

Abstract
Motivation: One of the most successful methods to date for recognizing protein sequences that are evolutionarily related has been profile hidden Markov models (HMMs). However, these models do not capture pairwise statistical preferences of residues that are hydrogen bonded in beta sheets. These dependencies have been partially captured in the HMM setting by simulated evolution in the training phase and can be fully captured by Markov random fields (MRFs). However, the MRFs can be computationally prohibitive when beta strands are interleaved in complex topologies. We introduce SMURFLite, a method that combines both simplified MRFs and simulated evolution to substantially improve remote homology detection for beta structures. Unlike previous MRF-based methods, SMURFLite is computationally feasible on any beta-structural motif.
Results: We test SMURFLite on all propeller and barrel folds in the mainly-beta class of the SCOP hierarchy in stringent cross-validation experiments. We show a mean 26% (median 16%) improvement in area under curve (AUC) for beta-structural motif recognition as compared with HMMER (a well-known HMM method) and a mean 33% (median 19%) improvement as compared with RAPTOR (a well-known threading method) and even a mean 18% (median 10%) improvement in AUC over HHPred (a profile–profile HMM method), despite HHpred's use of extensive additional training data. We demonstrate SMURFLite's ability to scale to whole genomes by running a SMURFLite library of 207 beta-structural SCOP superfamilies against the entire genome of Thermotoga maritima, and make over a 100 new fold predictions.
Availability and implementaion: A webserver that runs SMURFLite is available at: http://smurf.cs.tufts.edu/smurflite/
Contact: lenore.cowen@tufts.edu; bab@mit.edu
1 INTRODUCTION
Many researchers use hidden Markov models (HMMs) to annotate proteins according to homology, with popular systems such as Pfam (Finn et al., 2008) and Superfamily (Wilson et al., 2007) based on HMM methods integrated into UniProt. However, HMMs are limited in their power to recognize remote homologs because of their inability to model statistical dependencies between amino-acid residues that are close in space but far apart in sequence (Cowen et al., 2002; Lifson and Sander, 1980; Olmea et al., 1999; Steward and Thorton, 2002; Zhu and Braun, 1999).
For this reason, many have suggested (Lathrop and Smith, 1996; Liu et al., 2009; Menke et al., 2010; Peng and Xu, 2011; Thomas et al., 2008; White et al., 1994) that more powerful Markov random fields (MRFs) be used. MRFs employ an auxiliary dependency graph which allows them to model more complex statistical dependencies, including statistical dependencies that occur between amino-acid residues that are hydrogen bonded in beta sheets.
However, as the dependency graph becomes more complex, major design difficulties emerge. First, the MRF becomes more difficult to train. Second, it quickly becomes computationally intractable to find the optimal-scoring parse of the target to the model.
We have built a fully automated system, SMURFLite, that combines the power of MRFs with Kumar and Cowen's simulated evolution (Kumar and Cowen, 2010) (which offloads information about pairwise dependencies in beta sheets into new, artificial training data), in order to build the first MRF models that are computationally tractable for all beta-structural proteins, even those with limited training data. The SMURFLite system builds in part on the SMURF MRF (Menke et al., 2010), which uses multidimensional dynamic programming to simultaneously capture both standard HMM models and the pairwise interactions between amino acid residues bonded together in beta sheets. Unlike the full SMURF MRF, where the computational requirements of the random field become prohibitive on folds with deeply interleaved beta-strand pairs, such as barrels, SMURFLite is tractable on all beta-structural proteins. SMURFLite enables researchers to trade modeling power for computational cost by tuning an interleave threshold. The interleave threshold represents the maximum number of unrelated beta strands that can occur in linear sequence between the beta strands hydrogen bonded in a beta sheet while still being retained as pairwise dependencies in the MRF. As the interleave threshold increases, computation time increases, but so does the power of the MRF (see Fig. 1).
Fig. 1.

A closed beta barrel (PDB ID 1bw3, a Barwin domain) from the superfamily ‘Barwin-like endoglucanases’ to illustrate interleaving of strand pairs. Beta strands a and b, which close the barrel, have interleave 4, whereas strands c and d, which are adjacent in sequence, have interleave 1. Strands b and c have interleave 2.
We first test SMURFLite on all propeller and barrel folds in the mainly-beta class of the SCOP hierarchy in stringent cross-validation experiments. We show a mean 26% (median 16%) improvement in area under curve (AUC) for beta-structural motif recognition as compared with HMMER (a popular HMM method; Eddy, 1998) and a mean 33% (median 19%) improvement as compared with RAPTOR (a well-known threading method; Xu et al., 2003), and even a mean 18% (median 10%) improvement in AUC over HHPred (a profile–profile HMM method; Söding et al., 2005), despite HHpred's use of extensive additional training data. We demonstrate SMURFLite's ability to scale to whole genomes by running a SMURFLite library of 207 beta-structural SCOP superfamilies against the entire genome of T.maritima, and make over a 100 new fold predictions (available at http://smurf.cs.tufts.edu/smurflite). The majority of these predictions are for genes that display very little sequence similarity with any proteins of known structure, demonstrating the power of SMURFlite to recognize remote homologs.
We offer an online server (http://smurf.cs.tufts.edu/smurflite) for predicting remote homologs from our library of 207 mainly-beta superfamilies using SMURFLite. The online server sets the interleave threshold (the parameter that determines the complexity of the MRF) to 2; we have also shown that increasing the interleave number for SMURFLite can dramatically improve performance, but at a great computational cost. While the primary intent of using simulated evolution in conjunction with simplified MRFs is to compensate for the removal of highly-interleaved beta-strand pairs required for computational feasibility, we surprisingly find that simulated evolution can still improve full-fledged SMURF in cases of sparse training data. For instance, the 5-bladed beta propellers have only three superfamilies in SCOP, two of which contain only one family. We find that for the 5-bladed beta-propeller fold, combining SMURF and simulated evolution improves AUC from 0.73 for full SMURF alone to 0.89.
2 METHODS
2.1 Review of SMURF MRF framework
SMURF and SMURFLite rely on training data in the form of a multiple structure alignment with beta strand annotation. This alignment is created using the Matt program (Menke et al., 2008). Beta strand annotation is done on a structure-by-structure basis, where the beta-strand residue pairing is determined using the same algorithm implemented by the Rasmol (Sayle and Milner-White, 1995) visualization program. A post-processing step annotates those beta-strand residues that appear in more than half the structures in the alignment as beta-conserved. As gaps in beta strands would complicate training, this post-processing step makes beta-conserved template strands contiguous in the alignment exactly as in (Menke et al., 2010). The result at this stage is a sequence alignment (resulting from the Matt structural alignment) with conserved beta-strand pairs annotated according to the residue positions and conformation (buried or exposed to solvent).
The pairwise probability portion of the MRF is based on the beta probability tables that were computed by collecting a set of amphipathic beta sheets from the Protein Data Bank (PDB; Berman et al., 2000) and tabulating the frequencies of pairs of hydrogen-bonded residues in two tables, one for buried residues and one for residues exposed to solvent (Bradley et al., 2001; Menke et al., 2010). For each residue position, the most likely conformation (buried or exposed) is chosen based on whether that residue pairing is most probable from the buried or exposed beta-pairing tables.
Given a trained MRF, SMURF and SMURFLite align a query sequence to the MRF. The query phase of SMURF and SMURFLite computes the alignment of the sequence to the states of the MRF that maximizes the combined score:
In this combined score, the HMM score is the conditional probability of observing the sequence given the HMM portion of the model, and the pairwise score is the conditional probability of observing the paired beta-strand components of the sequence given the beta-pair portion of the model. Let the sequence have residues r1…rn, and the MRF have match states m1…ml, deletion states d1…dl and insertion states i1…il. Suppose that r1…rk and match states m1…ms have been assigned. Then, the probability of assigning rk to the next match state mj=ms+1 is:
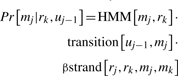 |
where uj−1 can be either dj−1, ij−1 or mj−1 depending on whether the current state is a deletion, insertion or match state. When the current state is a match state, the SMURFLite template replaces the transition[uj−1, mj] term with a value of 1. The β strand component set to be identically 1 unless the particular match state mj participates in a beta strand that is matched with a state mk earlier in the template. This component is the primary difference between our MRF and an ordinary HMM (Menke et al., 2010).
SMURFLite computes the maximum score of a sequence using multidimensional dynamic programming on the MRF. This dynamic programming resembles the classic Viterbi algorithm (Viterbi, 1967) used on HMMER's ‘plan7’ (Eddy, 1998) HMMs, except that some states are beta-strand states, which are required to be match states, and which are paired with other beta-strand nodes in the model. Because the pairwise component of the score can only be calculated for a given MRF node once it is determined what residue occupies the paired MRF node earlier in the sequence, each time the dynamic programming reaches a state in the MRF that corresponds to the first residue of the first beta strand in a set of paired beta strands, we need to keep track of multiple cases, depending on what residue in sequence is mapped to that state. SMURFLite uses a multidimensional array to memoize these possible subproblem solutions. A maximum gap size is set to the longest gap seen in the training data plus 20, for computational efficiency. When paired beta strands follow each other in sequence with no interleaving beta strands between them, the number of dimensions in the table for the dynamic programming is directly proportional to the maximum gap length. Let us call the last MRF state for the first of every pair of beta strands a ‘split’ state and the first MRF state for the second of that pair a ‘join’ state. Then, at every split state, the number of dimensions of the dynamic program will be multiplied by the maximum gap length, because the dynamic program must keep track of scores for each possible sequence position (up to the maximum gap length) that could be mapped to that state. At the corresponding join state, the number of dimensions will be reduced by the maximum gap length, because the scoring function can calculate all the pairwise probabilities of placing that residue into the join state, and then simply take the maximum of all ways to have placed its paired residue into the split state. However, when other beta strands are interleaved, the dynamic program must open additional multidimensional tables before clearing the previous ones from memory. Thus, the number of elements in the multidimensional table is never more than the sequence length times the maximum gap length raised to the interleaving number power.
2.2 Datasets
From SCOP (Murzin et al., 1995) version 1.75, we chose the folds ‘5-bladed Beta-Propellers’, ‘6-bladed Beta-Propellers’, ‘7-bladed Beta-Propellers’ and ‘8-bladed Beta-Propellers’. We also chose superfamilies from all of the mostly-beta folds containing the word ‘barrel’ in their description, whether open or closed, restricted to those superfamilies comprising at least four families (in order to facilitate leave-family-out cross-validation). These superfamilies were: ‘Nucleic acid-binding proteins’ (50249), ‘Translation proteins’ (50447), ‘Trypsin-like serine proteases’ (50494), ‘Barwin-like endoglucanases’ (50685), ‘Cyclophilin-like’ (50891), ‘Sm-like ribonucleoproteins’ (50182), ‘PDZ domain-like’ (50156), ‘Prokaryotic SH3-related domain’ (82057), ‘Tudor/PWWP/MBT’ (63748), ‘Electron Transport accessory proteins’ (50090), ‘Translation proteins SH3-like domain’ (50104), ‘Lipocalins’ (50814) and ‘FMN-binding split barrel’ (50475). Of these, we removed the superfamilies ‘Lipocalins’ and ‘Trypsin-like serine proteases,’ which were not structurally consistent enough to permit a multiple structure alignment for training HMMER or the SMURF variants, and which were broken into distinct superfamilies by (Daniels et al., 2012), with the result that 11 superfamilies containing barrels were selected. In addition, for the whole-genome search on T.maritima, out of 354 total superfamilies within the SCOP class ‘All beta proteins’, 288 (81%) of which contain at least two protein chains, 207 superfamilies (71%) were structurally consistent enough to be aligned using the Matt (Menke et al., 2008) structural alignment program. We built SMURFLite templates for these 207 superfamilies, and obtained from UniProt the protein sequences for T.maritima, comprising 1852 genes.
2.3 Training and testing process
For the beta-propeller folds, strict leave-superfamily-out cross-validation was performed. The propeller folds are structurally highly consistent (Menke et al., 2010), and thus high-quality Matt (Menke et al., 2008) multiple structure alignments were possible without descending to the superfamily level. For each propeller fold, its constituent superfamilies were identified. Each superfamily was left out, a training set was established from the protein chains in the remaining superfamilies, with duplicate sequences removed. An HMM (in the case of HMMER and HHPred) or MRF (in the case of SMURF and SMURFLite) were trained on the training set (HMMER parameter settings are discussed below). Protein chains from the left-out superfamily were used as positive test examples. Negative test examples were protein chains from all other folds in SCOP classes 1, 2, 3 and 4 (including propeller folds with differing blade counts), indicated as representatives from the non-redundant Protein Data Bank repository (nr-PDB) (Berman et al., 2000) database with non-redundancy set to a BLAST E-value of 10−7.
The beta propellers are atypical of most beta-structural SCOP folds, in that they structurally align well at the fold level of the SCOP hierarchy. For the beta-barrel superfamilies, strict leave-family-out cross-validation was performed. The barrel superfamilies are distinguished by strand number and shear as well as other structural features (Murzin et al., 1995), and so like most beta-structural motifs they do not align well structurally at the fold level. For this reason, the superfamily level was chosen for training. For each superfamily, its constituent families were identified. Each family was left out, a training set was established from the protein chains in the remaining families, with duplicate sequences removed. An HMM (in the case of HMMER and HHPred) or MRF (in the case of SMURF and SMURFLite) were trained on the training set. Protein chains from the left-out family were used as positive test examples. Negative test examples were protein chains from all other superfamilies in SCOP classes 1, 2, 3 and 4 (including other barrel superfamilies), indicated as representatives from the nr-PDB (Berman et al., 2000) database with non-redundancy set to a BLAST E-value of 10−7.
Each test example was aligned to the trained HMM (from HMMER and HHPred) and MRF, and was also threaded, using RAPTOR, against each individual chain in the training set (RAPTOR parameters are discussed below). The score reported for HMMER and HHPred was the output HMM score, and the score reported for SMURF and SMURFLite was the combined HMM and pairwise score from the MRF. For RAPTOR, the score reported for a test example was the highest score from all the scores resulting from threading that test example onto each chain in the training set. For each training set, the scores for each method were collected and a ROC curve (a plot of true positive rate versus false positive rate) computed. We report the area under the curve (AUC statistic) from this ROC curve (Sonego and Pongor, 2008).
2.4 P-values
SMURFLite computes the P-value for an alignment similarly to HMMER, using an extreme value distribution (EVD) (Eddy, 1998). An EVD is fitted to the distribution of raw scores over a random sampling of 5000 protein chains from across the SCOP hierarchy. The P-value is then simply computed as 1 –cdf(x ) for any raw SmurfLite score x, where cdf is the cumulative distribution function for the EVD.
2.5 SMURFLite augmented training data
(Kumar and Cowen, 2009, 2010) showed that ‘simulated evolution,’ augmenting limited training data with additional sequences produced by mutating the original sequences, improved the performance of HMMER at recognizing the same-superfamily level of homology. (Kumar and Cowen, 2010) used two types of simulated evolution: point-wise and pairwise. Here, we add only pairwise mutations based on beta-strand pairings, as we expect long-range interactions between beta strands to be highly conserved across similar structures. We postulated that the elimination of the beta-strand pairs SMURFLite must disregard because of computational complexity might be compensated for by augmenting the training data with artificial sequences based on likely mutations in those paired beta strands. This training data augmentation comes at insignificant runtime cost and is done before beta-strand pairs are removed from the template (but after their interleave number has been identified, where we define interleave number next below). The mutation frequencies are given by the Betawrap and SMURF (Bradley et al., 2001; Menke et al., 2010) pairwise probability tables. Using the same algorithm as (Kumar and Cowen, 2010), we generate 150 new artificial training sequences from each original training sequence. For each artificial sequence, we mutate at a 50 mutation rate per length of the beta strands. The parameters 150 and 50% were recommended by (Kumar and Cowen, 2010); we also evaluated a 10% mutation rate (a secondary peak according to their work) and performance was slightly worse (data available from the authors).
2.6 SMURFLite simplified random field
SMURFLite trains a MRF on a template built from a multiple structure alignment. Beta strands in the aligned set of structures are found by the program SmurfPreparse which is part of the SMURF package (Menke et al., 2010; Menke, 2009). The program not only outputs the positions of the consensus beta strands in the alignment, it also declares a position buried or exposed based on which of the two tables is the best fit to the amino acids that appear in that position in the training data. SMURFLite then assigns an interleave value to each beta-strand pair, as follows: any pairwise interaction between beta strands whose interleave value equals or exceeds the SMURFLite threshold is removed from the training data.
Consider three beta strands: A, B and C. Suppose strand A interacts with strand B and the (A,B) pair has an interleave value of 4, whereas strand B also interacts with strand C and that (B,C) pair has an interleave value of just 1. With a SMURFLite threshold of 2, the (A,B) pair would be discarded, but the (B,C) pair would remain in the training data. Thus, interleave numbers are properties of pairs of beta strands; a beta strand may be involved in multiple pairings, each of which may have a distinct interleave value. Discarding beta-strand pairs whose interleave value equals or exceeds the threshold guarantees that the MRF will have no beta-strand pairs greater than or equal to that threshold, and thus bounds the computational complexity, which is exponential in the maximum interleave value found in a training template.
Note that SMURFLite with an interleave threshold of 0, which will discard all beta-strand pair information, is simply an HMM.
2.7 HMMER implementation
SMURFLite was tested against HMMER version 3.0a2 with the ‘–seqZ 1’ and ‘–seqE 10000’ options applied to hmmsearch, and the ‘–symfrac 0.2’ and ‘–ere 0.7’ options applied to hmmbuild. The –seqZ 1 option ensures that E-values are comparable regardless of the size of the sequence database, whereas the –seqE 10000 option forces HMMER to return results for all query sequences. The –symfrac 0.2 option requires that only 20% of sequences need to be in agreement to cause a match state in a given column (the default is 50%). Given the remote homology at which we were performing experiments, 50% was an unreasonably high threshold that led to few match states being found. This option was also used by (Kumar and Cowen, 2009). The –ere option sets the minimum relative entropy per position target to 0.7 bits (the default is 0.59). Note that HMMER versions 3.0a2 and 3.0 both SAM sequence entropy (Karplus and Hu, 2001) by default. This entropy weighting scheme has been shown to be superior for remote homology detection tasks (Johnson, 2006; Kumar and Cowen, 2009).
HMMER 3.0a2 was used despite having been superseded by version 3.0, because it uniformly performs better on this task. This is because version 3.0 contains computational optimizations that cause it to reject a sequence (with no score provided) quickly if it does not appear to align well. These optimizations, however, cause nearly all query sequences outside the family level of homology to fail and return no score, with the result that HMMER version 3.0 never surpasses an AUC of 0.5.
2.8 RAPTOR implementation
SMURFLite was tested against RAPTOR, which was run with the options ‘-a nc’ indicating that the default threading algorithm described in the RAPTOR paper (Xu et al., 2003) was used. In addition, RAPTOR used the weighting parameters ‘weightMutation = 1.4009760151,’ ‘weightSingleton = 1,’ ‘weightLoopGap = 16.841836238,’ ‘weightPair = 0,’ ‘weightGapPenalty = 1’ and ‘weightSStruct = 3.0137849223.’ RAPTOR uses both sequence and structural features, and these options represent the recommended balance of these features (Xu et al., 2003).
2.9 HHPred implementation
SMURFLite was tested against HHPred version 1.5.1. HHPred HMMs for each SCOP family were downloaded from the HHPred web site, and queried using hhsearch. The score of the best-scoring family HMM within each superfamily was used in computing ROC curves.
2.10 Whole-genome search
All 1852 protein sequences from T.maritima were queried against beta-structural templates constructed from the nr-PDB (Berman et al., 2000) with non-redundancy determined by an E-value of 10−7, organized according to those 207 beta-structural superfamilies from SCOP that were able to be aligned using the Matt structural alignment program, using SMURFfLite with an interleave threshold of 2 and simulated evolution mutation rate of 50% on the residues that participate in beta strands. We computed P-values and alignments for all 1852×207 possible hits.
3 RESULTS
3.1 SMURFLite validation
SMURFLite's ability to recognize beta propellers and barrels was compared with HMMER (Eddy, 1998), RAPTOR (Xu et al., 2003) and HHPred (Söding et al., 2005) in a stringent cross-validation experiment. From SCOP (Murzin et al., 1995) version 1.75, we chose the folds ‘5-bladed Beta-Propellers’, ‘6-bladed Beta-Propellers’, ‘7-bladed Beta-Propellers’ and ‘8-bladed Beta-Propellers’. We also chose superfamilies from all of the mostly-beta folds containing the word ‘barrel’ in their description, whether open or closed, restricted to those superfamilies comprising at least four families (in order to facilitate leave-family-out cross-validation). These superfamilies were: ‘Nucleic acid-binding proteins’ (50249), ‘Translation proteins’ (50447), ‘Trypsin-like serine proteases’ (50494), ‘Barwin-like endoglucanases’ (50685), ‘Cyclophilin-like’ (50891), ‘Sm-like ribonucleoproteins’ (50182), ‘PDZ domain-like’ (50156), ‘Prokaryotic SH3-related domain’ (82057), ‘Tudor/PWWP/MBT’ (63748), ‘Electron Transport accessory proteins’ (50090), ‘Translation proteins SH3-like domain’ (50104), ‘Lipocalins’ (50814) and ‘FMN-binding split barrel’ (50475). Of these, we removed the superfamilies ‘Lipocalins’ and ‘Trypsin-like serine proteases,’ which were not structurally consistent enough to permit a multiple structure alignment for training HMMER or the SMURF variants, and which were broken into distinct superfamilies by (Daniels et al., 2012), with the result that 11 superfamilies containing barrels were selected.
SMURFLite was tested on these 5 propeller folds and 11 barrel superfamilies, with interleave thresholds of 1, 2 and 3, and with and without simulated evolution on the beta-strands (Kumar and Cowen, 2010). Here, the interleave threshold is a parameter of SMURFLite that trades off the computational complexity with the ability of the MRF to capture complicated long-range dependencies.
The balance between accuracy and computational efficiency is determined by the interleave threshold at which SMURFLite is run. In particular, we found that SMURFLite set to an interleave threshold of 3 or less was always fast. Thus, our first question is how SMURFLite with and without simulated evolution performs on our test set when the interleave threshold is set to 3 or less. We found that SMURFLite became extremely slow at an interleave threshold of 4, and essentially intractable at an interleave threshold of 5 or above. While SMURFLite with an interleave threshold of 1 or 2 requires roughly 1 s of wall-clock time on a 12-core 2.4 GHz AMD Opteron server, an interleave threshold of 4 raises this run-time requirement to 7–10 min. Restricting the interleave threshold to 3 or less has different impacts on the different folds in our test set. In particular, the beta strands in the propeller folds never have an interleave >3, which means that full SMURF, as we know, is tractable on these folds. However, we were still interested in how simplifying the random field to an interleave of 2 or 1 would impact performance, and also whether simulated evolution would help. In contrast, the barrel superfamilies in our test set contain a maximum beta-strand interleave of between 4 and 8. Interestingly, none of these barrels contained any beta strands with an interleave of 3 in the consensus Matt (Menke et al., 2008) alignment, so our restriction of running SMURFLite with an interleave threshold of 3 or less is equivalent, on the barrels, to running SMURFLite with an interleave threshold of 2.
SMURFLite with interleave threshold 2 and simulated evolution performs well on all propeller folds, with AUCs between 0.89 and 0.99. It always performs better than HMMER, and better than RAPTOR and HHPred except on the 7-bladed propellers (of which there are 39 non-redundant solved structures in 19 SCOP families), where HHPred achieves an AUC of 0.99 and RAPTOR achieves an AUC of 0.95 versus an AUC of 0.93 for SMURFLite with interleave threshold 2 and no simulated evolution (Table 1). Interestingly, on the 5-bladed propellers (of which there are only 14 non-redundant solved structures in 7 SCOP families), adding simulated evolution seems to greatly improve performance; even SMURFLite with an interleave threshold of 2 with simulated evolution outperforms full-fledged SMURF. While these results focus on the accuracy of the MRF score for the remote homolog decision problem, as opposed to the question of alignment quality, we note that SMURFLite with an interleave threshold of 1 or 2 produces highly similar alignments to full SMURF, particularly with respect to placing the ‘blades’ of the 6-, 7- and 8-bladed propellers.
Table 1.
AUC on beta-propeller folds. Best AUC for each structure is marked in bold
| HMMER | RAPTOR | HHPred | SMURF-Lite 1 | SMURF-Lite 1, SimEv | SMURF-Lite 2 | SMURF-Lite 2, SimEv | SMURF-Lite 3 | SMURF-Lite 3, SimEv | |
|---|---|---|---|---|---|---|---|---|---|
| 5-bladed | – | – | – | 0.75 | 0.89 | 0.73 | 0.89 | 0.73 | 0.89 |
| 6-bladed | 0.82 | 0.82 | 0.88 | 0.92 | 0.93 | 0.96 | 0.95 | 0.96 | 0.96 |
| 7-bladed | 0.89 | 0.95 | 0.99 | 0.92 | 0.91 | 0.93 | 0.91 | 0.93 | 0.91 |
| 8-bladed | – | 0.64 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 |
Note: for SmurfLite, the number (1,2,3) indicates the interleave threshold, and SimEv is simulated evolution. A dash (‘–’) in a result entry indicates the method failed on these structures, i.e. an AUC of <0.6.
For all 11 beta-barrel superfamilies, there is a maximum interleave number that ranges from 4 (as in the ‘Sm-like ribonucleoproteins’) to 8 (as in the ‘Cyclophilin-like’ superfamily). We find that for 6 of the 11 beta-barrel superfamilies, SMURFLite with an interleave of 2 and simulated evolution outperforms HMMER, RAPTOR and HHPred. For two of the remaining superfamilies, HMMER performs best; for two of the remaining superfamilies, RAPTOR performs best; and for one superfamily, HHPred performs best (Table 2).
Table 2.
AUC on beta-barrel superfamilies
| HMMER | RAPTOR | HHPred | SMURF-Lite 1 | SMURF-Lite 1, SimEv | SMURF-Lite 2 | SMURF-Lite 2, SimEv | |
|---|---|---|---|---|---|---|---|
| SMURFLite performs best | |||||||
| Translation proteins | – | – | 0.66 | 0.93 | 0.92 | 0.93 | 0.93 |
| Barwin-like endoglucanases | – | – | 0.75 | – | 0.77 | – | 0.63 |
| Cyclophilin-like | 0.67 | 0.61 | 0.7 | 0.82 | 0.85 | 0.82 | 0.83 |
| Sm-like ribonucleoproteins | 0.73 | 0.71 | 0.77 | 0.76 | 0.71 | 0.76 | 0.85 |
| Prokaryotic SH3-related domain | 0.81 | – | – | 0.83 | 0.82 | 0.83 | 0.83 |
| Tudor/PWWP/MBT | 0.78 | 0.74 | 0.67 | 0.83 | 0.77 | 0.83 | 0.79 |
| Nucleic acid-binding proteins | 0.75 | – | 0.67 | 0.76 | 0.89 | 0.76 | 0.92 |
| HHPred performs best | |||||||
| Translation proteins SH3-like | 0.83 | 0.81 | 0.86 | 0.62 | – | 0.62 | – |
| RAPTOR performs best | |||||||
| PDZ domain-like | 0.96 | 1.0 | 0.99 | 0.97 | 0.97 | 0.97 | 0.97 |
| FMN-binding split barrel | 0.62 | 0.82 | 0.61 | – | – | – | – |
| HMMER performs best | |||||||
| Electron Transport accessory proteins | 0.84 | – | 0.77 | 0.63 | – | 0.63 | 0.66 |
Note: for SmurfLite, the number (1,2) indicates the interleave threshold, and SimEv is simulated evolution. A dash (‘–’) in a result entry indicates the method failed on these structures, i.e. an AUC of <0.6.
As discussed above, SMURFLite begins to test the limits of computational tractability when interleave numbers of 4 are allowed. Since many barrel structures had beta-strand pairs with interleaves of 4, we wished to test if incorporating these more long-range pairwise dependencies into our MRF would improve performance. Some barrel superfamilies on which we tested have only strand pairs of interleave 1 or 2, excepting a pair of beta strands that close the barrel and thus have an interleave equivalent to the number of strands in the barrel. Certainly, including that last strand is beyond the computational power of SMURFLite. Other barrels, whether open or closed, have more complex strand topology and interleaves of 3 or 4 are common even in the middle of the barrels. We chose to run SMURFLite with an interleave of 4 on one of the barrel superfamilies of moderately complex topology, the ‘Barwin-like endoglucanase’ superfamily, of which an example appears in Figure 1. The ‘Barwin-like endoglucanase’ superfamily contains ‘Barwin,’ a protein that may be involved in a common defense mechanism in plants (Svensson et al., 1992).
On the ‘Barwin-like endoglucanase’ superfamily, we find an enormous improvement in performance from capturing that last strand pair, with AUC improving from 0.63 for SMURFLite with an interleave threshold of 2 and simulated evolution, to 0.94 for SMURFLite with an interleave threshold of 4 and simulated evolution (Fig. 2). Note that both HMMER and RAPTOR fail entirely on this superfamily.
Fig. 2.

Performance of SMURFLite compared with other methods on the ‘Barwin-like endoglucanases’ beta-barrel superfamily according to the AUC measure. For SMURFLite, the number (1,2,4) indicates the interleave threshold (indicating which strand pairs in the barrel participate in the MRF; note that interleave 3 is omitted since it is identical to interleave 2 for this fold), and SimEv indicates that simulated evolution was also performed on the beta strands in the training data. As the interleave threshold increases and the MRF becomes more powerful, performance tends to improve. Including simulated evolution also improves performance.
3.2 SMURFLite on whole genomes
We considered all 1852 genes from the bacterium T.maritima, a thermophilic organism that bears some similarity to Archaea and whose cell is wrapped in an outer membrane, or ‘toga’ (Huber et al., 1986). Out of 354 total superfamilies within the SCOP class ‘All beta proteins’, 288 (81%) of which contain at least two protein chains, 207 superfamilies (71%) were structurally consistent enough to be aligned using the Matt (Menke et al., 2008) structural alignment program. We built SMURFLite templates for these 207 superfamilies, and obtained from UniProt the protein sequences for each of 1852 genes. We predict 139 of the 1852 genes from T.maritima to belong to one of the 207 beta-structural SCOP superfamilies we consider, with a P-value of <0.005. Of the 139 genes about which we make predictions, 28 already have solved structures in the PDB, however, since there is a substantial time lag before new PDB structures are assigned to SCOP, only one of those structures was already given a SCOP assignment (and thus only one of these 28 structures potentially informed SMURFLite training). Thus, determining the correct SCOP asignments of the remaining 27 (an easy computational problem given full structural information) allows us to estimate the accuracy of SMURFLite predictions on these structures. Using the Matt (Menke et al., 2008) structural alignment program and the methodology of (Daniels et al., 2012), we computed SCOP superfamilies for all 27, and in 100% of the cases, Smurflite's predictions matched the structural alignments and hence SCOP superfamily assignments. We now survey the remaining 111 structures on which SMURFLite makes predictions, for which structural information is not yet available. In total, 8 of these 111 structures also had their SCOP superfamilies predicted in the study of Zhang et al. (2009) and in all 8 cases, our predictions are in agreement with the other study. We note that for most of these 111 structures, not only is not there solved structure, but there also is not close homology to proteins of solved structure. In particular, none have BLAST hits in UniProt with solved structure and >80% sequence identity, 18 have BLAST hits in UniProt with solved structure and between 30% and 80% sequence identity, and 4 have BLAST hits in UniProt with solved structure and <20% sequence identity. As an example, the gene Q9X087 shares only 20% sequence identity with its closest structurally-solved BLAST hit (Rhoptry protein from Plasmodium yoelii yoelii, which forms an alpha-helical structure) but we predict it to belong in the ‘beta-Galactosidase/glucuronidase domain’ SCOP superfamily with a P-value of 0.0006.
All models predicted can be found at http://smurf.cs.tufts.edu/smurflite/
4 DISCUSSION
We have presented SMURFLite, a method that combines long-range pairwise beta-strand interactions via a simplified MRF with simulated evolution, a method that augments training data to capture pairwise beta-strand interactions as well. SMURFLite in most cases performs considerably better than HMMER and RAPTOR; however, we examine those structures for which this is not so. We postulate that RAPTOR performs best in the case when there is significant structural conservation across families, whereas HMMER excels when there is a small but highly conserved sequence signature in members of a superfamily. In all four beta-barrel superfamilies on which RAPTOR achieves an AUC of <0.5, we see considerable structural variation in the protein backbones within each superfamily, according to the metric of (Daniels et al., 2012), as compared with the other barrel superfamilies. In contrast, the barrels on which RAPTOR performed best exhibited little structural variation. The cases in which SMURFLite performs poorly exhibit an interesting property: the structural alignment of the protein chains used in the training set preserves few, or sometimes none, of the beta strands as ‘consensus’ beta strands. When a significant number of beta strands are missing in this manner from the training data, SMURFLite exhibits poor specificity, scoring some non-homologous sequences comparably to homologous ones. The ‘Translation Proteins SH3-Like Domain,’ a superfamily in which HMMER significantly outperforms SMURFLite, is one in which the consensus alignment obtained from Matt retains zero beta strands, even though each individual structure has four strands. Thus, SMURFLite behaves like HMMER, except without HMMER's heuristic for quickly failing bad alignments, leading SMURFLite to report more false positives. The very premise of SMURFLite rests on the conservation of beta strands, and this finding emphasizes the importance of evolutionarily faithful structural alignments. In future work, we will also consider alternative structural aligners, such as TMalign (Zhang and Skolnick, 2005), in cases where they produce alignments that better conserve secondary structure.
We also compared SMURFLite to HHPred, though in a sense this is not an apples-to-apples comparison, because HHPred uses all of protein sequence space to build profiles for training; thus it can leverage a much larger training set than HMMER, RAPTOR, or SMURF or SMURFLite. Thus it is somewhat surprising that SMURFLite outperforms HHPred in median AUC on the propellers and barrels. We expect HHPred to excel in particular on superfamilies and folds with a high HHPred NEFF (Söding et al., 2005), where NEFF is the ‘number of effective families’ available for training the HHPred HMM.
In contrast, simulated evolution seems to help SMURFLite most on those structural motifs where the HHPred NEFF is lowest; i.e. it can generate diverse training data when diverse training data is lacking. A profile version of SMURFLite would be close in spirit to HHPred, and based on the previous discussions we would expect profiles might improve performance; this will be a subject for future investigation. We observed that simulated evolution either improves or does not affect AUC for beta-barrel superfamilies and beta-propeller folds with a HHPred NEFF of 20 or lower. The only cases in which we observed simulated evolution decreasing AUC were those cases where the NEFF was >20.
While the intent of using simulated evolution in conjunction with simplified MRFs is to compensate for the removal of highly-interleaved beta-strand pairs required for computational feasibility, we find that simulated evolution can still improve full-fledged SMURF in cases of sparse training data. For instance, the 5-bladed beta propellers have only three superfamilies in SCOP, two of which contain only one family. We find that for the 5-bladed beta-propeller fold, combining SMURF and simulated evolution improves AUC from 0.73 for full SMURF alone to 0.89.
We have demonstrated that SMURFLite is a powerful MRF methodology for beta-structural motif recognition that is computationally tractable enough to scale to whole genomes, requiring approximately 3 h to scan the T.maritima genome on a small compute cluster. We have also shown that increasing the interleave number for SMURFLite can have dramatic effects on performance, but at a great computational cost. Thus, looking at heuristic methods (Murphy et al., 1999; Smyth et al., 1997) that approximately compute the SMURF score more efficiently may add even more power to our approach in practice.
ACKNOWLEDGEMENTS
We thank A.Kumar, M.Menke and J.Xu.
Funding: N.M.D. and L.J.C. were funded in part by NIH grant 1R01GM080330. R.H. and B.B. were funded in part by NIH grant 1R01GM08187.
Conflict of Interest: none declared.
REFERENCES
- Berman H., et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley P., et al. Betawrap: successful prediction of parallel β-helices from primary sequence reveals an association with many microbial pathogens. Proc. Natl Acad. Sci. 2001;98:14819–14824. doi: 10.1073/pnas.251267298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L., et al. Predicting the beta-helix fold from protein sequence data. J. Comput. Biol. 2002;9:261–276. doi: 10.1089/10665270252935458. [DOI] [PubMed] [Google Scholar]
- Daniels N., et al. Touring protein space with Matt. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012;9:286–293. doi: 10.1109/TCBB.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Finn R., et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R., et al. Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90c. Arch. Microbiol. 1986;144:324–333. [Google Scholar]
- Johnson S. PhD Thesis. Washington University, St. Louis; 2006. Remote protein homology detection using hidden Markov models. [Google Scholar]
- Karplus K., Hu B. Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics. 2001;17:713–720. doi: 10.1093/bioinformatics/17.8.713. [DOI] [PubMed] [Google Scholar]
- Kumar A., Cowen L. Augmented training of hidden Markov models to recognize remote homologs via simulated evolution. Bioinformatics. 2009;25:1602–1608. doi: 10.1093/bioinformatics/btp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Cowen L. Recognition of beta-structural motifs using hidden Markov models trained with simulated evolution. Bioinformatics. 2010;26:i287–i293. doi: 10.1093/bioinformatics/btq199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop R., Smith T. Global optimum protein threading with gapped alignment and empirical pair scores. J. Mol. Biol. 1996;255:641–645. doi: 10.1006/jmbi.1996.0053. [DOI] [PubMed] [Google Scholar]
- Lifson S., Sander C. Specific recognition in the tertiary structure of β-sheets of proteins. J. Mol. Biol. 1980;139:627–629. doi: 10.1016/0022-2836(80)90052-2. [DOI] [PubMed] [Google Scholar]
- Liu Y., et al. Conditional graphical models for protein structural motif recognition. J. Comput. Biol. 2009;16:639–657. doi: 10.1089/cmb.2008.0176. [DOI] [PubMed] [Google Scholar]
- Menke M., et al. Matt: local flexibility aids protein multiple structure alignment. PLoS Comput. Biol. 2008;4:e10. doi: 10.1371/journal.pcbi.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke M., et al. Markov random fields reveal an n-terminal double beta-propeller motif as part of a bacterial hybrid two-component sensor system. Proc. Natl Acad. Sci. 2010;107:4069–4074. doi: 10.1073/pnas.0909950107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke M. PhD Thesis. Massachusetts: MIT; 2009. Computational approaches to modeling the conserved structural core among distantly homolgous proteins. [Google Scholar]
- Murphy K.P., et al. Proceedings of Uncertainty in AI. San Francisco, CA, USA: Morgan Kaufmann Publishers Inc.; 1999. Loopy belief propagation for approximate inference: an empirical study; pp. 467–475. [Google Scholar]
- Murzin A., et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Olmea O., et al. Effective use of sequence correlation and conservation in fold recognition. J. Mol. Biol. 1999;293:1221–1239. doi: 10.1006/jmbi.1999.3208. [DOI] [PubMed] [Google Scholar]
- Peng J., Xu J. A multiple-template approach to protein threading. Prot. Struct. Func. Bioinformatics. 2011;79:1930–1939. doi: 10.1002/prot.23016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayle R., Milner-White E. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- Smyth P., et al. Probabilistic independence networks for hidden markov probability models. Neural comput. 1997;9:227–269. doi: 10.1162/neco.1997.9.2.227. [DOI] [PubMed] [Google Scholar]
- Söding J., et al. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Suppl. 2):W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonego A.K., Pongor S. ROC analysis: applications to the classification of biological sequences and 3D structures. Brief. Bioinformatics. 2008;9:198–209. doi: 10.1093/bib/bbm064. [DOI] [PubMed] [Google Scholar]
- Steward R.E., Thorton J.M. Prediction of strand pairing in antiparallel and parallel β-sheets using information theory. Prot. Struct. Func. Bioinformatics. 2002;48:178–191. doi: 10.1002/prot.10152. [DOI] [PubMed] [Google Scholar]
- Svensson B., et al. Primary structure of barwin: a barley seed protein closely related to the C-terminal domain of proteins encoded by wound-induced plant genes. Biochemistry. 1992;31:8767–8770. doi: 10.1021/bi00152a012. [DOI] [PubMed] [Google Scholar]
- Thomas J., et al. Graphical models of residue coupling in protein families. IEEE/ACM Trans. Comp. Biol. Bioinf. 2008;5:183–197. doi: 10.1109/TCBB.2007.70225. [DOI] [PubMed] [Google Scholar]
- Viterbi A.J. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. IEEE Trans. Inf. Theory. 1967;13:260–269. [Google Scholar]
- White J., et al. Modeling protein cores with Markov random fields. Math. Biosci. 1994;124:149–179. doi: 10.1016/0025-5564(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Wilson D., et al. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res. 2007;35:D308–D313. doi: 10.1093/nar/gkl910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., et al. Raptor: optimal protein threading by linear programming. J. Bioinformatics Comput. Biol. 2003;1:95–117. doi: 10.1142/s0219720003000186. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–1549. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Braun W. Sequence specificity, statistical potentials and 3D structure prediction with self-correcting distance geometry calculations of beta-sheet formation in proteins. Protein Sci. 1999;8:326–342. doi: 10.1110/ps.8.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]