

Abstract
Motivation: Analysis of protein–protein interaction networks (PPINs) at the system level has become increasingly important in understanding biological processes. Comparison of the interactomes of different species not only provides a better understanding of species evolution but also helps with detecting conserved functional components and in function prediction.
Method and Results: Here we report a PPIN alignment method, called PINALOG, which combines information from protein sequence, function and network topology. Alignment of human and yeast PPINs reveals several conserved subnetworks between them that participate in similar biological processes, notably the proteasome and transcription related processes. PINALOG has been tested for its power in protein complex prediction as well as function prediction. Comparison with PSI-BLAST in predicting protein function in the twilight zone also shows that PINALOG is valuable in predicting protein function.
Availability and implementation: The PINALOG web-server is freely available from http://www.sbg.bio.ic.ac.uk/~pinalog. The PINALOG program and associated data are available from the Download section of the web-server.
Contact: m.sternberg@imperial.ac.uk
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
In recent years, there has been a substantial increase in the quantity of protein–protein interactions (PPIs) being detected by experimental methods. The analysis of PPI networks (PPINs) at the system level has become increasingly important in understanding biological processes. Comparison of the interactomes of different species not only provides a better understanding of species evolution but also helps with detecting conserved functional components and in function prediction.
Several methods have been developed to align interactomes, both globally and locally, most of which use sequence similarity or network topology or both in establishing the equivalence. PathBLAST (Kelley et al., 2003) marked the first local alignment method and was similar in principle to the BLAST search algorithm. Other local alignment methods include: NetworkBLAST (Sharan et al., 2005), which is the next generation of PathBLAST; MaWISH (Koyutürk et al., 2006), which adopts the evolutionary models of match, mismatch and deletion of the proteins; Graemlin 1.0 (Flannick et al., 2006), which infers an alignment from network modules; the Bayesian method (Berg and Lassig, 2006); the match and split algorithm (Narayanan and Karp, 2007); and Phunkee (Cootes et al., 2007), which aligns proteins based on sequence and context in the networks. The global alignment of networks proves to be more challenging due to the complexity and scale of the problem. Graemlin 2.0 (Flannick et al., 2009) formulates a model for protein duplication, deletion and mutation and aligns the network progressively using a hill-climbing algorithm. The Markov-random field-based method (Bandyopadhyay, 2006) and IsoRank (Singh et al., 2008) solve the problem by eigenvalue-based methods. GRAAL (Kuchaiev et al., 2010) uses only the network topological similarity to align the networks using a seed-and-extend method for graph alignment.
In many applications of PPIN alignment, the underlying objective is to equivalence proteins with related function and interacting partners. However, in most alignment methods only the sequence data or network topology are used to align the input networks despite the availability of other sources of information such as function annotation or gene expression. While sequences are informative in elucidating the orthologous relationships of proteins across species, they do not necessarily indicate functional similarity. The omission of function information could therefore result in many pairs of equivalenced proteins having little or no similarity of function. This in turn makes it less accurate in detecting conserved functional modules or predicting protein function. Recently, Ali and Deane (2009) have introduced functional similarity of proteins into a local alignment method. They used functional similarity in combination with sequence similarity in the local alignment match and split algorithm (Narayanan and Karp, 2007) and identified similar functional subnetworks. The recently developed method MI-GRAAL (Kuchaiev and Przulj, 2011) presents a global alignment algorithm in which different information can be incorporated such as topological features, sequence similarity and functional similarity. However, there has been no assessment on how the inclusion of functional similarity might influence the resulting alignments and any subsequent applications.
Here we describe PINALOG, a global network alignment algorithm. PINALOG forms the alignment between two PPINs based on the similarities of protein sequence and the protein function between the two networks. Functional similarity is formalized using GO (gene ontology) annotations. The main contributions in PINALOG are the use of communities in the networks to identify seed protein pairs and the scoring schemes used in the extension steps to include the neighbourhood similarity of mapped protein pairs. The benchmarking of PINALOG in comparison with other alignment methods shows that PINALOG obtains a good balance in including these features as it generates an alignment with consistently high number of conserved interactions, homologous aligned pairs and functionally similar protein pairs. Direct applications of PINALOG include protein complex prediction and protein function prediction. Complex prediction is performed by direct inheritance of protein complex data from a known species to an unknown one. PINALOG has been tested for complex prediction power using a human-yeast alignment. Protein function prediction is via direct transfer to an un-annotated protein from an annotated protein. The power of our function prediction is assessed in a cross-validated study.
2 METHODS
The design of the PINALOG alignment method is based on the observation that proteins are not uniformly distributed throughout PPINs. Instead there are some proteins that form well connected subnetworks. Furthermore, it has been shown by Brohee and van Helden (2006) that protein complexes and functional modules form highly connected components in the PPINs. Therefore, it would be more reliable and efficient to align two PPINs by first finding highly similar protein pairs (seed protein pairs) from these highly connected protein subnetworks (referred to as communities from now on) in the networks and then extending the alignment to other proteins in the neighbourhoods of seed protein pairs.
Let A and B be the PPINs for species A and B. ai and bj are the i-th and j-th proteins in PPIN A and B, respectively. CiA is the i-th community in PPIN A, and CjB is the j-th community in B. PINALOG aligns A and B in three steps as summarized in Figure 1: community detection, community mapping and extension mapping.
Fig. 1.

PINALOG global alignment method comprises of three main steps: (i) Community detection: identifies dense subnetworks of input networks using CFinder (e.g. red and blue 3, 4 and 5-node subnetworks in the lower part of the diagram). (ii) Community mapping: maps similar communities that have high similarity scores, i.e. containing many inter-species proteins with high similarity scores. In the first table (lower part of the diagram), mapped communities with high similarity scores are marked in red numbers. Similar protein pairs from mapped communities are extracted to form a list of core pairs. (iii) Extension mapping: maps proteins in the neighbourhood of the core protein pairs which are then added to the core, e.g. protein I mapped with X and H with Y in the second table in the lower part of the diagram. Extension mapping is repeated until no more pair is added.
Step 1: Community detection of input networks using CFinder
Communities in biological networks such as PPINs often indicate functional groupings of proteins in the network (Adamcsek et al., 2006; Lewis et al., 2010; Song and Singh, 2009). There are several methods to detect communities within a network such as the minimum-cut method, hierarchical clustering or the Girvan–Newman algorithm. In PINALOG, we employ CFinder (Palla et al., 2005), a clique percolation method, which is capable of detecting overlapping communities. Denote a k-clique as a subgraph of the network composed of k proteins where all pairs of proteins interact. Then a k-clique community is defined as union of all k-cliques that are reachable from each other through adjacent k-cliques (two adjacent k-cliques have k−1 proteins in common). CFinder constructs communities by merging adjacent k-cliques (see Supplementary Material for more details).
Node scoring scheme
The similarity between two proteins is a combination of sequence and functional similarity in the seed identification and with additional topological similarity in the extension mapping process. The sequence similarity of two proteins ai and bj is calculated based on their BLAST bit score as
| (1) |
S(ai, bj) is the BLAST bit score value when aligning ai and bj. Here, only pairs of proteins with an E-value <10−5 are used to calculate sequence similarity.
The functional similarity of two proteins annotated with GO terms is calculated by the method defined by Schlicker et al. (2006). Given two proteins ai and bj with two sets of associated GO terms g(ai)={t1, t2,…, tk} and g(bj)={t′1, t′2,…, t′l}, the protein functional similarity is calculated based on the semantic similarity (Lord et al., 2003) of terms in g(ai) and g(bj). Semantic similarity depends on the rarity of the terms in the annotation database as well as their distance in the ontology. The semantic similarity between each term in g(ai) and each term in g(bj) is calculated, resulting in a k×l table of semantic similarities. Let rowScore and colScore be the average of the row maxima and the average of column maxima in the table, respectively (see Supplementary Fig. SI-1), the Schlicker's similarity between ai and bj is then calculated as
| (2) |
The similarity of two proteins in Step (2) is defined as a linear combination of functional similarity and sequence similarity
| (3) |
In PINALOG, θ provides a relative weighting between sequence and functional similarity. The value of θ is automatically decided based on the number of reciprocal best BLAST hits in the protein sequences between two input species, reflecting the closeness of two species and thus the contribution of sequence similarity in the overall score. Details of how θ is calculated are given in Supplementary Material.
Step 2: Community mapping to obtain seed protein pairs
Community mapping is determined in two steps. The first step is to determine the highest score when equivalencing proteins in each community in species A with each community in species B. To this end, we define the similarity between two communities as the score F(CiA, CjB), the sum of similarities between protein pairs in the optimal equivalence (OptMap) of proteins in community CiA in species A with proteins in CjB in species B using the Hungarian method (Kuhn, 2005). The optimal equivalence is the mapping where the sum of protein pair similarities is largest. The Hungarian algorithm is a combinatorial optimization algorithm solving the assignment problem in polynomial time (see Supplementary Material).
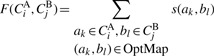 |
(4) |
The first step produces a community similarity matrix. This matrix is used in the second step to obtain the best equivalence of communities by maximizing the community scoring function:
| (5) |
The maximization of F(core) is achieved by Hungarian method based on the community similarity matrix. For each pair of mapped communities, the Hungarian mapping of proteins between them are obtained and added to the list of seed protein pairs. A filtering step is used to retain only the top 15% pairs of mapped proteins as the seeds for extension, known as the core equivalences.
Step 3: Extension mapping
Extension mapping is performed by considering neighbours of proteins in the core equivalence as candidates for adding to the alignment. In addition to protein sequence and functional similarity, topological similarity in the PPINs is included in the form of neighbourhood similarity. Let N(ai) and N(bj) be the set of all first neighbours (proteins separated by one interaction) and second neighbours (proteins separated by two interactions) of ai in A and bj in B. Let d(ak, al) denote the distance between ak and al in a network. The similarity between ai and bj in extension mapping is then defined as
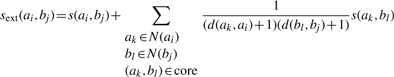 |
(6) |
By awarding the candidate pairs with a proportion of the score of neighbouring aligned pairs, PINALOG aims at mapping more neighbouring protein pairs. The Hungarian method is used to find the optimal equivalence of candidates. These candidates are then added to the equivalences in the core. The process is repeated until no more pairs are added (see Supplementary Fig. SI-1). As CFinder identifies communities that can be overlap, community mapping can result in many-to-many mappings in seed protein pairs, thus in the final alignment.
3 RESULTS
3.1 Alignment of human PPIN with other species
We have aligned different pairs of PPINs from human, yeast, fly, worm and mouse using: PINALOG; two state-of-the-art alignment methods Isorank and Graemlin 2.0, the integrative method MI-GRAAL and the naïve BLAST approach by reciprocal best BLAST hit (denoted as BLAST). The PPINs of these species were obtained from IntAct (Aranda et al., 2010) and aligned by both PINALOG and IsoRank. Graemlin 2.0 requires a training set to learn the parameters for the alignment. Thus comparison with Graemlin can only be made using the data set in the package, which provides PPINs of fly and yeast (denoted as fly2 and yeast2). As IsoRank and MI-GRAAL generate alignments with one-to-one mapping, for ease of comparison we reduce the final alignment of PINALOG to a one-to-one mapping (see Supplementary Material).
The aim of PINALOG is to obtain the best equivalence considering a balance of sequence homology, functional similarity and network topology. Thus one would expect differences in the alignment generated by a purely sequence-based approach and one from a network strategy such as PINALOG. However, there is no gold standard with which to compare the results, so we need to consider a range of metrics. Accordingly, in Table 1 and in Supplementary Material, we report NA, the number of aligned protein pairs; NC, the number of conserved interactions; NH, the number of protein pairs belonging to the same Homologene groups (Wheeler et al., 2005); NI, the number of interlogs (Walhout et al., 2000); and NF, the number of aligned protein pairs with functional similarity >0.5. Conserved interactions are interactions occurring in both species when two protein nodes forming an interaction in one species are equivalenced to two protein nodes which also form an interaction in the other species. NH is a common measure of alignment quality, counting the number of protein pairs belonging to the same homologous groups identified by the Homologene algorithm. ‘Interlog’ refers to an orthologous pair of interacting proteins between different species. We use the definition of interlog by Yu, et al. (2004) to quantify NI. Both NH and NI describe the sequence similarity of protein pairs in the alignments. If the functional similarity between two proteins is >0.5, their functions are considered related (Schlicker et al., 2006), hence NF.
Table 1.
Alignment results of different pairs of species by PINALOG (PA) (obtained with the automatically generated θ=0.871), IsoRank (IR), MI-GRAAL (MG) and BLAST (BL) for human/yeast
| Stat | PA | IR | MG | BL |
|---|---|---|---|---|
| NA | 5222 | 5674 | 5674 | 1818 |
| NC | 3319 | 717 | 4107 | 530 |
| NF | 3139 | 734 | 146 | 1347 |
| NH | 454 | 165 | 0 | 818 |
| NH/NA | 0.09 | 0.023 | 0 | 0.45 |
| NI | 460 | 136 | 0 | 465 |
| NI/NC | 0.14 | 0.19 | 0 | 0.88 |
The statistics (Stat) are NA, NC, NF, NH and NI which denote the number of aligned pairs of proteins, the number of conserved interactions, the number of protein pairs with functional similarity >0.5 and the number of Homologene pairs and interlogs. The ratios NH/NA and NI/NC are also given.
3.1.1 Human and yeast alignment results
We consider the results of aligning the two species with the most abundant PPI information, human and yeast. The values of NA and NC indicate the scale of the alignment. However, since some of these equivalences may not be biologically relevant, one cannot use NA or NC as an accuracy metric. The large difference between NC for PINALOG (3319) and IsoRank (717) highlights that markedly different alignments are obtained. This is the result of extending the alignment from the seed protein pairs in PINALOG. PINALOG and MI-GRAAL have similar values for NC. BLAST finds far less aligned pairs and conserved edges than PINALOG consistent with the objective of network alignment in establishing more equivalences than a purely sequence-based method.
Table 1 also shows that PINALOG finds far more pairs of aligned proteins with a functional similarity >0.5 than IsoRank, MI-GRAAL and BLAST. This is consistent with PINALOG including functional similarity in its equivalence. The histograms of functional similarity of mapped protein pairs in different methods (see Supplementary Fig. SI-3) shows the improved performance of PINALOG over MI-GRAAL in identifying functionally similar pairs.
The number of Homologenes (NH) is substantially higher using PINALOG (454 pairs) compared with IsoRank (165 pairs) and MI-GRAAL (0) although all methods have roughly the same number aligned pairs. Although a network-based alignment which considers interaction topology and function need not equivalence every Homologene pair, one would expect that many Homologene pairs are captured in the alignment. This suggests that more of the aligned pairs are biologically meaningful in PINALOG compared with IsoRank and MI-GRAAL. BLAST finds more Homologenes (NH) than PINALOG (and IsoRank) despite having less total equivalences (NA). Homologenes are identified mainly by two-way sequence similarity (blastp) of proteins in different species species together with other information such as phylogeny and synteny. Since network-based methods equivalence pairs of protein together with their interaction edge, one would expect that a sequence method would identify more Homologenes than a network-based approach.
The PINALOG alignment has 3.5 times as many interlogs (NI) as IsoRank (460 versus136) while MI-GRAAL fails to find any. This indicates that PINALOG finds many interlogs between species, which along with conserved interactions, might contribute to the functional similarity of PPINs across species. BLAST finds a few more interlogs than PINALOG (465 compared with 460). Considering one-to-one mapping of networks, we can take the NI of BLAST as the maximal number obtainable. Clearly PINALOG found most of these whereas IsoRank did not. However as PINALOG generated far more conserved interactions (NC) than IsoRank, it is helpful to consider the ratio of NI to NC. This ratio is 0.14 for PINALOG, 0.19 for IsoRank and 0.88 for BLAST. If the sole aim is to find interlogs, then BLAST is a superior approach to any of these three network alignment methods. However, the PINALOG alignment aims to establish equivalences based on function and interactions not just those from sequence.
We also assess whether these conserved edges produce large and dense connected subgraphs which is helpful in finding topologically similar areas of the aligned networks indicating functional similarity. A common connected subgraph (CCS) from the alignment is defined a connected subgraph of the conserved network. The largest CCS from the conserved graph obtained from PINALOG is composed of 1858 proteins connected by 2774 interactions; while that of IsoRank has only 44 proteins connected by 87 interactions (Supplementary Fig. SI-4). The conserved graph from PINALOG alignment does not only have more conserved interactions but is also similar in terms of function. Using a functional clustering method (see Supplementary Material) on the human conserved network from the PINALOG alignment, we identified several clusters of proteins with closely related functions. The corresponding yeast clusters are functionally similar to the human clusters. For example, cluster 12 in both species includes proteins that form different proteasome complexes; cluster 137 in both species contains proteins from the anaphase promoting complex. The average functional similarity of mapped clusters is 0.33, and 23% of the pairs of mapped clusters have average functional similarity >0.5.
Although the largest CCS of MI-GRAAL is large (3773 nodes, 3789 edges), it is not as dense as the largest CCS found by PINALOG-A (network density 0.001 versus 0.002, clustering coefficient 0.001 versus 0.091; see Supplementary Material). Functional clustering of the human conserved network of MI-GRAAL followed by mapping onto the yeast clusters shows that there is little or no functional similarity between corresponding clusters. The average functional similarity between corresponding clusters of MI-GRAAL is 0.16 as compared with PINALOG 0.33. The correspondence in function of mapped clusters obtained from the conserved graph of the PINALOG alignment indicates that the resulting alignment shows the actual biological equivalence between the two input networks.
3.1.2 Alignment of the human PPIN with other species
We have also applied PINALOG to align the networks of human-fly, human-worm and human-mouse. The PPINs of fly, worm and mouse are relatively sparse compared with the yeast interactome. Therefore, the alignment between human and species other than yeast are expected to have far fewer conserved interactions. However, as these species are closer to human in terms of evolution, the resulting alignment should produce a larger number of Homologenes. Taking into consideration these expected differences, the statistics for these comparisons (Supplementary Table SI-1) are broadly in keeping with the observations for human-yeast. For any comparison, PINALOG, IsoRank and MI-GRAAL identify a very similar number of aligned pairs (NA). In terms of conserved edges (NC), MA-GRAAL finds most, then PINALOG and finally IsoRank. In keeping with the use of functional similarity to establish the alignment, PINALOG finds more functional alignments (NF). PINALOG identifies more Homologene pairs than IsoRank and MI-GRAAL. In addition, PINALOG finds more interlogs that IsoRank and MI-GRAAL and for these comparisons the ratio NI/NC is at least double for PINALOG compared with IsoRank.
3.1.3 Yeast2-fly2 alignment and Graemlin's performance
In the alignment between yeast and fly interactomes, PINALOG and IsoRank perform comparably, apart from the PINALOG having more functional equivalences (NF). MI-GRAAL fails to find any Homologenes or interlogs. The values of NA and NC show that Graemlin's alignment is far smaller than those from PINALOG, IsoRank and MI-GRAAL. Graemlin only finds 23 Homologenes compared with 241 from PINALOG and 2211 from IsoRank. PINALOG, IsoRank and Graemlin find very similar number of interlogs (36, 34 and 36, respectively).
In summary, PINALOG generally performed better than IsoRank in most pairs of species in terms of conserved interactions, homologous pairs and interlogs. In the human-yeast alignment, although PINALOG finds more interlogs (NI), the fraction of conserved interactions which are interlogs is larger in IsoRank (see NI/NC). Very similar results are obtained in the fly2-yeast2 equivalences for PINALOG and IsoRank. Comparison with Graemlin 2.0 shows that PINALOG and IsoRank generate far larger alignments with more Homologenes. Although MI-GRAAL uses the same source of data including sequence and functional similarity, it does not produce alignments with similar protein pairs in terms of either function or sequence. We explored the use of different parameters in MI-GRAAL to improve performance, but were unable to obtain improvements over the default values. Further analysis on the functional similarity of aligned protein pairs (see Supplementary Material and Supplementary Fig. SI-5) shows the advantage of utilizing the available functional information in the alignment, which in turn provides a more biologically relevant mapping of the networks.
4 PROTEIN COMPLEX PREDICTION
One of the applications of network alignment is to use the available information on the protein complex in one species to predict the protein complex components in another species. To benchmark the protein complex prediction of PINALOG, we performed a comparison of PINALOG with network-based complex prediction methods, including MCODE (Bader and Hogue, 2003), MCL (Dongen, 2000), network alignment methods including IsoRank, MI-GRAAL and BLAST. Comparisons were made on the gold-standard set of yeast complexes MIPS CYDG (Güldener et al., 2005) comprising of 214 protein complexes. Predictions of the yeast complexes were made by PINALOG, IsoRank, MI-GRAAL and BLAST by transferring the protein complex information from human to yeast, with the human protein complexes information obtained from the MIPS CORUM (Ruepp et al., 2008) database.
An assessment of prediction methods was performed using a series of functions based on the overlapping score (OS, see Supplementary Material). The prediction p is said to match with complex m if OS(p,m) is >0.2. Given the scores of matching between predictions and known complexes, measures to assess the quality of prediction include: (i) Precision P is the fraction of predicted complexes p matching known complexes m; (ii) Recall R is the fraction of known complexes m matching predicted complexes p; and (iii) F-measure F1 is the harmonic mean of precision and recall, providing the overall performance of the prediction methods 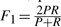 .
.
Figure 2 presents the precision, recall and F-measure of yeast protein complex prediction by PINALOG-A. PINALOG outperforms MCODE, MCL, IsoRank and MI-GRAAL in all three measures, with 37% recall at 21% precision and an F-measure of 26%. The performance of PINALOG is better than BLAST in precision but worse in recall. However, in the F-measure, which summarizes the overall performance, these two methods perform equally well.
Fig. 2.

Complex prediction results of PINALOG in comparison with other prediction methods based on recall, precision and F-measure. Complex predictions for yeast by these methods are compared with the gold-standard yeast complexes in MIPS CYDG. A prediction that has overlap with a true protein complex with an OS > 0.2 is considered matched. The number of matched complexes are used to calculate recall, precision and F-measure as defined in the main text.
It should be noted that the precision and recall are not high because the benchmark protein complexes are those identified by experiments that do not cover all protein complexes in yeast. Thus a high number of false positives (FPs) are expected. These FPs are candidates for protein complexes that could be studied experimentally. To assess whether these FPs are likely to be true protein complexes or not, we have examined the predicted clusters that do not match with known complexes. Using GOTermFinder (Boyle et al., 2004), we have identified significant GO term enrichment in many unmatched predicted clusters. For example, the 9 proteins of cluster 13 are enriched with terms related to chromatin modification, chromatin organization, and chromatin assembly and disassembly. Cluster 2, comprising 23 proteins, is enriched with terms relating to oxidation–reduction process. Another example is cluster 12 with 8 proteins that are related to M phase during cell division. The GOTermFinder analysis indicates that the putative FPs in the PINALOG prediction may well be true protein complexes.
BLAST performed as well as PINALOG and better than other single network-based methods. However, when inspecting the overlap in the predictions made by PINALOG and BLAST, we found that the overlap of the two predicted sets of clusters is not substantial, with only 50% of the predicted clusters matched. This suggests that PINALOG and BLAST could be used complementarily to predict protein complexes. This would provide a better coverage of the possible protein complexes. BLAST predicts clusters based on protein sequence, thus the prediction space is limited to sequence related proteins. In contrast, PINALOG predictions are made based on a combination of sequence, function and network topology, therefore the prediction space is extended to functionally related protein clusters that are reinforced by conserved interactions. We have combined the predictions made by PINALOG and BLAST into one predictor PINALOG+BLAST. This combination not only boosted the precision but also the recall rate, yielding a higher F-measure as compared with the individual methods (Fig. 2). We suggest that PINALOG and BLAST are used combination to help predicting protein complex more accurately.
5 BENCHMARKING OF PROTEIN FUNCTION PREDICTION
Another application of PINALOG is to predict the function of proteins by inheriting the annotation available of the aligned protein from the other species. PINALOG aims at providing function prediction (i) when there is no sequence homology; (ii) when there is a sequence homology between the un-annotated protein and the aligned annotated protein in the other species but the per cent sequence identity is low (typically <30%) and thus direct functional transfer can lead to misleading annotations (Tian and Skolnick, 2003; Todd et al., 2001). PINALOG is compared with the widely-used sequence-based method PSI-BLAST and to IsoRank.
A dataset of proteins to test the accuracy of function prediction was established with 415 GO annotated human proteins. These proteins had low per cent identity (<30%) to the closest annotated PSI-BLAST hit in the UniprotKB database. A 100-fold cross validation was performed. In each run of the cross validation, a set of proteins from the test set and had their GO terms hidden. The test proteins which were aligned with an annotated protein in the other species were identified and the function transferred. Over the 100 runs, a total of 169 proteins were equivalenced and their function predicted. Functions of mapped proteins in yeast represented by GO terms in two categories BP (biological process) and MF (molecular function) were directly transferred to the human proteins in the test set.
The results were analyzed in terms of precision and recall, where precision is defined as 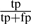 and recall as
and recall as 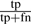 ; tp, fp and fn being the number of true positives, false positives and false negatives of the predictions made. Our method is a binary predictor without a variable cut-off parameter and thus no precision/recall curve can be produced. Table 2 shows that PINALOG outperforms PSI-BLAST prediction with a superior recall rate at similar level of precision with BP category. The McNemar test (McNemar, 1947) for the statistical significance of the difference in the performance was used based on the number of misclassifications in each method. The test indicates that PINALOG predictions are significantly different from PSI-BLAST in BP terms at the level of P=0.001 significance level. PINALOG predicts more BP terms with a recall twice that of PSI-BLAST (14% versus 7%) at the same level of precision (~28%). For the MF category, PINALOG and PSI-BLAST share very similar levels of recall (~28%) while the precision of PSI-BLAST is slightly better (43% versus 47%). The McNemar test suggests no significant difference between them at the P=0.001 significance level. On the other hand, PINALOG outperforms IsoRank in both BP (P=0.001) and MF (P=0.001) categories. For example, in the BP category, PINALOG has almost twice the recall compared with IsoRank (14% versus 8%) at higher precision (28% versus 20%, Table 2). To summarize, in the challenging area where sequence similarity does not contribute substantially to the prediction of protein function, PINALOG enhances the ability to predict function of these proteins.
; tp, fp and fn being the number of true positives, false positives and false negatives of the predictions made. Our method is a binary predictor without a variable cut-off parameter and thus no precision/recall curve can be produced. Table 2 shows that PINALOG outperforms PSI-BLAST prediction with a superior recall rate at similar level of precision with BP category. The McNemar test (McNemar, 1947) for the statistical significance of the difference in the performance was used based on the number of misclassifications in each method. The test indicates that PINALOG predictions are significantly different from PSI-BLAST in BP terms at the level of P=0.001 significance level. PINALOG predicts more BP terms with a recall twice that of PSI-BLAST (14% versus 7%) at the same level of precision (~28%). For the MF category, PINALOG and PSI-BLAST share very similar levels of recall (~28%) while the precision of PSI-BLAST is slightly better (43% versus 47%). The McNemar test suggests no significant difference between them at the P=0.001 significance level. On the other hand, PINALOG outperforms IsoRank in both BP (P=0.001) and MF (P=0.001) categories. For example, in the BP category, PINALOG has almost twice the recall compared with IsoRank (14% versus 8%) at higher precision (28% versus 20%, Table 2). To summarize, in the challenging area where sequence similarity does not contribute substantially to the prediction of protein function, PINALOG enhances the ability to predict function of these proteins.
Table 2.
Protein function prediction assessment by 100-fold cross validation, comparison with PSI-BLAST and IsoRank
| PINALOG-A |
PSI-BLAST |
IsoRank |
||||
|---|---|---|---|---|---|---|
| BP | MF | BP | MF | BP | MF | |
| Recall | 0.14 | 0.28 | 0.07 | 0.29 | 0.08 | 0.17 |
| Precision | 0.28 | 0.43 | 0.29 | 0.47 | 0.2 | 0.32 |
BP and MF stands for biological process and molecular function terms in GO.
PINALOG was used to predict functions for un-annotated human proteins without any PSI-BLAST hit in the human-yeast alignment. For the 60 such human proteins, PINALOG mapped 14 of them to the proteins in yeast and made predictions for human proteins from the yeast counterparts. The function of only nine proteins was predicted (and provided in Supplementary Table SI-2 in Supplementary Material) because the remaining five are mapped to proteins in yeast that have annotation only with very general GO terms. We compared our predictions to those from ffPred (Lobley et al., 2007), a web-server for function prediction of orphan or un-annotated proteins. Out of these nine proteins, ffPred provides functions for six proteins, three of which have very general GO terms, the rest of the predictions agreeing with our predictions. We have also run these nine proteins through the structure prediction server Phyre2 (Kelley and Sternberg, 2009) which detects templates which can be remote homologues not identifiable by PSI-BLAST. For six of the PINALOG predictions, the functional descriptions of these templates proteins match with or are similar to the PINALOG predictions. This strongly suggests that the PINALOG predictions are reliable and can help guide experimentalists to identify protein function.
6 DISCUSSION
Although many methods have been developed to align PPINs, comparing their performance remains difficult due to the absence of an unambiguous correct alignment, the complexity of the networks and the differing aims of the alignment methods. For local alignment methods, the difficulty is less severe as some local alignments can be evaluated by their agreement with known protein complexes. However, this approach is less useful to evaluate global alignment methods. Often assessment is performed based on the correctness of mapping orthologous protein pairs. However, this assessment measure would always penalize equivalences based on network topology when this conflicts with equivalences between orthologous pairs. Therefore this assessment method is not ideal in evaluating the performance of network alignment algorithms. Accordingly, we have also used a series of other measures.
PINALOG is flexible in allowing the use of sequence only alignment or sequence-function alignment depending on the requirement of the user. For the sequence-function alignment, parameters are automatically calculated from the input species and this will help non-experts. When aligning two species where one or both species are poorly annotated, alignment might be biased towards aligning well-annotated proteins whose functions are similar. Then it is advisable that sequence and network topology only are used to align the networks to avoid bias. We are developing a version of PINALOG to perform alignment of PPINs from multiple species. A web-server for PINALOG is available allowing users to upload necessary information and receive alignment results by email and by a link to the results file (http://www.sbg.bio.ic.ac.uk/~pinalog/). The computing time of the alignment process depends on the size of the input networks. The typical computing time is <24 h with the longest run in our assessment being the human-yeast alignment which takes 24 h on a computer with 2.8 GHz processor with 8 GB memory (see Supplementary Material). Given the importance of the resulting alignment, this time is acceptable. The code for PINALOG is available from the website, Download section.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Wellcome Trust for funding this project and Dr Mark Wass for valuable advice on function prediction.
Funding: Wellcome Trust
Conflict of Interest: MJES is a director and shareholder in Equinox Pharma Ltd which markets software and services in the bioinformatics and chemoinformatics area.
REFERENCES
- Adamcsek B., et al. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics. 2006;22:1021–1023. doi: 10.1093/bioinformatics/btl039. [DOI] [PubMed] [Google Scholar]
- Ali W., Deane C.M. Functionally guided alignment of protein interaction networks for module detection. Bioinformatics. 2009;25:3166–3173. doi: 10.1093/bioinformatics/btp569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda B., et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G., Hogue C. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S. Systematic identification of functional orthologs based on protein network comparison. Genome Res. 2006;16:428–435. doi: 10.1101/gr.4526006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J., Lassig M. Cross-species analysis of biological networks by Bayesian alignment. PNAS. 2006;103:10967–10972. doi: 10.1073/pnas.0602294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle E.I., et al. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohee S., van Helden J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics. 2006;7:488. doi: 10.1186/1471-2105-7-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cootes A.P., et al. The identification of similarities between biological networks: application to the metabolome and interactome. J. Mol. Biol. 2007;369:1126–1139. doi: 10.1016/j.jmb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Dongen S.V. PhD thesis. The Netherlands: University of Utrecht; 2000. Graph Clustering by Flow Simulation. [Google Scholar]
- Flannick J., et al. Graemlin: general and robust alignment of multiple large interaction networks. Genome Res. 2006;16:1169–1181. doi: 10.1101/gr.5235706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J., et al. Automatic parameter learning for multiple local network alignment. J. Comput. Biol. 2009;16:1001–1022. doi: 10.1089/cmb.2009.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldener U., et al. CYGD: the Comprehensive Yeast Genome Database. Nucleic Acids Res. 2005;33:D364–D368. doi: 10.1093/nar/gki053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley B.P., et al. Conserved pathways within bacteria and yeast as revealed by global protein network alignment. PNAS. 2003;100:11394–11399. doi: 10.1073/pnas.1534710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Sternberg M.J.E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Koyutürk M., et al. Pairwise alignment of protein interaction networks. J. Comput. Biol. 2006;13:182–199. doi: 10.1089/cmb.2006.13.182. [DOI] [PubMed] [Google Scholar]
- Kuchaiev O., et al. Topological network alignment uncovers biological function and phylogeny. J. Roy. Soc. Interf. 2010;7:1341–1354. doi: 10.1098/rsif.2010.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchaiev O., Przulj N. Integrative network alignment reveals large regions of global network similarity in yeast and human. Bioinformatics. 2011;27:1390–1396. doi: 10.1093/bioinformatics/btr127. [DOI] [PubMed] [Google Scholar]
- Kuhn H.W. The Hungarian method for the assignment problem. Nav. Res. Log. (NRL) 2005;52:7–21. [Google Scholar]
- Lewis A., et al. The function of communities in protein interaction networks at multiple scales. BMC Syst. Biol. 2010;4:100. doi: 10.1186/1752-0509-4-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobley A., et al. Inferring function using patterns of native disorder in proteins. PLoS Comput. Biol. 2007;3:e162. doi: 10.1371/journal.pcbi.0030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord P.W., et al. Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics. 2003;19:1275–1283. doi: 10.1093/bioinformatics/btg153. [DOI] [PubMed] [Google Scholar]
- McNemar Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika. 1947;12:153–157. doi: 10.1007/BF02295996. [DOI] [PubMed] [Google Scholar]
- Narayanan M., Karp R.M. Comparing protein interaction networks via a graph match-and-split algorithm. J. Comput. Biol. 2007;14:892–907. doi: 10.1089/cmb.2007.0025. [DOI] [PubMed] [Google Scholar]
- Palla G., et al. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435:814–818. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- Ruepp A., et al. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008;36:D646–D650. doi: 10.1093/nar/gkm936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker A., et al. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics. 2006;7:302. doi: 10.1186/1471-2105-7-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R., et al. Conserved patterns of protein interaction in multiple species. Proc. Natl Acad. Sci. USA. 2005;102:1974–1979. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., et al. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc. Natl Acad. Sci. USA. 2008;105:12763–12768. doi: 10.1073/pnas.0806627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Singh M. How and when should interactome-derived clusters be used to predict functional modules and protein function? Bioinformatics. 2009;25:3143–3150. doi: 10.1093/bioinformatics/btp551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W., Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J. Mol. Biol. 2003;333:863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Todd A.E., et al. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Walhout A.J.M., et al. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- Wheeler D.L., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–D45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., et al. Annotation transfer between genomes: protein–protein interologs and protein–DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.