

Abstract
Motivation: Many important aspects of evolutionary dynamics can only be addressed through simulations. However, accurate simulations of realistically large populations over long periods of time needed for evolution to proceed are computationally expensive. Mutants can be present in very small numbers and yet (if they are more fit than others) be the key part of the evolutionary process. This leads to significant stochasticity that needs to be accounted for. Different evolutionary events occur at very different time scales: mutations are typically much rarer than reproduction and deaths.
Results: We introduce a new exact algorithm for fast fully stochastic simulations of evolutionary dynamics that include birth, death and mutation events. It produces a significant speedup compared to direct stochastic simulations in a typical case when the population size is large and the mutation rates are much smaller than birth and death rates. The algorithm performance is illustrated by several examples that include evolution on a smooth and rugged fitness landscape. We also show how this algorithm can be adapted for approximate simulations of more complex evolutionary problems and illustrate it by simulations of a stochastic competitive growth model.
Contact: ltsimring@ucsd.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Natural evolution is an inherently stochastic process of population dynamics driven by mutations and selection, and the details of such evolutionary dynamics are increasingly becoming accessible via experimental investigation (Barrick et al., 2009; Chou et al., 2011; Finkel and Kolter, 1999; Pena et al., 2010; Ruiz-Jarabo et al., 2003). The importance of stochasticity comes from the fact that populations are always finite, mutations are random and rare, and at least initially, new mutants are present in small numbers. This realization prompted intensive studies of stochastic effects in evolutionary dynamics (Baake and Gabriel, 2000; Brunet et al., 2008; Desai et al., 2007; Gillespie, 1984; Hallatschek, 2011; Jain and Krug, 2007). Most of the models in these studies consider a reproducing population of individuals which are endowed with genomes that can mutate and thus affect either reproduction or death rate, as with the classical Wright–Fisher (Fisher, 1930; Wright, 1931) and Moran models (Moran, 1958) which describe a fixed population of replicating individuals. Specific models vary in the details of fitness calculation and mutation rules, but recent theoretical studies of even relatively simple models lead to non-trivial predictions on the rate of evolution as a function of the population size and the details of the fitness landscape (Brunet et al., 2008; Desai et al., 2007; Hallatschek, 2011; Kessler et al., 1997; Rouzine et al., 2003; Tsimring et al., 1996). However, the complexity of more realistic evolutionary models makes them analytically intractable and requires researchers to resort to direct numerical simulations in order to gain quantitative understanding of underlying dynamics.
On the most basic level, an evolutionary process is a Markov chain of discrete reactions of birth, deaths and mutations within a population of individuals. A direct and exact way of computing individual evolutionary ‘trajectories’ is to use the stochastic simulation algorithm (SSA; Gillespie, 1977) or its variants (Gibson and Bruck, 2000; Gillespie, 1976; Lu et al., 2004), in which birth, death and mutation events are treated as Markovian ‘reactions’. Unfortunately, for realistically large population sizes, direct stochastic simulation of even simple models becomes prohibitively expensive. Hence, there is an acute need for developing accelerated methods of stochastic simulations of evolutionary processes. Such methods usually involve approximations to the exact stochastic process based on certain small or large parameters that characterize the problem (for example, population size or mutation rates). Several approximate methods have been developed in recent years in the context of stochastic biochemical kinetics (Cao et al., 2005; Gillespie, 2001; Jahnke and Altintan, 2010; Rathinam and El Samad, 2007; Rathinam et al., 2003). Recently, Zhu et al. (Zhu et al., 2011) proposed an approximate hybrid algorithm suitable for simulation of evolutionary dynamics by combining the τ-leap algorithm (Gillespie, 2001) appropriate for abundant sub-populations that do not change their sizes much between individual events, and the direct SSA algorithm for small sub-populations. This method allows one to use large time steps during which multiple birth and death reactions may have occurred. However, it slows down dramatically after a new mutant has been produced, since the algorithm resorts to the direct SSA for all events in which the new mutants are involved until the population of the new mutant class reaches a pre-defined threshold.
Here, we develop a novel exact algorithm for simulation of evolutionary dynamics of a multi-species population undergoing asexual reproduction, death and mutation. Unlike the direct SSA, it only samples the evolutionary process at the times of mutations. Stochastic contributions from mutation, birth and death are included exactly, which is especially important for new species that initially contain a population size of one. We call this algorithm BNB (binomial-negative binomial), since as the name indicates, a population update requires sampling binomial and negative binomial pseudorandom variables with specific weights. This can be done efficiently using techniques similar to those used in the next reaction method (Gibson and Bruck, 2000).
If the mutations are rare compared with other (birth and death) events, this algorithm offers a significant speed advantage with respect to the SSA. Indeed, in most organisms, the mutation rate is much smaller than the birth and death rates, e.g. the probability of mutation per division for the genome in bacteria is μg ~ 10−3 (Drake et al., 1998). Thus, only a small (compared to the population size) number of new mutants appear in each generation. Even in viruses that generally are characterized by a high mutation rate μg ~ 1, most mutations are neutral and thus do not strongly influence the population dynamics.
In the following, we begin with a general approach to the stochastic simulation of a system of reactions that are arbitrarily divided into ‘fast’ and ‘slow’ reactions. We then specialize to the evolutionary model in which the mutation rate is assumed to be much smaller than the birth and death rates. We present examples that illustrate the accuracy and power of the proposed algorithm for models describing evolution of a population regulated by serial dilution. Then we discuss a modification of the algorithm that allows for its use in more complex situations when the exact algorithm is not applicable. Finally, we illustrate the approximate method by a simple example of co-evolving species competing for a common nutrient source.
2 ALGORITHM
The BNB algorithm is a stochastic updating rule for the state of an evolving set of species, which are defined by their internal state (‘genotype’) that in turn determines the birth, death and mutation rates for each species. This algorithm is exact when the birth, death and mutation rates (not the propensities!) remain constant between consecutive mutations. A single iteration of the BNB algorithm updates the state of the system to the time just after the next mutation has occurred. By applying this updating rule multiple times, the dynamics of the evolving system can be sampled by ‘jumping’ from one mutation to the next. In case when the rates are changing slowly between mutations, an approximate variant of the BNB can be applied (see below).
The core of the BNB algorithm is based on an exact solution for a stochastic model of dividing, dying and mutating discrete populations of cells. A single iteration of the BNB algorithm uses this solution to rapidly perform the following steps: (i) determine from which species and at what time a new mutant cell is generated; (ii) update the populations of all species to the time just prior to this mutation; (iii) generate a new mutant cell that either establishes a new species in the simulation or is added to a species already contained in the simulation; and (iv) update the time of the simulation to the time of this mutation.
This section contains the derivation and the detailed description of the BNB algorithm.
2.1 Stochastic simulation of a two-scale stochastic process
We consider the general case of a continuous time and discrete state stochastic system that is subject to a set of reactions among which some are ‘fast’ and some are ‘slow’. We designate them as fast and slow operationally, for a given state of the system (e.g. abundances of each species) at a given time. Typically, the mean time interval between two consecutive fast reactions will be much smaller than the mean time interval between two consecutive slow reactions. Our goal is to jump directly from one slow reaction event to the next and exactly sample the state of the system at the time of slow reaction.
Let us lump all slow reactions into one that we call ‘mutation’ and consider the dynamics of the system between two consecutive mutations. For simplicity, we assume that the propensities for each possible mutation are proportional to each other for a given system state, such that we can select the type of mutation independently of when a mutation occurs. If the probability of mutations were zero, the probability pi(t) for being at state i at time t satisfies the master equation that only includes fast reactions
| (1) |
Now, suppose that mutations occur with rate μi at state i. We can introduce the probability Pi(t) that the system is at state i at time t and a mutation has not yet occurred. It is easy to see that Pi(t) satisfies the ‘leaky’ master equation
| (2) |
The probability Yi(t) that at least one mutation has occurred while the system was at state i before time t satisfies the following equation
| (3) |
Note that Yi(t)=0 at initial time t=0. Define the probability P(t)≡∑i Pi(t) for no mutation to have occurred by time t and Y(t)≡∑i Yi(t) for some mutation to have occurred at least once at any state by the time t. By construction, Y(t)+P(t)=1, and therefore
| (4) |
Thus, P(t) is strictly non-increasing in time, as expected. Knowledge of P(t) allows us to sample time to the next mutation tm. We also need to know which state of the system is mutated. It is easy to show that the probability ρi(t) that the system is at state i at the time of a mutation is
| (5) |
Thus, assuming we can solve for Pi(t), we can formulate the following algorithm for updating the stochastic system at mutation times:
Algorithm 1 —
Define the initial state of the system i0, i.e. define Pi(0)=δii0 (where δij is the Kronecker symbol).
Solve for Pi(t), which provides functions P(t) and ρi(t) [Equations (4)–(5)].
Sample the next mutation time according to the cumulative probability P(t). This can be done via the inversion method, such that the next time tm=P−1(r), where r is a uniform random variable between 0 and 1.
Add tm to the current time.
Sample the distribution ρi(tm) to generate the new state im just before the mutation (slow reaction).
Choose the specific mutation according to their relative propensities and update the state of the system after the state update in Step 5.
Return to Step 1 until finished.
Of course, to complete this algorithm, we should be able to solve for or otherwise compute the dynamics of the probabilities Pi according to Equation (2). While this may be difficult in general to do analytically, it may still be much simpler that solving the full system. In particular, as we discuss in the following section, the problem can be solved exactly when the fast reactions include only birth and death whereas the slow reactions include only mutations.
2.2 Generating function solution for a single-species birth/death/mutation model
There exists a vast literature on the analysis of statistical properties of the so-called linear birth–death processes. The analytical treatments usually involve solving the corresponding master equation via the generating function method (Bartlett, 1955; Cox and Miller, 1965). Exact solutions have been found for several models including pure birth–death systems as well as systems with immigration and emigration (Crawford and Suchard, 2011; Ismail et al., 1988; Karlin and Mcgregor, 1958; Novozhilov et al., 2006). Here, we will follow the same general approach, but since we are interested in the statistics of mutating species, we will add the mutation ‘reaction’ in the model which manifests itself through leakage of probability. We begin with the case of a single class of species. The number of individuals n can fluctuate due to statistically independent birth, death and mutation reactions. Birth has propensity gn, death has propensity γn and mutation has propensity μn. As before, we are only interested in the interval of time between two subsequent mutations, so the resultant state of the mutated individual is irrelevant. Thus the mutation is simply defined as the creation and subsequent departure of a single individual from the class.
Define Pn(t) to be the probability that the system is at state n at time t and that a mutation has not yet occurred. The generating function G(s, t)=∑n=0∞ Pn(t)esn can be computed for an initial population n0 at time t=0 by (see Supplementary Material for details)
| (6) |
with
| (7) |
| (8) |
| (9) |
| (10) |
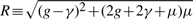 and W = g+γ+μ. Using a uniform random number r distributed between 0 and 1, the next mutation time is then
and W = g+γ+μ. Using a uniform random number r distributed between 0 and 1, the next mutation time is then
| (11) |
which exists for
| (12) |
When Equation (12) is not satisfied, this indicates that the population will go extinct before a mutation occurs if the population is unperturbed for infinite time.
The time to extinction, tx, can then be sampled by inversion of the extinct state probability P0(t),
| (13) |
2.3 BNB expansion
After computing the time to the next mutation, we need to generate a sample number of individuals at the time of mutation. The number of individuals conditional on no mutation at time t is distributed according to the generating function G(s, t) given by Equation (6). Here, we show that this seemingly complicated distribution can be exactly sampled by drawing two random numbers—one binomial, and one negative binomial. Many popular software packages, e.g. (Press et al., 2007), contain fast algorithms for generating these random numbers (note that negative binomials can be generated by Poisson random variates with a Gamma-distributed parameter).
Equation (6) can be recast via a binomial expansion
 |
(14) |
Since an integer power of a geometric generating function corresponds to a negative binomial generating function, Equation (14) can be interpreted as a generating function of a process in which the system either has mutated by time t with probability 1−pM(t)n0, or if the system has not yet mutated, then it is in a state ñ whose distribution is a binomial superposition of n0 negative binomial distributions. While Equation (14) does not directly provide the probability to be in a particular state at the time of a mutation, it provides the probability Pn(t) at an arbitrary time t conditional on no mutation. We can then generate a sample of the population ñ conditional on no mutation at time t by the following procedure.
Algorithm 2 —
Generate a binomial random number
, with success probability 1−(pE(t)/pM(t)) and n0 terms.
If
, then the system at time t is in the extinct state ñ=0.
Otherwise, generate the new state variable ñ:
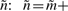
, where
is a negative binomial number of order
and probability of success pB(t).
We are also interested in the probability ρn(t) for a system to be in the state n at the mutation time. It is easy to see that ρn(t) ∝ μn Pn(t) ∝ nPn(t) [see Equation (5)]. To compute these probabilities, we introduce the corresponding generating function Gρ(s, t)=∑n=0∞ ρn(t)esn. After straightforward algebra, we obtain from Equation (6)
(15) which has the binomial expansion
(16) Equation (16) has the same form as Equation (14), and thus, ρn can be also sampled. Specifically, the algorithm for computing the state of the system just before the next mutation (at time tm) for the single species reads as follows.
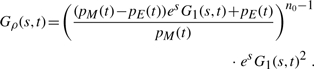
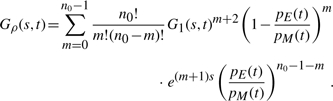
Algorithm 3 —
Generate a binomial random number
, with success probability 1−(pE(tm)/pM(tm)) and n0 − 1 terms.
Generate the updated state ñ at the mutation time:
, where
is a negative binomial number of order
and probability of success pB(t).
Note that the system will never be in the extinct state, which reflects that an extinct population cannot mutate.
2.4 Simulating multiple co-evolving species: first mutation method
In this section, we return to the original problem of an evolving population of multiple species. We enumerate species by index i, with ni(t) individuals in each species. We are interested in sampling the set {ni(tm)} at mutation times tm. We assume that the system parameters (birth, death and mutation rates) do not change between mutations unless the algorithm is ended early between two mutations. At the time of mutation, one individual is created from mutating class im and, depending on the type of mutation, is either added to one of the other existing classes (if such a class already exists) or becomes the founding member of a new class.
The algorithm for generating a sample stochastic evolution trajectory, which we call First Mutation BNB, is as follows.
Algorithm 4 —
Initialize the system with N classes of species at time t=0. Specify populations of all classes ni, i=1,…, N. Each class has its own set of birth, death and mutation rates gi, γi, μi.
For each class, generate N random numbers ri uniformly distributed between 0 and 1. For each i=1,…, N, generate a time ti to the next mutation by Equation (11). When Equation (12) is not satisfied, set ti=∞.
Find the minimum mutation time tm=min(ti) and the corresponding class im. Update the time t→t+tm.
Update the population for the mutated class im using two random numbers (one binomial and another negative binomial) according to the Algorithm 3.
Update the populations of all other classes according to Algorithm 2.
Select the specific mutation that occurs. If the mutation generates a member of a non-existent class, create a new class N+1 with nN+1=1 and its own set of parameters gN+1, γN+1, μN+1. Otherwise, add 1 to the corresponding existing class.
One or several of the non-mutated classes may have zero population and are thus extinct. Remove extinct classes from the list and reduce the number N of classes accordingly.
Return to Step 2 until the algorithm has completed.
To end the algorithm at a specific time rather than at a mutation event, all populations can be updated according to Algorithm 2 with the time duration t*−t, where t is the current time, and t* is the prescribed end time. This update would be done just after Step 2 when t*<t+min(ti) first occurs. Ending at a specific time is useful for a number of purposes, such as if the population is reported or modified at fixed time intervals, or if rates are adjusted at fixed time intervals.
The Algorithm 4 is analogous to the first reaction method used for stochastic simulation of reaction networks (Gillespie, 1976), in that the simulation of a system with N classes of co-evolving species generates 3N random numbers in order to step to the next mutation. This algorithm can thus become inefficient as the number of classes becomes large. To remedy this shortcoming, an optimized and only slightly more complex version of this algorithm is presented in the next section.
2.5 Simulating multiple co-evolving species: next mutation method
In fact, the number of random variables generated for each mutation in Algorithm 4 is excessive. Different species evolve independently between mutations, and even at the mutation time, only two classes are coupled, due to the mutating population generating and then contributing a single member to another species class. If this mutational coupling did not exist, the dynamics of species would be statistically independent at all times, and we could simulate all species independently using only three random numbers per mutation event.
This line of reasoning leads to a similar but optimized algorithm (see Supplementary Material for the algorithm and further justification), where the populations and next mutation times of species are re-sampled only for the two species that are coupled via a mutation event, whereas population sizes and next mutation times of all other classes are not re-sampled. Validity of the algorithm hinges on the statistical independence of species that are uncoupled by a mutation. The method is analogous to the next reaction method (Gibson and Bruck, 2000), so we label the algorithm Next Mutation BNB.
The optimized scheme reduces the typical number of new random variables required per mutation to only six after the first iteration, independently of the total number of classes N. Only initialization and finalization of the algorithm have a computational cost of order N, so efficiency of the algorithm primarily depends on how frequently the algorithm is restarted, as is the case whenever the whole population is sampled for observation.
The Next Mutation BNB algorithm is always as fast or faster than the First Mutation BNB. We thus use Next Mutation BNB (or just BNB) exclusively for the simulation examples of this article.
2.6 Approximate simulation method using BNB
One major benefit of the BNB algorithm is that binomials and negative binomials rapidly generate an update for the evolving system with linear propensities for birth, death and mutation in a non-interacting population. While this situation is typically the case for cells kept in log-phase growth, the cases when species are interacting or when propensities deviate from a linear law are also of interest. Because of this, we outline how the BNB algorithm can be adapted to approximately, but accurately, simulate more complicated systems.
The basis of the BNB algorithm is the generating function solution Equation (6), and it is straightforward to show from the short time form of this generating function that the BNB algorithm applied for sufficiently short time increments, during which birth, death and mutation rates are considered constant, can simulate systems with population-dependent rates. Between BNB updates, all of these rates can be updated in a state-dependent manner. This approach is similar to the τ-leap approximation to stochastic systems, which is often used to accelerate simulations of chemical reaction networks (Gillespie, 2001). The basis of τ-leap is that the propensities for reactions can be considered approximately constant during some time interval, such that the update scheme for τ-leap assumes each reaction occurs a Poisson-distributed number of times. Simulation error magnitude in τ-leap is closely associated with how well propensities are kept constant during a given time interval, and based on this connection, a few prescriptions for the step size have been suggested (Cao et al., 2006, 2007; Gillespie and Petzold, 2003). In contrast, BNB as an approximate updating scheme assumes that the propensities are approximately linear with respect to population, i.e. having constant rates. Deviation from the linear law is the primary factor influencing simulation error in BNB updating.
An important aspect of an approximate BNB updating method is that large and small species populations are treated uniformly, such that the same updating scheme applies to both situations with equal speed and relative accuracy. This may be contrasted to τ-leap methods, which due to large relative fluctuations of the propensity for small populations are no longer valid except for very short time steps. Zhu et al., 2011 introduced a hybrid τ-leap method which simulates species lower than a given population (the ‘cutoff’) using direct Gillespie algorithm. The tradeoff for the increased accuracy is a much-increased workload, since Gillespie algorithm simulates each reaction event individually. New species, which start as single cells, or species that naturally exist in low abundances are especially susceptible to an increase in workload for finite cutoff.
3 RESULTS
3.1 Exact simulations
In this section, we will apply the BNB algorithm to examples that can be exactly simulated using BNB. These examples deal with modeling the evolution of heterogenous cell populations in a hypothetical bioreactor designed to maintain exponentially growing cultures. We illustrate several phenomena that have been explored previously in analogous situations, e.g. for populations of fixed size, though we pursue these phenomena in the regime where large fluctuations in total population size (10-fold in most of our simulations) are routine.
The following models assume that cells are kept sufficiently dilute in culture such that limiting nutrients and other cell–cell interactions are not a factor. These cells thus grow and divide freely. The bioreactor prevents cell cultures from growing too dense by measuring the population size periodically (after every time duration Δt) and diluting the culture by binomial sampling to the mean population size nmin once the population has exceeded the population size nmax. In the simulations, we advance time directly from one mutation to the next or until the system has evolved longer than the maximal time duration Δt, at which point cells may be diluted if the population has exceeded nmax. It is also straightforward to simulate a bioreactor that continuously dilutes cultures to stem population growth, where the rate of media turnover and, correspondingly, cell ‘death’ is controlled, but we do not consider such an case here. An analysis in the Supplementary Material demonstrates that performance of BNB for these situations can far exceed that for direct Gillespie and τ-leap methods.
Abrupt dilution events can greatly enhance the effect of stochasticity, since there is a corresponding reduction in genetic diversity associated with each sub-sampling of the population. The smaller population after a dilution event will be heavily influenced by the particular individuals retained, leading to a form of the founder effect (Templeton, 1980). Even in light of this fact, we show that many phenomena found for fixed population sizes, e.g. wave behavior for population fitness, also occur using a dilution protocol that might occur experimentally.
3.1.1 Linear fitness model
Suppose that species are characterized by a positive integer index m that is a measure of fitness. Birth rate gm is a linear function of m, gm=1+ϵ(m−1). Death rate γm is constant across species. Mutation rate is proportional to growth rate (faster growing species also mutate faster), μm=ηgm. During a mutation of species with index m, a new member of species with index m−1 or m+1 is created, as chosen uniformly at random. If a species with index m=1 mutates, a new member of the species with index 2 is always created.
It has been demonstrated for ϵ>0 in the case of a constant total population that evolution on a linear fitness landscape leads to traveling population waves (Brunet et al., 2008; Desai et al., 2007; Hallatschek, 2011; Kessler et al., 1997; Rouzine et al., 2003; Tsimring et al., 1996), such that the mean fitness of the population linearly grows in time. However, the finite-size stochastic system can only be treated heuristically (Kessler et al., 1997; Tsimring et al., 1996), asymptotically (Brunet et al., 2008; Desai et al., 2007; Rouzine et al., 2003), or under certain specific modeling assumptions (Hallatschek, 2011). Thus, exact numerical simulations of large evolving populations in linear fitness landscapes are useful for testing the existing theories. Simulations indeed produce wave-like behavior (Fig. 1). The wave velocity scales linearly with the logarithm of the population size, as predicted (Fig. 2).
Fig. 1.

Simulations of the linear fitness model with Δt=0.1, ϵ=10−3, η=10−3 and nmin=nmax/10. The instantaneous distributions of the populations over the species index normalized by nmax as a function of time are shown for nmax=104 (A) and nmax=105 (B). Wave-like behavior is evident in both cases, though the smaller population leads to a noisier and slower wave. Panels (C) and (D) show the corresponding probabilities averaged over 800 realizations. The wave velocity, by a least squares linear fit to the ensemble mean fitness, is 0.93×10−3 and 2.1×10−3 indices per unit time for (C) and (D), respectively.
Fig. 2.

(A) The wave velocity (indices per unit time) of the linear fitness system has a slow (logarithmic) dependence on the population size set by nmax, in agreement with theoretical results (Brunet et al., 2008; Desai et al., 2007; Hallatschek, 2011; Kessler et al., 1997; Rouzine et al., 2003; Tsimring et al., 1996) (parameters are the same as in Fig. 1). Blue dots represent individual velocity measurements based on least squares fitting of a line to the last half of the mean index trajectory. Red line shows the least squares fit of the velocity as a linear function of ln nmax over the range nmax>104. The velocities from Figure 1C and D are plotted as green squares and diamonds, respectively. (B) Same as (A) for ϵ=10−4 and η=10−2. The weaker fitness gradient leads to a noisier distribution of velocities. (For a colour version of this Figure see Supplementary Data online).
We used similar simulations to study the effects of quenched fitness fluctuations on the propagation of traveling evolution waves. This problem is qualitatively analogous to the models of transport in systems with quenched disorder that are known to exhibit phase transitions (Bouchaud et al., 1990; Monthus and Bouchaud, 1996), and we expect similar behavior for evolution in a linear model with quenched disorder in the growth rate law. We assumed that the fitness as a function of the species index m has a fluctuating piece in addition to the linear dependence. Specifically, we consider growth rates that vary as 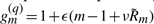 , where ν≥0 provides the scaling of noise, and
, where ν≥0 provides the scaling of noise, and 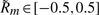 are independent uniform random numbers. In the case when ν<1, an increase in m always leads to an increase in growth rate, and wave propagation should proceed but with moderately reduced velocity. The case with ν>1 is qualitatively different, since an increase in m need not imply an increase in fitness. In this regime, it is possible to form rare but wide barriers due to fluctuations in the fitness, and these barriers when they exist can trap the system for an exponentially large time. This case can be contrasted against a potential with similar but deterministic variation gm(a)=1+ϵ(m−1+ν((m mod 2)−0.5)), which for ν>1 has fitness barriers only a single species wide. Figure 3 shows that quenched disorder exhibits substantially different behavior than the case when fitness contains regular variation. The system with quenched disorder in particular exhibits a sharp decrease in wave velocity as disorder is increased to ν>1, akin to a phase transition.
are independent uniform random numbers. In the case when ν<1, an increase in m always leads to an increase in growth rate, and wave propagation should proceed but with moderately reduced velocity. The case with ν>1 is qualitatively different, since an increase in m need not imply an increase in fitness. In this regime, it is possible to form rare but wide barriers due to fluctuations in the fitness, and these barriers when they exist can trap the system for an exponentially large time. This case can be contrasted against a potential with similar but deterministic variation gm(a)=1+ϵ(m−1+ν((m mod 2)−0.5)), which for ν>1 has fitness barriers only a single species wide. Figure 3 shows that quenched disorder exhibits substantially different behavior than the case when fitness contains regular variation. The system with quenched disorder in particular exhibits a sharp decrease in wave velocity as disorder is increased to ν>1, akin to a phase transition.
Fig. 3.

Ruggedness of the fitness landscape impacts speed of evolution in a linear fitness model. Shown are apparent wave velocities (blue dots) derived by least-square fitting of the mean index 〈m〉 across species as a function of time. The model with deterministic alternating fitness and nmax=104 (A) or nmax=105 (B) leads to a smooth decay of wave velocity with respect to the perturbation amplitude ν. In contrast, a model with quenched disorder in fitness and nmax=104 (C) or nmax=105 (D) exhibits an abrupt decrease in wave velocity suggesting a phase transition. In all cases, nmin=nmax/10, η=10−3, ϵ=10−2, γm=0.1, and Δt=0.02. The red curve indicates trend lines generated by a Savitzky–Golay filter. (For a colour version of this Figure see Supplementary Data online).
3.1.2 NK model simulations
Due to the general way the BNB algorithm treats mutations, it can be applied to more complicated evolutionary models. We used a variant of the NK model (Kauffman and Levin, 1987) to simulate evolution on fitness landscapes with various degrees of ruggedness. Despite large fluctuations in population, we could reproduce classical results for NK models, including state-dependent wave speed for smooth fitness landscapes, and punctuated evolution for rugged landscapes. Results and analysis of this model are found in the Supplementary Material.
3.2 BNB as an approximate algorithm: evolution in nutrient-limited environments
BNB can also be applied as an approximate algorithm for systems with state-dependent growth rates. Propensities may deviate from the linear law assumed in the BNB algorithm, but the BNB algorithm may still approximate a system with non-constant birth, death and mutation rates by evolving the system with a BNB step restricted to a short duration τ. Rates are then updated using the new populations before integrating the system with another BNB step, and so on. Validity of this process depends on self-consistency of the assumptions in the BNB algorithm, especially that propensities for reactions are independent of other species and proportional to population (see Supplementary Material for details).
We checked performance of this approximate algorithm for a system in which several species compete for a common nutrient that is supplied at a constant rate. Different species can consume this nutrient with different effectiveness, which provides selective pressure. Specifically, we suppose a linear fitness model for species, gm=am(1+∑ℓ
aℓ
nℓ/K0)−1, γm=0.1, μm=ηgm, am=1+ϵ(m−1), with species index m, η=10−3, and a scaling factor ϵ=1. In contrast to the other simulations in this text, birth rates are coupled in such a way that the total population in the system autonomously relaxes on average to a fixed value 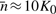 without the need of dilution events triggered by the population. The evolution of the system is linked to the ratio of growth rates gm1/gm2=am1/am2, which indicates that species with a higher index m tend to grow faster than those with lower index. Due to this effect, the system exhibits wave-like behavior (Fig. 4).
without the need of dilution events triggered by the population. The evolution of the system is linked to the ratio of growth rates gm1/gm2=am1/am2, which indicates that species with a higher index m tend to grow faster than those with lower index. Due to this effect, the system exhibits wave-like behavior (Fig. 4).
Fig. 4.

Wave behavior for the evolution in a model with competition, simulated using BNB as an approximate algorithm with time step τ=1. (A) A single realization of the species distribution as a function of time for initial population 100. (B) The mean population distribution for an ensemble of 800 simulations.
The recurrent creation and subsequent growth of new species in the competition model suggests that BNB could maintain better accuracy than τ-leaping schemes, since BNB faithfully simulates arbitrarily small populations and also exponential growth. We tested this for short-time simulations, and we found that in this context that BNB can provide consistently increased accuracy when compared to a hybrid τ-leap algorithm (Fig. 5).
Fig. 5.

Simulation accuracy for the model with competition. Using BNB (red), hybrid τ-leap (dashed blue) or direct SSA (light green), the model (with K0=1000 and initial population =100) was simulated over 105 realizations. As a measure of error, statistics of the population of the first mutant (index=2) were examined at time t=50. (A) The histogram (bin width=250) of this population for simulations using step size τ=5. BNB matches direct simulation closely, while hybrid τ-leap with cutoff 10 suffers from major inaccuracies. (B) L2 error between the histogram of direct SSA simulation and that of either BNB or the hybrid τ-leap normalized by the minimal expected statistical deviation, see Supplementary Material for details. (C) Same as (B), but as a function of the cutoff value for the hybrid τ-leap algorithm with τ=5. (D) Mean workload of the hybrid τ-leap and the approximate BNB algorithms, normalized by the workload for the BNB algorithm, as a function of the cutoff value. (For a colour version of this Figure see Supplementary Data online).
4 DISCUSSION
In this article, we have proposed an algorithm, which can be used to sample exactly co-evolving species that do not interact between mutations, and faithfully approximate the evolution of weakly-interacting species. BNB algorithm not only accounts for the stochastic fluctuations that arise due to the random nature of genetic mutations, but it also accounts for the small-number fluctuations due to the growth of new species that are spawned as single cells. Each iteration of the BNB algorithm generates the time of the next mutation and the abundances of all species just after the mutation. This algorithm is exact when the birth, death and mutation rates do not change between consecutive mutations. Although similar in spirit to approximate leaping schemes developed for modeling stiff stochastic chemical kinetics (Cao et al., 2005; Gillespie, 2001; Jahnke and Altintan, 2010; Rathinam and El Samad, 2007; Rathinam et al., 2003; Zhu et al., 2011), it differs significantly by providing an exact sampling at (irregular) intervals corresponding to mutational events. The method yields a substantial speed advantage over a straightforward SSA when the mutations are rare compared with birth and death events. The method is accessible, since the central part in implementing BNB is constructing fast methods that generate binomial and negative binomial pseudorandom numbers, both of which are available in standard code libraries (Press et al., 2007). More generally, the BNB algorithm is applicable to the simulations of systems in which underlying reactions are all first order and their rates remain unchanged between coarse-grained simulation steps.
Using the exact BNB algorithm, we simulated several evolution models for a hypothetical bioreactor that performs abrupt dilutions of cell culture when the total cell population exceeds a prescribed value. An analogous experimental bioreactor would periodically reduce the total number of cells, replenish nutrients and remove wastes in order to maintain log-phase growth of bacterial populations. In contrast to the classical theoretical setting, where the total number of cells is often kept constant, our model bioreactor maintained periodic 10-fold variations in the total number of cells. Despite these wild fluctuations in total population size, most phenomena and population size scaling were preserved. We found the classical scaling laws of adaptation velocity with the population size, as well as the evidence of a phase transition in the case of rugged linear models.
Real cell cultures almost always exhibit some degree of interaction within and among species, and so we showed how the BNB algorithm can also be extended to an approximate algorithm that is competitive with τ-leap and hybrid schemes adapted for evolutionary dynamics simulations (Zhu et al., 2011). A practical advantage of the approximate BNB algorithm is its uniformity; a BNB step is implemented with identical code for all population sizes. A specific model for species competing for common nutrients was introduced to test BNB, and BNB was found to readily provide good accuracy with minimal workload when compared to analogous τ-leap simulations. We anticipate the advantage of BNB to be maintained in the case where simulations require accurate and fast simulation of exponential growth of species that routinely are found at low population counts, as is the case when new fitter species grow to overtake the population. It should be noted, however, that even though the BNB algorithm can be used to simulate rather general systems, there are systems where BNB performs comparably to or even worse than τ-leap.
The present work presents the foundation for the BNB algorithm, but there exist several immediate directions for future refinement. We anticipate that simple modification of the BNB algorithm should enhance the accuracy for a wide variety of models with interacting species, analogously to a proposed midpoint method for τ-leaping (Anderson et al., 2010). Similarly straightforward modifications may also lead to a BNB formalism that approximates time-dependent birth, death and mutation rates, as needed for externally driven metabolic networks, e.g. the GAL network (Bennett et al., 2008). A less trivial extension would be to remove the assumption that birth, death and mutation rates are constant across species. Experimentally, cells within a common species exhibit variability in their cellular state (Elowitz et al., 2002), which could lead to a distribution of growth rates within a single species. Such a modified BNB could then be useful for answering questions concerning how species evolution couples to cellular state.
Funding: National Institutes of Health, grants P50GM085764 [W.H.M.]; RO1GM069811 [J.H.]; and R01GM089976 [L.S.T.].
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Anderson D.F., et al. Error analysis of tau-leap simulation methods. 2010 arXiv:0909.4790v2. [Google Scholar]
- Baake E., Gabriel W. Biological evolution through mutation, selection, and drift: an introductory review. Ann. Rev. Comp. Phys. 2000;7:203–264. [Google Scholar]
- Barrick J.E., et al. Genome evolution and adaptation in a long-term experiment with escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Bartlett M. An Introduction Stochastic Processes with Special Reference to Methods and Applications. Cambridge, United Kingdom: Cambridge University Press; 1955. [Google Scholar]
- Bennett M.R., et al. Metabolic gene regulation in a dynamically changing environment. Nature. 2008;454:1119–1122. doi: 10.1038/nature07211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchaud J.P., et al. Classical diffusion of a particle in a one-dimensional random force-field. Ann. Phys. 1990;201:285–341. [Google Scholar]
- Brunet E., et al. The stochastic edge in adaptive evolution. Genetics. 2008;179:603–620. doi: 10.1534/genetics.107.079319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., et al. The slow-scale stochastic simulation algorithm. J. Chem. Phys. 2005;122:014116. doi: 10.1063/1.1824902. [DOI] [PubMed] [Google Scholar]
- Cao Y., et al. Efficient step size selection for the tau-leaping simulation methods. J. Chem. Phys. 2006;124:044109. doi: 10.1063/1.2159468. [DOI] [PubMed] [Google Scholar]
- Cao Y., et al. Adaptive explicit-implicit tau-leaping method with automatic tau selection. J. Chem. Phys. 2007;126:224101. doi: 10.1063/1.2745299. [DOI] [PubMed] [Google Scholar]
- Chou H.-H., et al. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D., Miller H. The Theory of Stochastic Processes. New York: Wiley; 1965. [Google Scholar]
- Crawford F.W., Suchard M.A. Transition probabilities for general birth–death processes with applications in ecology, genetics, and evolution. J. Math. Biol. 2011 doi: 10.1007/s00285-011-0471-z. doi: 10.1007/s00285-011-0471-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M., et al. The speed of evolution and maintenance of variation in asexual populations. Curr. Biol. 2007;17:385–394. doi: 10.1016/j.cub.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W., et al. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz M.B., et al. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Finkel S.E., Kolter R. Evolution of microbial diversity during prolonged starvation. Proc. Natl Acad. Sci. USA. 1999;96:4023–4027. doi: 10.1073/pnas.96.7.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. The Genetical Theory of Natural Selection. Clarendon Press; 1930. [Google Scholar]
- Gibson M.A., Bruck J. Efficient exact stochastic simulation of chemical systems with many species and many channels. J. Phys. Chem. A. 2000;104:1876–1889. [Google Scholar]
- Gillespie D.T. General method for numerically simulating stochastic time evolution of coupled chemical-reactions. J. Comput. Phys. 1976;22:403–434. [Google Scholar]
- Gillespie D.T. Exact stochastic simulation of coupled chemical-reactions. J. Phys. Chem. 1977;81:2340–2361. [Google Scholar]
- Gillespie D.T. Approximate accelerated stochastic simulation of chemically reacting systems. J. Chem. Phys. 2001;115:1716–1733. [Google Scholar]
- Gillespie J. Molecular evolution over the mutational landscape. Evolution. 1984;38:1116–1129. doi: 10.1111/j.1558-5646.1984.tb00380.x. [DOI] [PubMed] [Google Scholar]
- Gillespie D.T., Petzold L.R. Improved leap-size selection for accelerated stochastic simulation. J. Chem. Phys. 2003;119:8229–8234. [Google Scholar]
- Hallatschek O. The noisy edge of traveling waves. Proc. Natl Acad. Sci. 2011;108:1783. doi: 10.1073/pnas.1013529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail M.E.H., et al. Linear birth and death models and associated Laguerre and Meixner polynomials. J. Approx. Theory. 1988;55:337–348. [Google Scholar]
- Jahnke T., Altintan D. Efficient simulation of discrete stochastic reaction systems with a splitting method. BIT Numer. Math. 2010;50:797–822. [Google Scholar]
- Jain K., Krug J. Deterministic and stochastic regimes of asexual evolution on rugged fitness landscapes. Genetics. 2007;175:1275–1288. doi: 10.1534/genetics.106.067165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S., Mcgregor J. Linear growth, birth and death processes. J. Math. Mech. 1958;7:643–662. [Google Scholar]
- Kauffman S., Levin S. Towards a general-theory of adaptive walks on rugged landscapes. J. Theor. Biol. 1987;128:11–45. doi: 10.1016/s0022-5193(87)80029-2. [DOI] [PubMed] [Google Scholar]
- Kessler D.A., et al. Evolution on a smooth landscape. J. Stat. Phys. 1997;87:519–544. [Google Scholar]
- Lu T., et al. Cellular growth and division in the Gillespie algorithm. Syst. Biol. 2004;1:121–128. doi: 10.1049/sb:20045016. [DOI] [PubMed] [Google Scholar]
- Monthus C., Bouchaud J.P. Models of traps and glass phenomenology. J. Phys. A, Math. Gen. 1996;29:3847–3869. [Google Scholar]
- Moran P. Random processes in genetics. Math. Proc. Cambridge Phil. Soc. 1958;54:60–71. [Google Scholar]
- Novozhilov A.S., et al. Biological applications of the theory of birth-and-death processes. Brief. Bioinform. 2006;7:70–85. doi: 10.1093/bib/bbk006. [DOI] [PubMed] [Google Scholar]
- Pena M.I., et al. Evolutionary fates within a microbial population highlight an essential role for protein folding during natural selection. Mol. Syst. Biol. 2010;6:387. doi: 10.1038/msb.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press W.H., et al. Numerical Recipes: The Art of Scientific Computing. 3rd. New York: Cambridge University Press; 2007. [Google Scholar]
- Rathinam M., El Samad H. Reversible-equivalent-monomolecular tau: a leaping method for ‘small number and stif’ stochastic chemical systems. J. Comput. Phys. 2007;224:897–923. [Google Scholar]
- Rathinam M., et al. Stiffness in stochastic chemically reacting systems: the implicit tau-leaping method. J. Chem. Phys. 2003;119:12784–12794. doi: 10.1063/1.1763573. [DOI] [PubMed] [Google Scholar]
- Rouzine I.M., et al. The solitary wave of asexual evolution. Proc. Natl Acad. Sci. USA. 2003;100:587–592. doi: 10.1073/pnas.242719299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Jarabo C.M., et al. Synchronous loss of quasispecies memory in parallel viral lineages: a deterministic feature of viral quasispecies. J. Mol. Biol. 2003;333:553–563. doi: 10.1016/j.jmb.2003.08.054. [DOI] [PubMed] [Google Scholar]
- Templeton A.R. The theory of speciation via the founder principle. Genetics. 1980;94:1011–1038. doi: 10.1093/genetics/94.4.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimring L.S., et al. RNA virus evolution via a fitness-space model. Phys. Rev. Lett. 1996;76:4440–4443. doi: 10.1103/PhysRevLett.76.4440. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T., et al. Efficient simulation under a population genetics model of carcinogenesis. Bioinformatics. 2011;27:837–843. doi: 10.1093/bioinformatics/btr025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.