch for transgene expression effectively prevents or minimizes leaky expression in non-targeted cells while enables strong expression from the well-characterized CAG promoter in targeted cells, a clear advantage over the specific promoter-driven single transgenic approach in which the promoters used could have variable (sometimes unknown or uncharacterized) expression in both targeted and non-targeted cells. Third, because of the efforts we and others have taken to systematically characterize expression in Cre-driver lines 8, 9, especially when similar reporter lines are used, documented and publicly available information about Cre-recombination patterns (e.g. http://connectivity.brain-map.org/transgenic/search/basic) can advise researchers about the cell-type specificity of expression expected when using a particular Cre driver with these reporter lines. This is of great importance also considering that unintended ectopic Cre expression may exist to varied degrees in some Cre lines, and informed choice about appropriate Cre (or inducible Cre) lines for specific cell types need to be made. Finally, this proven expression system will also facilitate the rapid incorporation of newly engineered optogenetic variants, and once validated, apply them to all the Cre lines. This presents a “one-for-all” opportunity than expressing one opsin in one cell type at a time, and will further increase the range of optogenetic capabilities for investigating neural circuits and brain function.
Methods
All experimental procedures related to the use of mice were approved by the Institutional Animal Care and Use Committees of the Allen Institute for Brain Science, the Howard Hughes Medical Institute, Boston University, Seattle Children’s Research Institute, or Rutgers University, in accordance with NIH guidelines.
Gene targeting in ES cells and generation of knock-in Cre reporter mice
Targeting constructs were generated using a combined gene synthesis (GenScript) and molecular cloning approach. ChR2(H134R)-tdTomato was synthesized based on the hChR2 sequence 26, and ChR2(H134R)-EYFP was synthesized based on the ChR2 sequence 3. Both fragments, as well as the ss-Arch-EGFP-ER2 6 and the eNpHR3.0-EYFP 23 fragments, were each cloned into a Rosa26-pCAG-LSL-WPRE-bGHpA targeting vector 9, in between LSL and WPRE sequences. LSL sequence contains specifically LoxP – Stop codons – 3x SV40 polyA – LoxP.
The targeting vectors were linearized and transfected into the 129/B6 F1 hybrid ES cell line G4 44. G418-resistant ES clones were first screened by PCR using primers spanning the 1.1 kb 5’ genomic arm (forward primer: 5’-gggctccggctcctcagaga-3’, reverse primer: 5’-atgccaggcgggccatttac-3’), and then confirmed by Southern blot analysis of HindIII digested DNA, which was probed with a 1.1 kb genomic fragment from immediately upstream of the 5’ arm. Positive ES clones were injected into C57BL/6J blastocysts to obtain chimeric mice following standard procedures. Chimeric mice were bred with C57BL/6J mice to obtain germline transmitted F1 mice. The reporter mice can be bred with the Rosa26-PhiC31 mice (JAX Stock # 007743) 45 to delete the PGK-neo cassette in the germline of the mice.
Expression characterization
Reporter mice were crossed to various Cre lines, including Emx1-Cre (JAX Stock # 005628), Camk2a-CreERT2 (JAX # 012362), Pvalb-IRES-Cre (JAX # 008069) and Chat-IRES-Cre (JAX # 006410). Expression of the reporter genes was assessed by both native fluorescence (without antibody staining) on perfused, microtomed sections and by ISH.
For ISH, the Allen Institute-established pipelines for tissue processing, probe hybridization, image acquisition, and data processing were used. The procedures were previously described 9, 46 and can be found at the Transgenic Mouse database (http://help.brain-map.org/display/mouseconnectivity/Documentation). Expression levels were analyzed with 2-tailed, paired or unpaired Student’s t-test at an alpha level of 0.05.
Electrophysiology and photostimulation on Ai27 and Ai32 brain slices
P30 – P60 mice were used. Recordings on 300-μm thick barrel cortical slices were performed in the presence of 5 μM (R)-CPP (Tocris) and 10 μM NBQX (Tocris). Electrophysiology and stimulus conditions were as described 26. A series of 473-nm laser pulses (Crystal Laser) of 1-ms duration and 30-1770 μW (at specimen) for photostimulation was delivered through an air objective (4x; 0.16 NA; UPlanApo, Olympus) with a beam diameter of 6-20 μm (scattering in the tissue was not taken into account). Photostimulation marched through an 8×16 grid with 50 μm spacing. The laser stimuli were given in a spatial sequence designed to avoid consecutive stimulation to neighboring spots to minimize desensitization 47. Data were acquired using Ephus (available at www.ephus.org).
Inflection point analysis
APs triggered in axons could be distinguished from those triggered in somata and dendrites by their waveforms. Dendritic APs have a charging phase that precedes reaching the AP threshold (set to be 15 mV) at the inflection points. To determine the charging phase and the inflection point, we calculated the first derivative of an AP trace. The AP was identified as a derivative peak larger than 25 mV/ms; the charging phase as a derivative peak before AP and larger than 0.5 mV/ms; and the inflection point as the lowest derivative point between the charging peak and the AP peak. APs with an inflection point higher than 15mV were scored as somatic/dendritic; otherwise as axonal.
In utero electroporation
In utero electroporation (IUE) was done as previously described 26, 28. In brief, the plasmids for electroporation contained ChR2-mVenus (2 μg/μl) and cytoplasmic mCherry at 3:1 molar ratio. E16 timed-pregnant C57BL/6J mice were deeply anesthetized using an isoflurane-oxygen mixture. The uterine horns were exposed and 0.2-0.5 μl of DNA solution with Fast Green dye (Sigma) was pressure injected (Picospritzer, General Valve) through a pulled glass capillary tube (Warner Instruments). The head of each embryo was placed between custom-made tweezer-electrodes, with the positive plate contacting the right side of the head. For E16 animals, the transfection was restricted to layer 2/3 cortical cells in the electroporated hemisphere. IUE mice were used for slice physiology at ages of P14-P21.
Electrophysiology and photostimulation on Ai35 and Ai39 brain slices
Mice 7-21 weeks old were used to prepare coronal slices (neocortex experiments, 350-400 μm thick; hippocampus experiments, 400-500 μm thick) for intracellular whole cell recordings. Illumination of brain slice preparations was supplied by a 593-nm, 50-mW yellow laser (LaserSight Technologies), a 640-nm, 50-mW diode red laser (Opto Engine), or a Luxeon 5 watt white LED (Lumileds Lighting). Light sources were coupled to a 200-μm, 0.22-NA optical fiber. Light output was calibrated with a Thorlabs PM100 meter and S130A detector (Thorlabs).
Hippocampal network recordings
Young adult mice (P35-50, Fig. 5) and adult mice (6 months, Supplementary Fig. 6) were used. Extracellular KCl was elevated from 3 to 8 mM over a 20-min period to initiate spontaneous bursting in the hippocampal slice. Experiments began 10 min after KCl reached 8 mM. Extracellular recordings were made from areas CA1 and CA3 using glass electrodes filled with ACSF (<2 MΩ). An optical fiber (200-μm diameter) connected to a computer driven white LED source (Fig. 5) or a 640-nm laser (Supplementary Fig. 6) was positioned over CA3 or CA1. Population bursting and the integration of population bursts were recorded prior to, during and following the light stimulation. Integration of the population bursts was conducted in real-time using an in-line integrator (time constant = 200 ms; James Franck Institute electronic shop). Instantaneous frequency and amplitude of integrated bursts were analyzed post hoc using Clampfit 10. Differences were determined using 2-tailed unpaired t-test between two means, or one-way repeated measures ANOVA followed by multiple comparisons testing (Dunnett’s comparison) among three or more means.
In vivo photostimulation and extracellular recording in awake, head-fixed mice
Recordings in awake head-fixed mice (3-7 months old) were performed as previously described 6, 32. Briefly, under isoflurane anesthesia, a plastic head-plate was implanted over the cortex. Once the mouse was recovered from surgery, recordings were made while the mouse was awake and head fixed, using linear multi-contact silicone electrodes (NeuroNexus). To avoid light-induced artifact on the silicone electrodes, we also used borosilicate glass microelectrodes filled with saline. The glass microelectrodes have an impedance of ~7 MΩ. Optical fibers were coupled to the electrodes (100-μm fiber coupled to silicone electrode, and 200-μm fiber to glass electrode), with the tip positioned 500-900 μm above the recording sites. The optical fiber was connected to a green laser (532 nm) or a blue laser (473 nm), with tunable power (Shanghai Laser Corp). Lasers were controlled by a function generator (Agilent Tech). Light intensity was measured with a power meter PM100D (Thorlabs). Data acquisition was performed with a multichannel Omniplex system (Plexon) for the NeuroNexus silicone electrode, or with a Multiclamp 700B amplifier and digitized with a Digidata 1440 digitizer (Molecular Device).
Spikes were sorted with Offline Sorter 3.0 (Plexon). Neurons modulated by light were identified by performing a paired t-test, for each neuron, between the baseline firing rate before light onset and firing rate during light illumination, across all trials for that neuron, thresholding at p < 0.05 significance level as previously described 6, 32. Instantaneous firing rate histograms were computed by averaging the instantaneous firing rate with a time bin of 10 ms for Ai35 and Ai39 neurons or 5 ms for Ai32 and Ai27 neurons.
Both green and blue light illumination of the silicone multi-contact electrodes produced significant slow artifacts as previously observed with Tungsten electrodes 32. The blue light-induced artifact was particularly strong on the silicone electrodes, which sometimes saturated the data acquisition amplifier at the onset of each light pulse, thus we excluded the first 20 ms after light onset for calculations for all Ai27 and Ai32 neurons. The green light-induced artifact on silicone electrodes was much smaller in magnitude, and never saturated the amplifier system, allowing us to determine the latency of light-induced neural modulation in Ai35 and Ai39 mice. Light did not produce any optical artifact on glass electrodes. Latency was defined as the time from light onset to the time at which firing rate was significantly different from baseline for the following 30 ms.
In vivo photostimulation and extracellular recording in behaving Pvalb-IRES-Cre;Ai32 mice
Construction of fiber-based optoelectronic probes was done as previously described 34. The silicon probes have 4 shanks (Buzsaki32 from NeuroNexus). The shanks are 200 μm apart from each other and bear 8 recording sites each (160 μm2 each site; 1-3 MΩ impedance) arranged in a staggered configuration (20 μm vertical separation) 40. As light guides, we used multi-mode optical fibers (105 μm in diameter; AFS 105/125; Thorlabs). To restrict light activation only to the brain volume monitored by the silicon probe, the fiber was etched by dipping into concentrated hydrofluoric acid until the desired 5-20 μm diameter was achieved. The fiber was positioned on the silicon shank at ~100 μm above the uppermost recording site, and the rest of the fiber was glued to the shank. Light modulation was provided by DPSS laser (473 nm; SDL-473-050T; Shanghai Dream Lasers Technology). The intensity of light at the tip of the etched fiber could be varied. Typical stimulus intensity varied between 1 to 5 mW.
Recordings were performed either in anesthetized (isofluorane) or waking freely behaving mice (6-8 weeks old). In the chronic mice, the optoelectronic probe assembly was fixed to a micromanipulator and lowered into the brain by slow steps. In mice with light activation of the reticular nucleus, etched optic fibers were placed in the reticular nucleus unilaterally or bilaterally. Cortical activity was monitored by epidural recordings, using #000 screws, driven into the bone. Recording sessions typically lasted for 1 hour, during which the animal’s behavior alternated between periods of walking and immobility. Neurophysiological signals were amplified and multiplexed by a miniature head-stage of a 256-channel multiplexed amplifier system and the multiplexed signals were directly recorded by the computer. After recovery, the animals could move relatively unconstrained due to the low weight and small size of the head-stage as well as the decreased number of connecting wires. Neuronal activity was sampled at 20 kHz per channel at 16 bit resolution, while the overall gain of the multiplexer system was 400x (KJE-1000; Amplipex). Spike sorting was performed semi-automatically, using KlustaKwik, followed by manual adjustment of the clusters 48.
Considerations for light intensity measurement in different experimental paradigms
The amount of depolarization or hyperpolarization generated in a given cell is corresponding to the summation of the photocurrents generated from all the activated opsin molecules. The latter is proportional to irradiance (light intensity per unit area) at the target multiplied by the illuminated surface area of the target cell (including cell body and dendritic/axonal processes, without considering further heterogeneity in opsin distribution and local membrane excitability). Therefore illumination by a diffused light (e.g. directly from a LED or through a regular optical fiber) with lower irradiance but covering a larger surface area of the cell could have the same effect as illumination by a high intensity and highly focused, therefore high irradiance, light beam (e.g. laser light through the objective lens) on a smaller surface area of the cell. Furthermore, even highly focused light becomes scattered as it enters the brain tissue, and it is impossible to estimate the actual irradiance at the target without knowing the location/depth of the cell or which part of it is illuminated. For these reasons, different methods were employed to report the light intensities in our study. In brain slice photosilencing experiments (Figs. 3-5) irradiance (in mW mm-2) of the relatively diffused light at the surface of the brain slice was presented. In brain slice photoactivation experiments (Fig. 2) radiant flux (i.e. light intensity, in mW) of the focused light beam (6-20 μm wide) at the surface of the brain slice was presented. In the in vivo photostimulation of cortical excitatory neurons (Fig. 6), the irradiance at the recorded cell level was estimated by Monte Carlo simulation based on known distance between the tip of the optical fiber (100-200 μm wide) and the electrode. In the in vivo photostimulation of hippocampal and thalamic Pvalb-positive neurons (Fig. 7), radiant flux at the tip of the etched optical fiber (5-20 μm wide) that was attached to the silicon probe was reported. All these types of descriptions can be considered appropriate for cross-experimental comparisons in each of their respective paradigm where the light delivery approach is consistent.
Supplementary Material
Acknowledgments
We are grateful for the technical support of the Atlas Production Team, led by Paul Wohnoutka, and the Technology Team, led by Chinh Dang, at the Allen Institute. We thank Andras Nagy for providing the G4 ES cell line, and Karl Deisseroth for providing the eNpHR3.0 construct. This work was funded by the Allen Institute for Brain Science and the Howard Hughes Medical Institute, NIH grant (DA028298) to H.Z., NIH grants (MH90478 and MH093667) to E.T., NIH grant (MH085944) and Alfred P. Sloan Foundation to X.H, NIH grants (NS034994 and MH54671) and NSF to G.B., and a Marie Curie Fellowship (EU FP7 PEOPLE 2009 IOF 254780) to A.B. The authors wish to thank the Allen Institute founders, Paul G. Allen and Jody Allen, for their vision, encouragement, and support.
Footnotes
Author Contributions
L.M., J.K. and H.G. generated the Cre reporter mouse lines. T.M., B.M.H. and K.S. conducted the slice physiology study on Ai27 and Ai32 mice. H.K., Y-W.A.H., A.J.G., S.Z., J.M.R. and E.E.T. conducted the slice physiology study on Ai35 and Ai39 mice. J-M.Z., X.G., Y.M. and X.H. conducted the in vivo cortical recordings. A.B., S.F. and G.B. conducted the in vivo hippocampal and thalamic recordings. A.R.J. provided institutional support. E.S.B. provided the Arch-ER2 construct. L.M., T.M., H.K., J-M.Z., A.B., S.F., E.S.B., G.B., X.H., E.E.T. and H.Z. analyzed data and wrote the paper.
Publisher's Disclaimer: Open public access: All 4 Cre-reporter mouse lines have been deposited to the Jackson Laboratory for distribution (JAX Stock Numbers for Ai27: 012567, Ai32: 012569, Ai35: 012735, Ai39: 014539). All 4 gene-targeting DNA constructs have been deposited to Addgene, a plasmid repository, for distribution. All ISH expression data will be available at http://connectivity.brain-map.org/transgenic/search/basic.
References
- 1.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 2.Li X, et al. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci USA. 2005;102:17816–17821. doi: 10.1073/pnas.0509030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- 6.Chow BY, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegemann P, Moglich A. Channelrhodopsin engineering and exploration of new optogenetic tools. Nat Methods. 2011;8:39–42. doi: 10.1038/nmeth.f.327. [DOI] [PubMed] [Google Scholar]
- 8.Gong S, et al. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taniguchi H, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhlman SJ, Huang ZJ. High-resolution labeling and functional manipulation of specific neuron types in mouse brain by Cre-activated viral gene expression. PLoS ONE. 2008;3:e2005. doi: 10.1371/journal.pone.0002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arenkiel BR, et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron. 2007;54:205–218. doi: 10.1016/j.neuron.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci USA. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagglund M, Borgius L, Dougherty KJ, Kiehn O. Activation of groups of excitatory neurons in the mammalian spinal cord or hindbrain evokes locomotion. Nat Neurosci. 2010;13:246–252. doi: 10.1038/nn.2482. [DOI] [PubMed] [Google Scholar]
- 16.Chuhma N, Tanaka KF, Hen R, Rayport S. Functional connectome of the striatal medium spiny neuron. J Neurosci. 2011;31:1183–1192. doi: 10.1523/JNEUROSCI.3833-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren J, et al. Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron. 2011;69:445–452. doi: 10.1016/j.neuron.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 18.Tsunematsu T, et al. Acute Optogenetic Silencing of Orexin/Hypocretin Neurons Induces Slow-Wave Sleep in Mice. J Neurosci. 2011;31:10529–10539. doi: 10.1523/JNEUROSCI.0784-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao S, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katzel D, Zemelman BV, Buetfering C, Wolfel M, Miesenbock G. The columnar and laminar organization of inhibitory connections to neocortical excitatory cells. Nat Neurosci. 2011;14:100–107. doi: 10.1038/nn.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao S, et al. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol. 2008;36:141–154. doi: 10.1007/s11068-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36:129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gradinaru V, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han X, et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:1. doi: 10.3389/fnsys.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagel G, et al. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 26.Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 27.Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis TL, Jr, Mao T, Arnold DB. A role for myosin VI in the localization of axonal proteins. PLoS Biol. 2011;9:e1001021. doi: 10.1371/journal.pbio.1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peron S, Svoboda K. From cudgel to scalpel: toward precise neural control with optogenetics. Nat Methods. 2011;8:30–34. doi: 10.1038/nmeth.f.325. [DOI] [PubMed] [Google Scholar]
- 30.Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009;12:568–576. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 32.Han X, et al. Millisecond-timescale optical control of neural dynamics in the nonhuman primate brain. Neuron. 2009;62:191–198. doi: 10.1016/j.neuron.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishizuka N, Weber J, Amaral DG. Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat. J Comp Neurol. 1990;295:580–623. doi: 10.1002/cne.902950407. [DOI] [PubMed] [Google Scholar]
- 34.Royer S, et al. Multi-array silicon probes with integrated optical fibers: light-assisted perturbation and recording of local neural circuits in the behaving animal. Eur J Neurosci. 2010;31:2279–2291. doi: 10.1111/j.1460-9568.2010.07250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Csicsvari J, Hirase H, Czurko A, Mamiya A, Buzsaki G. Oscillatory coupling of hippocampal pyramidal cells and interneurons in the behaving Rat. J Neurosci. 1999;19:274–287. doi: 10.1523/JNEUROSCI.19-01-00274.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halassa MM, et al. Selective optical drive of thalamic reticular nucleus generates thalamic bursts and cortical spindles. Nat Neurosci. 2011 doi: 10.1038/nn.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 38.Klausberger T, et al. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- 39.Sirota A, et al. Entrainment of neocortical neurons and gamma oscillations by the hippocampal theta rhythm. Neuron. 2008;60:683–697. doi: 10.1016/j.neuron.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujisawa S, Amarasingham A, Harrison MT, Buzsaki G. Behavior-dependent short-term assembly dynamics in the medial prefrontal cortex. Nat Neurosci. 2008;11:823–833. doi: 10.1038/nn.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu XB, Murray KD, Jones EG. Low-threshold calcium channel subunit Ca(v) 3.3 is specifically localized in GABAergic neurons of rodent thalamus and cerebral cortex. J Comp Neurol. 2011;519:1181–1195. doi: 10.1002/cne.22567. [DOI] [PubMed] [Google Scholar]
- 42.Tanahira C, et al. Parvalbumin neurons in the forebrain as revealed by parvalbumin-Cre transgenic mice. Neurosci Res. 2009;63:213–223. doi: 10.1016/j.neures.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Lima SQ, Hromadka T, Znamenskiy P, Zador AM. PINP: a new method of tagging neuronal populations for identification during in vivo electrophysiological recording. PLoS ONE. 2009;4:e6099. doi: 10.1371/journal.pone.0006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.George SH, et al. Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:4455–4460. doi: 10.1073/pnas.0609277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond CS, Soriano P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE. 2007;2:e162. doi: 10.1371/journal.pone.0000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 47.Shepherd GM, Pologruto TA, Svoboda K. Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron. 2003;38:277–289. doi: 10.1016/s0896-6273(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 48.Harris KD, Henze DA, Csicsvari J, Hirase H, Buzsaki G. Accuracy of tetrode spike separation as determined by simultaneous intracellular and extracellular measurements. J Neurophysiol. 2000;84:401–414. doi: 10.1152/jn.2000.84.1.401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.