

Abstract
Machado-Joseph disease (MJD), also known as Spinocerebellar Ataxia type 3, is an inherited dominant autosomal neurodegenerative disorder. An expansion of CAG repeats in the ATXN3 gene is translated as an expanded polyglutamine domain in the disease protein, ataxin-3. Selective neurodegeneration in MJD is evident in several subcortical brain regions including the cerebellum. Mitochondrial dysfunction has been proposed as a mechanism of neurodegeneration in polyglutamine disorders. In this study, we used different cell models and transgenic mice to assess the importance of mitochondria on cytotoxicity observed in MJD. Transiently transfected HEK cell lines with expanded (Q84) ataxin-3 exhibited a higher susceptibility to 3-nitropropionic acid (3-NP), an irreversible inhibitor of mitochondrial complex II. Increased susceptibility to 3-NP was also detected in stably transfected PC6-3 cells that inducibly express expanded (Q108) ataxin-3 in a tetracycline-regulated manner. Moreover, cerebellar granule cells from MJD transgenic mice were more sensitive to 3-NP inhibition than wild-type cerebellar neurons. PC6-3 (Q108) cells differentiated into a neuronal-like phenotype with nerve growth factor (NGF) exhibited a significant decrease in mitochondrial complex II activity. Mitochondria from MJD transgenic mouse model and lymphoblast cell lines derived from MJD patients also showed a trend towards reduced complex II activity. Our results suggest that mitochondrial complex II activity is moderately compromised in MJD, which may designate a common feature in polyglutamine toxicity.
Keywords: Machado-Joseph disease, ataxin-3, protein aggregation, mitochondria, cell death, succinate dehydrogenase
1. Introduction
Machado-Joseph disease (MJD), also known as spinocerebellar ataxia type 3, is an inherited autosomal dominant neurodegenerative disorder. MJD is the most common hereditary ataxia in the world, accounting for 21-28% of the autosomal-dominant inherited cerebellar ataxias [1,2]. Worldwide, it affects 1 or 2 individuals per 100 000 people, but its prevalence can increase to 1% of the population in some regions of the globe. Clinically, MJD is characterized by progressive gait and limb ataxia, dysarthria and a variable combination of other symptoms including pyramidal signs, dystonia, oculomotor disorders, faciolingual weakness, neuropathy, progressive sensory loss and parkinsonian features [3]. The first symptoms usually become apparent in adulthood (third or fourth decade of life), although they can manifest earlier in life in severe cases. Symptoms and neurological deficits progressively worsen over time, culminating in patients' death 20 to 30 years later. In MJD, neurodegeneration and astrogliosis are restricted to subcortical brain regions and are particularly prevalent in the pontine nuclei, the dentate nucleus, subthalamic nucleus and spinal cord [3]. The mechanisms leading to neuronal dysfunction and cell death are still poorly understood.
MJD belongs to the polyglutamine (polyQ) expansion disorders, a group of ten diseases that share the same genetic alteration, a pathological increase in the number of CAG codon repeats. In MJD, the CAG expansion is present in exon 10 of ATXN3 gene, located on chromosome 14 (14q32.1) [4,5]. Normal individuals have 12 to 42 CAG repeats, but in MJD carriers, the repeat range extends from 52 to 84 [2]. The ATXN3 gene codes for the ubiquitously expressed 42 kDa protein ataxin-3, and thus, the pathological CAG-expanded gene is translated into ataxin-3 with an extended polyQ tract in its C-terminus [6,7,8,9]. Although its full biological role remains elusive, several studies have demonstrated that ataxin-3 binds and cleaves polyubiquitin chains, exhibiting a deubiquitinating-like activity [10,11,12,13,14]. The presence of an expanded polyQ region promotes protein misfolding and destabilization of the expanded ataxin-3 structure, leading to protein deposition and formation of intracellular inclusions [15,16,17,18]. Intracellular aggregates are found in the nuclei of neurons from degenerated areas of MJD patient brains, as well as in nuclei and cytoplasm of cell lines expressing expanded ataxin-3 [19,20,21,22]. Ataxin-3 localizes to the cytoplasm and nucleus, but the presence of expanded ataxin-3 in the nucleus seems to be important for the manifestation of disease in MJD transgenic mice [23].
Several studies have reported altered mitochondrial structure and a consistent deregulation of mitochondrial activity and function in polyQ disorders. Mitochondria from Huntington's disease (HD) patients and transgenic mice show a decrease in membrane potential and in the ability to retain Ca2+ [24,25]. Severe defects in mitochondrial complex II and III activities and deficiency in complex IV have also been reported to be correlated to neuronal death in HD [26,27,28]. PolyQ proteins also impair mitochondrial transport in neuronal processes and induce caspase activation through mitochondrial pathways [29,30,31]. Moreover, polyQ disease proteins and polyQ-containing protein fragments are able to induce mitochondrial dysfunction and mitochondrial swelling in vitro [29]. Although much data has been gathered for other polyQ disorders [28, 32], there is a lack of information regarding altered mitochondrial activity in MJD and the role of mitochondria in the progress of this pathology. Thus, in this study we determined the susceptibility of cells expressing expanded ataxin-3 to selective mitochondrial inhibitors and assessed the activity of mitochondrial complexes in MJD transgenic mouse brain and human lymphoblasts. The results show a significant trend towards impairment in complex II in different models of MJD and human MJD peripheral cells.
2. Materials and Methods
2.1. Materials
Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute's medium (RPMI) were acquired from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Neurobasal medium and B-27 supplement, fetal bovine serum (FBS), horse serum were purchased from GIBCO (Paisley, UK). Hygromycin, blasticidin were from Invitrogen (Paisley, UK). Nerve growth factor (NGF) was purchased from Alomone Labs (Jerusalem, Israel). Primary antibodies against ataxin-3 1H9 (1:1000; Chemicon, USA) and microtubule associated protein 2 (MAP-2) were obtained from Chemicon (Temecula, CA, USA). Anti-coilin was from BD biosciences (San Jose, CA, USA) and anti-promyelocytic leukemia protein (PML) was from Santa Cruz Biotechology (Santa Cruz, CA, USA). Secondary antibodies for immmunocytochemistry anti-mouse Alexa-fluor 594, anti-rabbit Alexa-fluor 488 and Hoechst 33342 were from Molecular Probes (Eugene, OR, USA), while secondary antibodies for western blotting were acquired from GE healthcare (Uppsala, Sweden). Superfect reagent was purchased from Qiagen (Hilden, Germany). Unless otherwise stated, all other reagents were from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and were of analytical grade.
2.2. Constructs, cell lines culture and transfections
Enhanced green fluorescent protein (EGFP)-ataxin-3 (Q28) and EGFP-ataxin-3 (Q84) plasmids encode a fluorescent fusion-protein composed of full length human ataxin-3 (MJD1a isoform) with EGFP attached to its N-terminus [22]. These constructs were transfected into HEK 293 cells with Superfect reagent, according to manufacturer's instructions. HEK 293 cells were maintained in DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) streptomycin/penicillin. Stably transfected, doxycycline-inducible PC6-3 wild type (Q28) or expanded (Q108) cell lines were kept in RPMI supplemented with 5% (vol/vol) FBS, 10% (vol/vol) horse serum, hygromycin, blasticidin and 1% (vol/vol) streptomycin/penicillin. To induce neural differentiation, PC6-3 cells were plated on poly-L-lysine-coated multiwell plates and incubated with 100 ng/ml NGF for 7 days, in low serum-containing medium (1% (vol/vol) FBS) to reduce cell proliferation. Expression of human ataxin-3 was controlled through the addition of doxycycline to the media. Normal and 2 MJD (JMMA and JMJW have 28/68 and 27/78 repeat alleles, respectively) lymphoblastic cell lines were grown in RPMI supplemented with 10% (vol/vol) FBS, 250 mg/L glucose, 10 mM HEPES, 1 mM sodium pyruvate and 1% (vol/vol) streptomycin/penicillin. All cell lines were kept in a 95% air and 5% CO2 atmosphere at 37°C.
2.3. Culture of cerebellar granule cells isolated from MJD transgenic mice
Cerebellar granule cells from 6-7 day-old MJD transgenic mouse [33] pups or wild-type littermates were prepared according to the protocol for the isolation of cerebellar granule cells from rat pups previously described [34], with slight modifications attending to the different species of origin. Cells were plated on poly-L-lysine-coated multiwell plates or glass coverslips. Cells were maintained in Neurobasal medium supplemented with 2% (vol/vol) B-27, 0.2 mM glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin and 24 mM KCl. After the first day in culture, 10 μM cytosine arabinose was added to the media to prevent glial cell proliferation. Old medium was partially removed and new medium was added to granule cells every fourth day. Cerebellar granule cells were kept in a humidified atmosphere of 95% air and 5% CO2, at 37°C, and used after 7-8 days in vitro. Isolated granule cells from expanded ataxin-3 transgenic mice were as healthy in culture as the neurons from the wild-type littermates.
2.4. Cytochemistry and immunocytochemistry
Transfected HEK 293 cells expressing EGFP-human ataxin-3 for 24 or 48 hours had their media removed and were washed 2 times with phosphate buffered solution (PBS, in mM: 137 NaCl, 2.7 KCl, 1.4 K2HPO4, 4.3 Na2HPO4, pH 7.4). The cells then were fixed with methanol:acetone (1:1) solution for 15 minutes in ice. After 3 washes with PBS cells were incubated with Hoechst 33342 (1 μg/μl) in PBS, for 5 minutes. The coverslips were further washed with PBS and mounted in DAKO solution.
PC6-3 cells and murine cerebellar granule cells were also washed 2 times with PBS before fixation with 4% (wt/vol) paraformaldehyde (PFA) in PBS, for 15 minutes. Then, cells were rinsed 3 times with PBS and opsonized with 5% (vol/vol) goat serum in PBT (phosphate-buffered saline plus 0.1% (vol/vol) Triton X-100) for 1 hour at room temperature. Cells were stained with the primary antibodies: anti-ataxin-3 1H9 (1:1000), anti-coilin (1:350), anti-PML protein (1:350) and anti-MAP-2 (1:200) diluted in 5% (vol/vol) goat serum in PBT, for one hour at room temperature. Cells were then washed 2 times with PBS and incubated with the secondary antibodies: anti-mouse Alexa-fluor 594 (1:250) and anti-rabbit Alexa-fluor 488 (1:250) in 5% (vol/vol) goat serum in PBT for one hour at room temperature. The cells were stained with Hoechst 33342 (1 μg/μl) in PBS for 5 minutes before being mounted in DAKO solution.
Immunofluorescence was visualized with an Axioplan fluorescence microscope (Zeiss, Thornwood, NY) using 20, 40, and 63× objectives. Images were captured digitally with a Zeiss MRM AxioCam camera and assembled in Photoshop 7.0 (Adobe Systems, Mountain View, CA). For each antibody, fluorescence intensity was controlled using identical capture time between genotypes, and images were captured on a linear scale.
2.5. Cell viability assay
HEK293 cells transfected with EGFP-ataxin-3 (Q28) or (Q84) for 48 hours and transgenic or wild-type cerebellar granule cells after 7 days in culture were incubated with mitochondrial inhibitors for a 24 hour-period. A large concentration range was tested for each of two inhibitors: rotenone (10 nM - 20 μM), an inhibitor of complex I, and 3-nitropropionic acid (3-NP, 10 μM - 10 mM), an irreversible inhibitor of succinate dehydrogenate. Cell viability was evaluated by following the reduction of (3-[4-5-dimethylthiazol-2- ]-2,5-diphenyltetrazolium bromide (MTT). MTT (0.5 μg/ml) diluted in Krebs buffer (in mM: 120.9 NaCl, 4.8 KCl, 1.2 KH2PO4, 25.5 NaHCO3, 13 glucose, 10 HEPES, pH 7.4) was added to the cell cultures and MTT reduction was carried on for 2 hours. The resultant formazan crystals were dissolved in a HCl-containing solution (0.04 M in isopropanol). The optical density was measured at 570 nm.
2.6. Evaluation of cell membrane integrity
Extracellular medium and intracellular samples of HEK 293 cell lines expressing EGFP-ataxin-3 for 48 hours and undifferentiated PC6-3 neural cell lines expressing human ataxin-3 were collected and stored at -80°C. Intracellular extracts were obtained through a double cycle of cell freezing and thawing in a HEPES-buffered solution (10 mM HEPES + 1% (vol/vol) Triton), followed by cell scraping. Intracellular and extracellular samples were centrifuged at 20 800 × g for 10 minutes, at 4°C to eliminate cell debris and the resulting supernatant was used for the determination of lactate dehydrogenase (LDH) activity. LDH activity was assessed spectrophotometrically by following the conversion rate of NADH to NAD+ at 340 nm in a Perkin Elmer lambda 2 UV/VIS spectrophotometer. LDH leakage was expressed as percentage of the total activity (% of LDH released = extracellular LDH/extracellular LDH + intracellular LDH) [35].
2.7. Mitochondrial fractionation
Mitochondrial crude fractions of PC6-3 and lymphoblastic cell lines were prepared in a sucrose medium (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, pH 7.4), at 4°C. Briefly, cells were washed twice and scraped in ice-cold sucrose medium, followed by homogenization in a potter (120 strokes). The homogenates were centrifuged in a refrigerated table-top centrifuge at 2300 rpm for 12 minutes at 4 °C to precipitate cell debris and nuclei. The resulting supernatants were collected and stored at -80°C.
Brain mitochondria were isolated from wild-type and MJD homozygous transgenic mice with 4-5 months according to a method previously described [36], with the slight modifications, using 0.02% (wt/vol) digitonin to free mitochondria from the synaptosomal fraction. In brief, mice were euthanized with a ketamine/xylazine solution, followed by a brain perfusion with PBS to reduce blood contamination. The whole brain was rapidly removed, washed, minced, and homogenized at 4°C in 10 ml of isolation medium (225 mM mannitol, 75 mM sucrose, 5 mM Hepes, 1 mM EGTA, 1 mg/ml bovine serum albumin, pH 7.4) containing 5 mg of the bacterial protease. Single brain homogenates were brought to 30 ml and then centrifuged at 750 × g (Beckman Coulter Avanti J-26 XP1 Centrifuge) for 5 minutes. Mitochondria were recovered from the supernatant by centrifugation at 12 000 × g for 8 minutes. The pellet, including the fluffy synaptosomal layer, was resuspended in 10 ml of the isolation medium containing 0.02 (wt/vol) % digitonin and centrifuged at 12 000 × g for 8 min. The brown mitochondrial pellet without the synaptosomal layer was then resuspended again in 10 ml of medium and re-centrifuged at 12 000 × g for 10 minutes. The mitochondrial pellet was resuspended in 300 ml of resuspension medium (225 mM mannitol, 75 mM sucrose, 5 mM Hepes, pH 7.4) and stored at -80°C.
2.8. Mitochondrial respiratory chain complex activities
NADH–ubiquinone oxidoreductase assay – Complex I activity was determined at 340 nm by following the decrease in NADH absorbance due to ubiquinone reduction to ubiquinol [37]. Complex I activity was expressed in nanomoles/minute/milligram protein and corresponds to the rotenone sensitive rate. The enzyme activity was corrected for citrate synthase activity. Succinate–ubiquinone oxidoreductase assay – Complex II activity was monitored at 600 nm by following the reduction of 6,6-dichlorophenolindophenol (DCPIP) by the ubiquinol resulting from this reaction [38]. Complex II activity was expressed in nanomoles/minute/milligram protein and corresponds to the thenoyltrifluoroaceton (TTFA) sensitive rate. The enzyme activity was corrected for citrate synthase activity.
Ubiquinol–cytochrome c reductase assay – Complex III activity was monitored at 550 nm by following the ubiquinol reduction of cytochrome c. The assay was started by adding the sample to the reaction mixture (in mM: 35 K2HPO4, pH 7.2, 1 EDTA, 5 MgCl2, 1 KCN, 5 μM rotenone) containing 15 μM cytochrome c and 15 μM ubiquinol, at 30°C. Complex III activity was expressed in rate constant (k) per minute per milligram of protein and corrected for citrate synthase activity.
Cytochrome c oxidase assay – Complex IV activity was determined at 550 nm by measuring the oxidation of reduced cytochrome c by cytochrome c oxidase [39]. Complex IV activity was expressed in rate constant (k) per minute per milligram of protein and corrected for citrate synthase activity.
Citrate synthase assay - Citrate synthase (CS) activity was performed at 412 nm following the reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) in the presence of acetyl-CoA and oxaloacetate [40]. CS activity was expressed in nanomoles/minute/milligram of protein.
2.9. SDS-PAGE and immunoblotting
Total extracts from transfected HEK 293 cells, PC6-3 cell lines or primary cerebellar granule cells were collected and SDS-PAGE was performed in 10% polyacrylamide gels. Next, the proteins were transferred onto polyvinylidene difluoride membranes (Hybond-P, GE healthcare), which were further blocked in a 5% (wt/vol) non-fat milk solution for 60 minutes. Membranes were incubated with anti-ataxin-3 monoclonal (1H9, 1:1000) overnight at 4°C with agitation. After washing, the blots were incubated with alkaline phosphatase-conjugated anti-mouse secondary antibody and developed with enhanced chemifluorescence. The membranes were scanned in a Biorad VersaDoc Imaging System Model 3000.
2.10. Statistical analysis
Data were expressed as mean ± SEM of the number of experiments indicated in the figure legends. Comparisons among multiple groups were analyzed with two-way analysis of variance (ANOVA) followed by the Bonferroni post-hoc test, as indicated in the figure legends. Comparisons between two groups/conditions were performed with two-tailed unpaired Student's t-test. Significance was defined as p<0.05.
3. Results
3.1. EGFP-ataxin-3 protein aggregation in a transient expression cell model
We initiated our study in a transient expression cell model expressing wild-type or expanded ataxin-3. HEK 293 cells were transfected with mammalian expression vector encoding EGFP fusion protein of wild-type (Q28) or expanded (Q84) ataxin-3 (Figure 1A). EGFP was located at the N-terminus of ataxin-3, near the globular Josephin domain containing the catalytic site. The expression of ataxin-3 fusions was confirmed by western blotting (Figure 1B) and fluorescence microscopy. Both expression vectors were highly expressed with similar transfection efficiency (Figure 1D). Transfected cells displayed a strong EGFP signal 24 hours after transfection (Figure 1Ca, 1Cc). Confocal microscopy revealed different patterns of intracellular localization for wild-type and expanded ataxin-3 (Figure 1C). EGFP-ataxin-3 (Q28) was distributed homogenously throughout the cell (although more abundant in the cytoplasm), regardless of the length of time (24 or 48 hours) after transfection or the expression levels in the cell (Figure 1Ca, 1Cb). In contrast, EGFP-ataxin-3 (Q84) was initially diffusely distributed in the cytoplasm and nucleus of most cells (Figure 1Cc). However, in a small number of cells and at a higher magnification, small sites of EGFP-ataxin-3 (Q84) accumulation with a perinuclear distribution were already detectable (Figure 1Ce). At 48 hours of expression, EGFP-ataxin-3 (Q84) formed readily detectable cytoplasmic and perinuclear aggregates (Figure 1Cd). The aggregates were not present in all transfected cells, and were more frequent in the cells exhibiting higher levels of ataxin-3 expression. Interestingly, in cells exhibiting protein aggregates, nearly all of the intracellular pool of expanded ataxin-3 localized to the aggregate.
Figure 1.
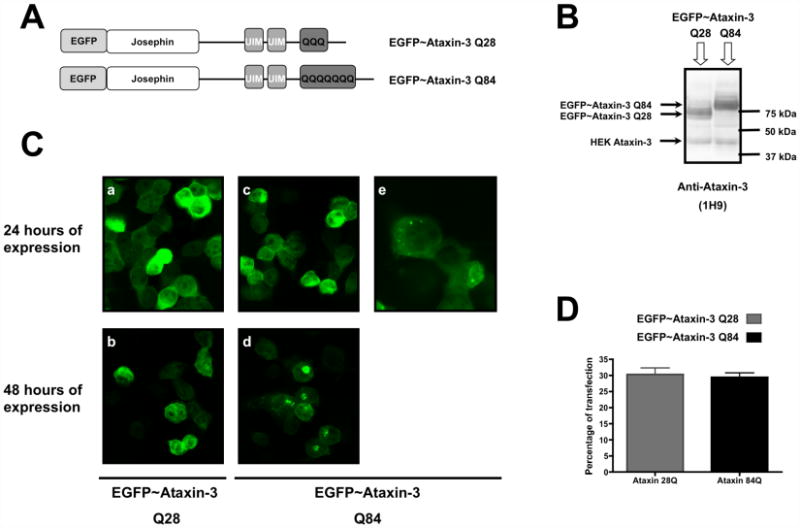
EGFP-ataxin-3 Q84 fusion protein aggregates in a transiently expressed MJD cell model. HEK 293 cells were transfected with EGFP-ataxin-3 Q28 or EGFP-ataxin-3 Q84 plasmid constructs and their expression was carried out for 24 or 48 hours. (A) Representative schemes of the wild-type (Q28) and expanded (Q84) ataxin-3 fusion protein being expressed. (B) Total extracts prepared from HEK293 cells transfected with EGFP-ataxin-3 (Q28) or (Q84) were analysed through western blotting for ataxin-3 to assess the expression of the plasmid constructs. (C) Representative confocal fluorescent microscopy images of HEK293 cells expressing EGFP-ataxin-3 Q28 (a, b) or EGFP-ataxin-3 Q84 (c, d, e) for 24 (a, c, e) or 48 (b, d) hours. (e) Higher magnification of HEK cells transfected with EGFP-ataxin-3 Q84 for 24 hours. (D) HEK293 cells expressing EGFP-ataxin-3 Q28 or EGFP-ataxin-3 Q84 fusion proteins were fixed, nuclear stained with Hoechst 33342 and quantified through fluorescent microscopy. The graph plots the percentage of cells expressing wild-type (Q28) or expanded (Q84) ataxin-3 fluorescent fusion proteins in three independent transfections.
The aggregates were not SDS-insoluble, consistent with earlier reports [41,42]. The characteristic high molecular weight bands of SDS-resistant aggregates immunopositive for ataxin-3 or retention of aggregates in the stacking gel or in the interface between the stacking and the running gels were not detected by western blotting (data not shown). Moreover, longer periods of expression of EGFP-ataxin-3 (Q84) (72, 96 hours) did not result in an increase in the number or size of aggregates (data not shown). For this reason, all other experiments using this cell model were carried out at 48 hours of expression.
3.2. Inducibly expressed expanded ataxin-3 accumulates in subnuclear compartments
To compare the formation of ataxin-3 aggregates between dividing cells and neuronal-like cells in the same cellular environment, we used stably transfected PC6-3 cell lines (a PC12 sub-clone [43] expressing wild-type (Q28) or expanded (Q108) ataxin-3 under a tetracycline-regulated promoter (Figure 2). These cell lines stably overexpress human wild-type or expanded ataxin-3 when doxycycline is administered to the culture media. Moreover, after NGF treatment, PC6-3 cells lose their normal growth round shape to develop long cellular processes resembling neurites, assuming a neuronal-like morphology. After differentiation, they exhibit an excitable polarized cell membrane sensitive to depolarization with potassium, not responding to glutamate or acetylcholine stimuli (not shown). Low concentrations of doxycycline (0.1 μg/ml) were efficient at inducing maximal expression of ataxin-3 in these cell lines with no further increase in expression observed for higher dosages of the antibiotic (Figure 2A). There were also no significant differences in the ataxin-3 levels produced in PC6-3 cells expressing wild-type or expanded ataxin-3. To study the ataxin-3 distribution in cells with a neuronal-like phenotype, PC6-3 cells were incubated with NGF for 7 days in culture, before induction of wild-type or expanded human ataxin-3 expression for 24, 48, 72 and 96 hours. Fluorescent microscopic images of PC6-3 cells immunostained for ataxin-3 revealed that the cellular distribution of wild-type ataxin-3 did not alter over time and no aggregation was detected along the time of expression tested (Figure 2B), as previously observed in HEK293 cells (Figure 1Ca, 1Cb). However, in PC6-3 cells, wild-type ataxin-3 (Q28) had more affinity for the nuclear compartment than the EGFP-ataxin-3 Q28 fusion protein. On the other hand, the expanded form of ataxin-3 (Q108) exhibited a propensity to aggregate in PC6-3 cells, which was visible already at 24 hours after the induction of expression (Figure 2B). Some PC6-3 cells expressing expanded ataxin-3 (Q108) developed visible ataxin-3 positive aggregates, primarily nuclear. These aggregates were SDS-soluble, as no higher molecular weight bands or aggregates in the stacking gel were observed on western blots (data not shown). Interestingly, the formation of intranuclear ataxin-3 aggregates was observed only in PC6-3 cells differentiated with NGF and therefore committed to a neuronal intracellular environment; protein aggregates were not visible in dividing cells (not shown). In cells without visible aggregates, expanded ataxin-3 displayed a nuclear-enriched distribution, similar to wild-type ataxin-3. When present, aggregates condensed almost all ataxin-3 fluorescent signal into a discrete intranuclear distribution, resembling the localization of some subnuclear compartments. In order to investigate this possibility, we co-immunostained PC6-3 cells for ataxin-3 and various signature proteins of subnuclear structures. The aggregates co-localized with promyelocytic leukemia protein (PML) and were often juxtaposed to coilin-positive bodies (Figure 2C, D). Nuclear PML-enriched bodies are known to co-localize with aggregates of polyQ proteins and the specific association between the Cajal (coiled) bodies and expanded ataxin-3 has been previously observed [44].
Figure 2.
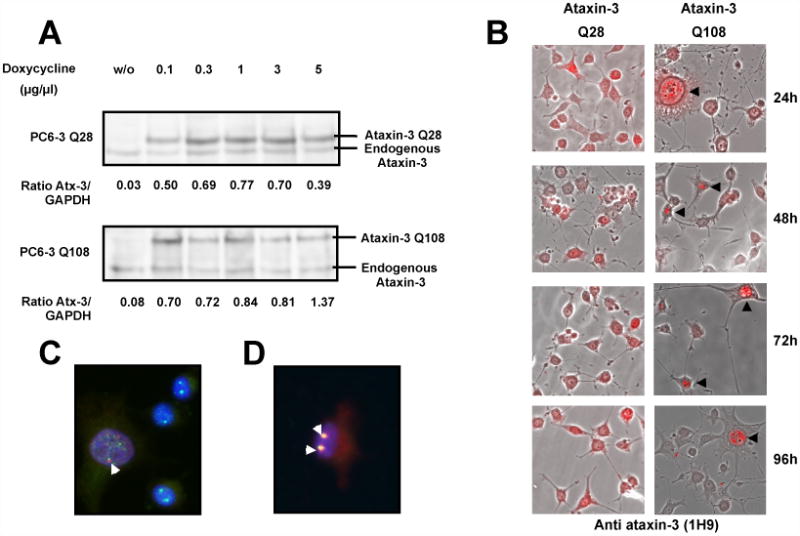
Aggregates of human expanded ataxin-3 in PC6-3 ataxin-3 Q108 cells are primarily nuclear. (A) Total extracts were prepared from PC6-3 ataxin-3 Q28 cells or PC6-3 ataxin-3 Q108 cells incubated in the absence or presence of increasing concentrations (0.1, 0.3, 1, 3 and 5 μg/μl) of doxycycline for 24 hours and subsequently probed for ataxin-3 on a western blot. (B) Merged representative images of fluorescent and optical differential interference contrast microscopy of PC6-3 cells incubated with doxycycline (1 μg/μl) for expression of human ataxin-3 Q28 or human ataxin-3 Q108 during 24, 48, 72 or 96 hours and immunostained for ataxin-3 (in red). (C, D) Representative images of PC6-3 ataxin-3 Q108 cells expressing human expanded ataxin-3 for 48 hours, which were fixed and immunostained for ataxin-3 (in red) and coilin (in green) (C) or PML protein (in green) (D). Yellow shows co-localization between proteins. Nuclei were stained with Hoechst 33342 (1 μg/ml).
3.3. Ataxin-3 aggregation in neuronal cultures from a transgenic mouse model
We also investigated the intracellular localization of expanded ataxin-3 in a neuronal context, using primary cultures of cerebellar granule cells isolated from a human expanded ataxin-3 transgenic mouse model [33]. This MJD transgenic mouse model mimics several pathological features of MJD in humans [33]. In this model, the expression of the human expanded ataxin-3 (Q71) is under the control of the mouse prion promoter, which drives expression of the transgene throughout the brain. Intranuclear inclusions immune-positive for ataxin-3 are detected in several subthalamic regions of the brain and in the spinal cord of the adult mouse [33].
Western blotting of both wild-type and transgenic total brain extracts was performed to characterize expanded ataxin-3 expression (Figure 3B). Immunostaining for ataxin-3 was higher in the nucleus both for cultured neurons derived from non-transgenic and transgenic mice, however, there were differences in the intranuclear distribution of wild-type murine ataxin-3 and expanded human ataxin-3 (Figure 3A). Fluorescent microscopy images revealed intranuclear foci of expanded ataxin-3 similar to what we observed in the previous cell models. In wild-type neurons, nuclear ataxin-3 had a diffuse and homogeneous disposition, but in the nucleus of transgenic neurons, several discrete foci were easily detectable in almost all cells. The small size and the lack of higher molecular weight bands on western blots suggest that these ataxin-3 positive nuclear foci are not mature, SDS-insoluble aggregates. No differences in nuclear distribution of expanded ataxin-3 were observed in cultured neurons carrying one versus two copies of the transgene (i.e. hemizygous versus homozygous transgenic mice; data not shown). However, both homozygous and hemizygous transgenic mice at a later age develop intranuclear inclusions in the adult mouse brain [33].
Figure 3.
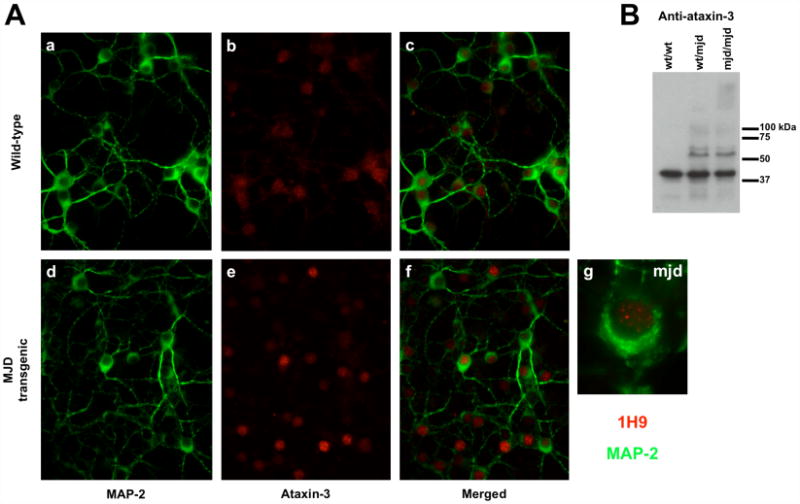
Aggregation in the human expanded ataxin-3 transgenic mouse. (A) Cerebellar granule cells were isolated from 7 days wild-type (a, b, c) or transgenic (d, e, f) pups and kept in culture for 7 days. Cells were stained for MAP-2 (a, c, d, f - in green) and ataxin-3 (b, c, e, f - in red) and visualised through fluorescence microscopy. (g) Higher magnification of transgenic cerebellar granule cell immunostained for MAP-2 (in green) and ataxin-3 (in red). (B) Western blotting for ataxin-3 was performed in total brain extracts of wild-type (wt/wt), or transgenic (one copy (wt/mjd) or two copies (mjd/mjd) of the transgene) 4 month old mice to evaluate the expression of the transgene and the aggregation of human expanded ataxin-3.
3.4. Overexpression of expanded ataxin-3 enhances cell death upon mitochondrial complex II inhibition
Non-transfected HEK cells and cells expressing EGFP-ataxin-3 (Q28) or EGFP-ataxin-3 (Q84) for 48 hours were exposed to increasing concentrations of inhibitors of mitochondrial respiratory chain complexes I and II, rotenone and 3-NP respectively, for 24 hours. Incubation with rotenone or 3-NP induced a dose-dependent decrease in MTT reduction in HEK cells (Figure 4A). Similar levels of cell death were observed for ataxin-3 (Q28) and ataxin-3 (Q84) expressing HEK cell lines subjected to rotenone treatment (Figure 4A), suggesting that expression of expanded ataxin-3 does not affect cellular susceptibility to inhibition of mitochondrial complex I. In contrast, a statistically significant difference between the survival of the two ataxin-3 expressing HEK cell lines was detected in the presence of 3-NP (Figure 4A). For the lower concentrations of 3-NP (100 μM and 1 mM) tested, cells expressing EGFP-ataxin-3 (Q84) were more vulnerable than cells expressing EGFP-ataxin-3 (Q28) cells, although statistical significance was only detected in the presence of 100 μM 3-NP. Untransfected cells had a similar response to 3-NP exposure as EGFP-ataxin-3 (Q28) cell line, indicating that the differences in cell viability observed at low 3-NP concentrations were not due to transfection procedures, nor to the overexpression of ataxin-3 per se (Figure 4A). Antimycin A, an inhibitor of complex III, was also tested in the HEK cell lines, but there were no differences in cell survival between wild-type and expanded ataxin-3 expressing cells (Figure S1 – supplementary data).
Figure 4.
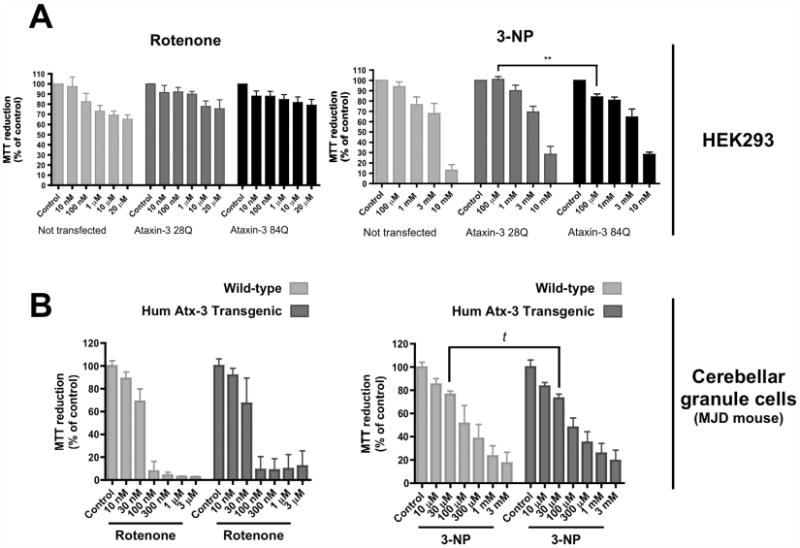
Overexpression of expanded ataxin-3 (Q84) increases cell susceptibility to low concentrations of 3-nitropropionic acid (3-NP). HEK 293 cells transfected with EGFP-ataxin-3 (Q28) or (Q84) (A) or cerebellar granule cells from wild-type or transgenic mice pups (B) were incubated with rotenone (10 nM - 20 μM) or 3-NP (10 μM - 10 mM), for 24 hours. After this incubation period, cell viability was assessed by MTT (0.5 μg/ml) reduction. Graphs summarize the mean ± SEM of the percentage of control for each cell type of 4-8 independent experiments, run in duplicates. Statistical analysis: ** p<0.01, compared to HEK 293 EGFP-ataxin-3 Q28 treated with 100 μM 3-NP, two-way ANOVA followed by Bonferroni multiple comparison test; t p<0.05, compared to wild-type cerebellar granule cells treated with 30 μM 3-NP (Student's t-test).
Cerebellar granule neurons from MJD transgenic or non-transgenic mice were also exposed to inhibitors of mitochondrial complexes I and II, rotenone and 3-NP respectively, for 24 hours. In comparison to HEK 293 cells, the cerebellar granule neurons were more susceptible to rotenone or 3-NP (Figure 4). At 1 mM 3-NP, around 25% of the cerebellar granule cells survived, in contrast to 75% survival of HEK cells. Non-transgenic and MJD neurons responded similarly to rotenone inhibition (Figure 4B). On the contrary, transgenic cerebellar granule cells exhibited a higher susceptibility for 30 μM 3-NP, when compared to wild-type neurons (Figure 4B).
3.5. Transient expression of expanded ataxin-3 promotes loss of membrane integrity
3-NP-induced cell death was also analysed by following LDH leakage in transfected HEK cells. Increasing 3-NP dosage induced higher levels of extracellular LDH. Statistically significant differences between these two transfected cell lines were observed for low 3-NP concentrations (Figure 5A). At 100 μM 3-NP, the EGFP-ataxin-3 (Q84) line showed higher extracellular LDH levels than EGFP-ataxin-3 (Q28) expressing cells, supporting the lower cell viability at this concentration, as observed before. Interestingly, the EGFP-ataxin-3 (Q84) cell line exhibited an tendency towards a higher basal LDH release, which was statistically different from the EGFP-ataxin-3 (Q28) expressing cells (Figure 5A), suggesting that expression of expanded ataxin-3 might decrease membrane integrity. Adenosine triphosphate (ATP) levels, adenosine diphosphate (ADP) levels and caspase-3-like activity were also measured in HEK cells expressing ataxin-3 under the same experimental conditions described above. Neither the incubation with 3-NP nor the expression of expanded ataxin-3 altered any of these parameters in these cells (data not shown). The increased loss of membrane integrity (LDH release) and the absence of activated caspase-3 in western blotting and fluorimetric assays suggested that EGFP-ataxin-3 cell lines were most certainly dying through necrosis or a caspase-3 independent pathway.
Figure 5.
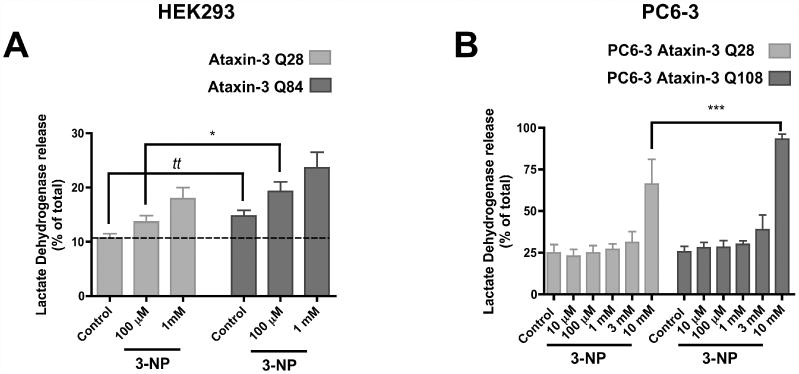
Expanded ataxin-3 induces higher levels of cell death. HEK 293 cells expressing EGFP-ataxin-3 Q28 or Q84 (A) and PC6-3 cells expressing wild-type (Q28) or expanded (Q108) human ataxin-3 (B) were exposed to increasing concentrations of 3-NP (100 μM and 1 mM for HEK cells (A); 10 μM - 10 mM for PC6-3 cells (B)), for 24 hours. Total extracts and the extracellular media of every sample were collected and analysed for LDH activity to evaluate the degree of cell death. Graphs plot the mean ± SEM of the percentage of extracellular LDH activity of the total LDH activity, for each sample. 4-15 independent experiments were conducted. Statistical analysis: (A) tt p<0.01 compared to HEK 293 EGFP-ataxin-3 Q28 (Student's t-test); * p<0.05 compared to HEK 293 EGFP-ataxin-3 Q28 treated with 100 μM 3-NP, two-way ANOVA followed by Bonferroni multiple comparison test. (B) *** p<0.001 compared to PC6-3 Ataxin-3 Q28 treated with 10 mM 3-NP, two-way ANOVA followed by Bonferroni multiple comparison test.
We also examined the susceptibility to complex II inhibition in NGF-differentiated PC6-3 cells expressing wild-type (Q28) or expanded (Q108) ataxin-3. Figure 5B shows that concentrations of 3-NP from 10 μM to 3 mM do not change significantly the release of LDH. However, there was a strong increase in the LDH release for both cell lines in the presence of 10 mM 3-NP (Figure 5B). The expanded ataxin-3 PC6-3 cells displayed statistically significant higher levels of extracellular LDH for this concentration, compared to wild-type ataxin-3 (Q28).
3.6. Expression of expanded ataxin-3 affects mitochondrial complex II activity
The increased susceptibility to 3-NP observed in different MJD cell models led us to hypothesise that expanded ataxin-3 overexpression could affect the mitochondria at the level of mitochondrial complex II. Therefore, we next determined the activities of all the mitochondrial respiratory complexes in several MJD models with the purpose of detecting any other effects expanded ataxin-3 might have on mitochondrial electron transport chain. To accomplish this, we collected mitochondrial crude samples from undifferentiated and NGF-differentiated PC6-3 cell lines expressing human ataxin-3, wild-type and MJD transgenic adult mouse brains, and three human lymphoblastic cell lines. Mitochondria from whole mouse brain were obtained from non-transgenic or homozygous transgenic mice at 4-5 months of age. At this age, homozygous MJD mice manifest clear symptoms of abnormal phenotype characteristic of MJD [33]. Three human lymphoblast cell lines were established after immortalization of lymphoblasts collected from two MJD patients and one healthy individual [6], with the two MJD lymphoblastic cell lines containing 27/78 (JMJW) and 28/68 (JMMA) CAG repeat alleles.
Measurement of the activity of individual mitochondrial complexes revealed a common trend throughout the three MJD models tested (Figure 6). In all models, no alterations were detected in the activities of complexes I, III and IV in the presence of expanded ataxin-3.
Figure 6.
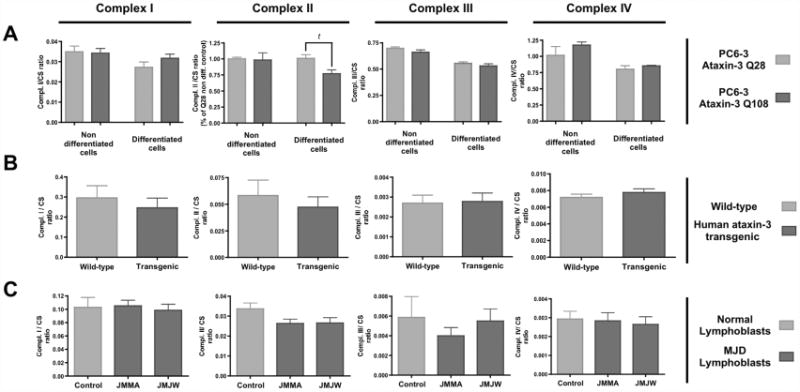
Expanded ataxin-3 expression impairs mitochondrial complex II activity. The activities of complexes I, II, III and IV of the mitochondrial respiratory chain were determined spectrophotometrically in (A) mitochondrial extracts from undifferentiated and NGF-differentiated PC6-3 ataxin-3 Q28 and Q108 cell lines, (B) isolated brain mitochondria of 4-5 months wild-type or MJD homozygous transgenic mice, and (C) human lymphoblastic cell lines derived from MJD patients (JMMA, JMJW) or control individuals. Graph shows the mean ± SEM of 4-9 independent measurements of the mitochondrial complexes activities. The activities of mitochondrial complexes were normalised for citrate synthase activity. Statistical analysis: t p<0.05, compared to NGF-differentiated PC6-3 Ataxin-3 Q28 (Student's t-test).
Nevertheless, strong tendencies, and in some cases significant differences, were observed for the activity of mitochondrial complex II (Figure 6). Statistically significant differences were observed in NGF-differentiated PC6-3 cell lines expressing expanded versus wild-type human ataxin-3 (Figure 6A). PC6-3 ataxin-3 (Q108) cells exhibited an approximately 25% reduction in complex II activity, compared to PC6-3 ataxin-3 (Q28). A decrease in complex II activity was also determined for the MJD human lymphoblastic cell lines, although statistically significance was not achieved. Accordingly, the protein levels of complex II subunits (ShdA and ShdB) were not altered in mitochondrial extracts of MJD lymphoblastic cell lines (Figure S2 – supplementary data).
4. Discussion
Human ataxin-3 has been shown to have an inherent capacity to self-aggregate, even the wild-type variant [45]. The presence of a pathological polyQ tract in the ataxin-3, however, introduces an additional step in the aggregation process, resulting in the formation of highly stable and SDS-insoluble aggregates [41]. The presence of protein aggregates containing expanded ataxin-3 and other proteins in specific areas of the brain of MJD patients is a pathological hallmark of this neurological disease [19,46,47]. In agreement with this, ataxin-3 protein aggregation was observed in the cell and animal models used in this study and occurred selectively in the expanded ataxin-3-expressing models. Analysis of mitochondrial susceptibility in ataxin-3 expressing models revealed that the mitochondrial inhibitor producing the strongest response throughout the different models was 3-NP, an irreversible complex II inhibitor. Indeed, most of the MJD cell models showed a tendency towards increased susceptibility to 3-NP treatment and decreased activity of complex II.
Higher levels of protein and the expression of a more aggregation-proned isoform (MJD1a) of expanded ataxin-3 may account for the larger aggregates observed in HEK EGFP-ataxin-3 (Q84) cells. Although in some in vitro studies wild-type ataxin-3 has been shown to aggregate [41,45], in cells we only observed aggregation of expanded ataxin-3.
Aggregation of expanded EGFP-ataxin-3 (Q84) in HEK 293 cell lines was associated with deleterious effects, increasing the susceptibility to mitochondrial complex II inhibitor (3-NP) treatment. Indeed, expression of expanded ataxin-3 alone was sufficient to increase cell death in this model. Aggregation of expanded ataxin-3 in PC6-3 ataxin-3 (Q108) cells decreased cell survival only in the presence of high 3-NP concentrations, compared to HEK EGFP-ataxin-3 (Q84) cells or MJD cerebellar granule cells, which may account for a lower expression of the transgene in these cells. The aggregates in PC6-3 cells and transgenic cerebellar granule cells are discrete nuclear structures and smaller than the bulky clumps observed in HEK 293 cells. Steric interference caused by the larger aggregates in HEK 293 cells over intracellular movement of proteins and organelles, might contribute for the increased cytotoxicity in these cells. The nuclear localization of aggregates in PC6-3 ataxin-3 (Q108) cells was not random. They were intimately associated with PML bodies and closely juxtaposed to coiled or Cajal bodies. PML bodies have been described as sites of protein degradation in the nucleus, while Cajal bodies are part of a subnuclear organelle implicated in RNA splicing [48,49,50]. The close location of expanded ataxin-3 aggregates to nuclear sites of protein degradation may represent a cellular effort to degrade the accumulated protein. Additionally, these specific nuclear sites may gather specific proteins or factors that trigger the seeding stages of expanded ataxin-3 deposition.
Although expanded ataxin-3 is expressed throughout the body, only neurons of some subcortical regions of the brain degenerate in MJD patients. Interestingly, the cells from these regions also represent some of the main sites of ataxin-3 aggregation [46]. In accordance, the formation of aggregates selectively occurs in PC6-3 (Q108) cells committed to a neuronal phenotype (i.e., upon differentiation with NGF) and is associated with increased susceptibility to high concentrations of the mitochondrial complex II inhibitor, 3-NP. Accumulation of human expanded ataxin-3 in discrete subnuclear sites was also observed in neuronal cultures derived from MJD transgenic mice. Even though the cerebellar granule cells were collected from 7 day-old mouse pups, nuclear foci of accumulated ataxin-3 were already detectable, suggesting that at least the subnuclear localization of expanded ataxin-3, may start early in development, well before the first signs of neuropathology. These early stages of expanded ataxin-3 accumulation were accompanied by a mild mitochondrial susceptibility of cerebellar MJD neurons. Expanded ataxin-3 inclusions in this mouse model have only been reported in adulthood, starting at 2.5 months of age [33]. Therefore, the expanded ataxin-3 nuclear foci detected in 7 day-old transgenic neurons may represent the initial stages of ataxin-3 accumulation not yet capable of exerting a full toxic effect. Although the discussion regarding the protective versus pathological role of polyQ aggregates is not fully resolved [51], in our experimental models, aggregates of expanded ataxin-3, which may be dimers, oligomers, or higher order aggregates, were associated with increased cell death.
Activation of mitochondrial apoptotic pathways and reduction in antioxidant enzymes activity followed by increased mitochondrial DNA damage have been reported for cellular models of MJD, establishing a mitochondrial role in MJD pathology [42,53]. We have not observed apoptotic events or energy deficits upon expression of expanded ataxin-3 in HEK293 cells. However, expanded ataxin-3 may exert its pathological effects by interfering and disrupting additional biochemical pathways. Notably, mitochondrial complex II or succinate dehydrogenase is a biochemical converging point between the mitochondrial electron chain and the citric acid or Krebs cycle. Interestingly, we observed that complex II was the only mitochondrial complex to exhibit a consistent tendency towards decreased activity in the presence of expanded ataxin-3, particularly in differentiated PC6-3 cells expressing ataxin-3 (Q108) and in lymphoblastic cell lines derived from MJD patients. Complex II activity in brain mitochondria from MJD transgenic mice showed to be more resilient to human expanded ataxin-3 toxicity. The capacity to sustain a satisfactory complex II activity may be associated to an absence of major neurodegeneration, previously described in this MJD transgenic model [33]. On the other hand, the mild decrease in complex II activity in dividing, peripheral cells such as MJD lymphoblastic cell lines is a rather interesting result. Indeed, more studies need to be carried out to verify this direct relationship between expression of expanded ataxin-3 and the drop in complex II activity. Inhibition of complex II was previously linked to production of reactive oxygen species (ROS) [54, 55]. Thus, increased oxidative stress in cells expressing expanded ataxin-3 would explain the increased susceptibility to ROS induced by the extra complex II inhibition provided by 3-NP treatment. Nevertheless, complex I and complex III inhibitions did not increase cell susceptibility of expanded ataxin-3 expressing cells, suggesting the possibility that oxidative stress should not be the only mechanism underlying ataxin-3 toxicity. Although complex II does not contribute to the mitochondrial proton gradient, disruption of normal mitochondrial calcium handling might be another mechanism involved in ataxin-3 pathology. It is worth noting that a significant decrease in complex II activity was observed in a cell model committed to a neuronal phenotype, the NGF-treated PC6-3 ataxin-3 (Q108) cells.
These cells show aggregates of expanded ataxin-3 occurring primarily in the nucleus, despite the low levels of expanded ataxin-3 expression. Although, complex II is the only mitochondrial complex of the respiratory chain that is exclusively nuclear-encoded and expanded ataxin-3 has been reported to interfere with gene expression in the nucleus [17], we have not found alterations in protein levels of complex II subunits in MJD lymphoblasts (Figure S2 - supplementary data). Nevertheless, the activity of complex II is not solely determined by changes in the expression of its subunits. Alterations in regulatory proteins involved in phosphorylation and acetylation of complex II [56], mitochondrial import of subunits and/or the activity of chaperones responsible for complex II assembly could also interfere with its activity, without altering the protein levels. One or more of these phenomena may contribute to mitochondrial dysfunction in MJD.
Ataxin-3 is a deubiquitinating enzyme that has been implicated in the ubiquitin-proteasome pathway and in cellular protein quality control. Recent studies have described a crosstalk between the mitochondria and the proteasome [57]. Expression of expanded ataxin-3 may impair the ubiquitin-proteasome system or disrupt the crosstalk between the proteasome and mitochondria. Similarly to what was found upon expression of mutant huntingtin [58], macroautophagy malfunction may also occur in the context of expanded ataxin-3 expression due to a failure on the engulfment of cytosolic components in autophagosomes, precluding an adequate elimination of aggregated proteins and/or dysfunctional mitochondria. Indeed, impaired protein degradation pathways may delay protein turnover, promoting the accumulation of altered mitochondrial complexes with reduced activity. An impairment of mitochondrial complex II in MJD cell and animal models is particularly intriguing considering that selective vulnerability to complex II inhibitors and defects in complex II activity have been reported for other polyQ diseases, especially HD [26,59]. Mitochondrial complex II impairment might be central to some of the biochemical changes elicited by various polyQ expanded proteins, making this mitochondrial complex an important target in polyQ toxicity.
Supplementary Material
Figure S1- Cell susceptibility to antimycin A is not altered by human expanded ataxin-3 expression. Untransfected HEK293 cells and HEK293 cells transfected with EGFP-ataxin-3 (Q28) or (Q84) were exposed to increasing concentrations of antimycin A (10 nM – 30 μM) for 24 hours, before cell viability assessment through MTT (0.5 μg/ml) assay. Graph summarizes the mean ± SEM of the percentage of control for each cell type of 4 independent experiments, run in duplicates.
Figure S2- Protein levels of mitochondrial complex II subunits are not changed in MJD lymphoblastic cell lines. (A) 40 μg of protein from mitochondrial extracts of normal and MJD (JMMA, JMJW) lymphoblastic cell lines were run in SDS-PAGE and mitochondrial complex II (succinate dehydrogenase) subunits (SdhA and SdhB) were detected through immunoblotting with anti-SdhA and anti-SdhB antibodies. Quantification of SdhA (B) and SdhB (C) protein levels in MJD lymphoblastic cell lines relatively to the control line in 3 independent experiments, run in triplicates.
Research Highlights.
We evaluated mitochondrial dysfunction in models of Machado-Joseph disease.
Expanded ataxin-3 accumulation occurs in the cytoplasm and nuclei of cells.
Nuclear ataxin-3 aggregates are correlated to Cajal and PML bodies distribution.
Expanded ataxin-3 induced an increased susceptibility to complex II inhibition.
Machado-Joseph disease models show a decreased mitochodrial complex II activity.
Acknowledgments
The plasmid constructs for EGFP-ataxin-3 (Q28) and EGFP-ataxin-3 (Q84) were generously provided by Prof. Patricia Maciel (U. Minho, Braga, Portugal). Portuguese Foundation for Science and Technology (FCT), scholarship SFRH/BD/17275/2004 and NIH grant R01NS038712 (HLP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albrecht M, Hoffman D, Evert BO, Scmitt I, Wullner U, Lengauer T. Structural modelling of ataxin-3 reveals distant homology to adaptins. Proteins. 2003;50:355–370. doi: 10.1002/prot.10280. [DOI] [PubMed] [Google Scholar]
- 2.Rüb U, Brunt ER, Del Turco D, de Vos RA, Gierga K, Paulson H, Braak H. The nucleus raphe interpositus in spinocerebellar ataxia type 3 (Machado-Joseph disease) J Chem Neuroanat. 2003;25:115–127. doi: 10.1016/s0891-0618(02)00099-6. [DOI] [PubMed] [Google Scholar]
- 3.Rüb U, de Vos RA, Schultz C, Brunt ER, Paulson H, Braak H. Spinocerebellar ataxia type 3 (Machado-Joseph disease): severe destruction of the lateral reticular nucleus. Brain. 2002;125:2115–2124. doi: 10.1093/brain/awf208. [DOI] [PubMed] [Google Scholar]
- 4.Kawagushi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–8. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 5.Riess O, Rub U, Pastore A, Bauer P, Schols L. SCA3: Neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–137. doi: 10.1007/s12311-008-0013-4. [DOI] [PubMed] [Google Scholar]
- 6.Paulson HL, Das SS, Crino PB, Perez MK, Patel SC, Gotsdiner D, Fischbeck KH, Pittman RN. Machado-Joseph Disease gene product is a cytoplasmic protein widely expressed in brain. Ann Neurol. 1997;41:453–462. doi: 10.1002/ana.410410408. [DOI] [PubMed] [Google Scholar]
- 7.Trottier Y, Cancel G, An-Gourfinkel I, Lutz Y, Weber C, Brice A, Hirsch E, Mandel JL. Heterogeneous intracellular localization and expression of ataxin-3. Neurobiol Dis. 1998;5:335–347. doi: 10.1006/nbdi.1998.0208. [DOI] [PubMed] [Google Scholar]
- 8.Ichikawa Y, Goto J, Hattori M, et al. The genomic structure and expression of MJD, the Machado-Joseph disease gene. J Hum Genet. 2001;46:413–422. doi: 10.1007/s100380170060. [DOI] [PubMed] [Google Scholar]
- 9.Schmitt I, Evert BO, Khazneh H, Klockgether T, Wüellner U. The human MJD gene: genomic structure and functional characterization of the promoter region. Gene. 2003;314:81–88. doi: 10.1016/s0378-1119(03)00706-6. [DOI] [PubMed] [Google Scholar]
- 10.Berke SJ, Paulson HL. Protein aggregation and the ubiquitin protesome pathway: gaining the UPPer hand on neurodegeneration. Curr Opin Genet Dev. 2003;13:253–261. doi: 10.1016/s0959-437x(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 11.Burnett B, Li F, Pittman RN. The polyglutamine neurodegenrative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet. 2003;12:3195–3205. doi: 10.1093/hmg/ddg344. [DOI] [PubMed] [Google Scholar]
- 12.Donaldson KM, Li W, Ching KA, Batalov S, Tsai CC, Joazeiro CA. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamines aggregates. Proc Natl Acad Sci USA. 2003;100(15):8892–8897. doi: 10.1073/pnas.1530212100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chai Y, Berke SS, Cohen RE, Paulson HL. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J Biol Chem. 2004;279:3605–3611. doi: 10.1074/jbc.M310939200. [DOI] [PubMed] [Google Scholar]
- 14.Winborn BJ, Travis SM, Todi SV, Scaglione KM, Xu P, Williams AJ, Cohen RE, Peng J, Paulson HL. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitina chains. J Biol Chem. 2008;283(39):26436–26443. doi: 10.1074/jbc.M803692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bevivino AE, Loll PJ. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta-fibrils. Proc Natl Acad Sci. 2001;98:11955–11960. doi: 10.1073/pnas.211305198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cemal CK, Carroll CJ, Lawrence L, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet. 2002;11:1075–1094. doi: 10.1093/hmg/11.9.1075. [DOI] [PubMed] [Google Scholar]
- 17.Evert BO, Vogt IR, Vieira-Saecker AM, Ozimek L, de Vos RA, Brunt ER, Klockgether T, Wüllner U. Gene expression profiling in ataxin-3 expressing cell lines reveals distinct effects of normal and mutant ataxin-3. J Neuropathol Exp Neurol. 2003;62:1006–1018. doi: 10.1093/jnen/62.10.1006. [DOI] [PubMed] [Google Scholar]
- 18.Masino L, Nicastro G, Menon RP, Dal Piaz F, Calder L, Pastore A. Characterization of the structure and the amyloidogenic properties of the josephin domain of the polyglutamine-containing protein ataxin-3. J Mol Biol. 2004;344:1021–1035. doi: 10.1016/j.jmb.2004.09.065. [DOI] [PubMed] [Google Scholar]
- 19.Paulson HL, Perez MK, Trottier T, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19(2):333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 20.Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]
- 21.Evert B, Wüllner U, Schulz JB, Weller M, Groscurth P, Trottier Y, Brice A, Klockgether T. High level expression of expanded full-length ataxin-3 in vitro causes cell death and formation of intracellular inclusions in neuronal cells. Hum Mol Genet. 1999;8:1169–1176. doi: 10.1093/hmg/8.7.1169. [DOI] [PubMed] [Google Scholar]
- 22.Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci. 2002;99:9310–9315. doi: 10.1073/pnas.152101299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bichelmeier U, Schmidt T, Hubener J, et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J Neurosci. 2007;27:7418–7428. doi: 10.1523/JNEUROSCI.4540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 25.Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT. In vitro effect of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington's disease. Arch Biochem Biophys. 2003;410:1–6. doi: 10.1016/s0003-9861(02)00585-4. [DOI] [PubMed] [Google Scholar]
- 26.Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39(3):385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 28.Solans A, Zambrano A, Rodríguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPOS complexes II and III. Hum Mol Gen. 2006;15(20):3063–3081. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- 29.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 30.Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai HF, Tsai HJ, Hsieh M. Full-length expanded ataxin-3 enhances mitochondrial-mediated cell death and decreases bcl-2 expression in human neuroblastoma cells. Biochem Biophys Res Commun. 2004;324:1274–1282. doi: 10.1016/j.bbrc.2004.09.192. [DOI] [PubMed] [Google Scholar]
- 32.Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- 33.Goti D, Katzen SM, Mez J, Kurtis N, et al. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J Neurosci. 2004;24(25):10266–10279. doi: 10.1523/JNEUROSCI.2734-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courtney MJ, Lambert JJ, Nicholls DG. The interactions between plasma membrane depolarization and glutamate receptor activation in the regulation of cytoplasmic free calcium in cultured cerebellar granule cells. J Neurosci. 1990;10:3873–3879. doi: 10.1523/JNEUROSCI.10-12-03873.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rego AC, Santos MS, Oliveira CR. Influence of the antioxidants vitamin E and idebenone on retinal cell injury mediated by chemical ischemia, hypoglycemia or oxidative stress. Free Radic Biol Med. 1999;26:1405–1417. doi: 10.1016/s0891-5849(98)00337-2. [DOI] [PubMed] [Google Scholar]
- 36.Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S. Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab. 1987;7:752–758. doi: 10.1038/jcbfm.1987.130. [DOI] [PubMed] [Google Scholar]
- 37.Ragan CI, Wilson MT, Darley-Usmar VM, Lowe PN. In: Subfractionation of mitochondria, and isolation of the proteins of oxidative phosphorylation, in Mitochondria, a practical approach. Darley-Usmar VM, Rickwood D, Wilson MT, editors. IRL Press; London: 1987. pp. 79–112. [Google Scholar]
- 38.Hatefi Y, Stiggall DL. Preparation and properties of succinate: ubiquinone oxidoreductase (complex II) Methods Enzymol. 1978;53:21–27. doi: 10.1016/s0076-6879(78)53008-5. [DOI] [PubMed] [Google Scholar]
- 39.Wharton DC, Tzagotoff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- 40.Coore HG, Denton RM, Martin BR, Randle PJ. Regulation of adipose tissue pyruvate dehydrogenase by insulin and others hormones. Biochem J. 1971;125:115–127. doi: 10.1042/bj1250115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem. 2006;281(25):16888–16896. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 42.Wong SL, Chan WM, Chan HY. Sodium dodecyl sulfate-insoluble oligomers are involved in polyglutamine degeneration. FASEB J. 2008;22(9):3348–3357. doi: 10.1096/fj.07-103887. [DOI] [PubMed] [Google Scholar]
- 43.Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13(9):3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Xu H, Neggi S, Sbramony SH, Hebert MD. Differential effects of polyglutamine proteins on nuclear organization and artificial reporter splicing. J Neurosci Res. 2007;85(11):2306–17. doi: 10.1002/jnr.21369. [DOI] [PubMed] [Google Scholar]
- 45.Gales L, Macedo-Ribeiro S, Arsequell G, Valencia G, Saraiva MJ, Damas AM. Towards a structural understanding of the fibrillization pathway in Machado-Joseph's disease: trapping early oligomers of non-expanded ataxin-3. J Mol Biol. 2005;353(3):642–654. doi: 10.1016/j.jmb.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt T, Landwehrmeyer GB, Schmitt I, et al. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998;8:669–679. doi: 10.1111/j.1750-3639.1998.tb00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada M, Hayashi S, Tsuji S, Takahashi H. Involvement of the cerebral cortex and autonomic ganglia in Machado-Joseph disease. Acta Neuropathol (Berl) 2001;101:140–444. doi: 10.1007/s004010000277. [DOI] [PubMed] [Google Scholar]
- 48.Morris GE. The Cajal Body. Biochim Biophys Acta. 2008;1783(11):2108–2115. doi: 10.1016/j.bbamcr.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 49.St-Germain JR, Chen J, Li Q. Involvement of PML nuclear bodies in CBP degradation through the ubiquitin-proteasome pathway. Epigenetics. 2008;3(6):342–349. doi: 10.4161/epi.3.6.7203. [DOI] [PubMed] [Google Scholar]
- 50.Wojcik C, DeMartino GN. Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003;35(5):579–589. doi: 10.1016/s1357-2725(02)00380-1. [DOI] [PubMed] [Google Scholar]
- 51.Uchihara T, Iwabuchi K, Funata N, Yagishita S. Attenuated nuclear shrinkage in neurons with nuclear aggregates - a morphometric study on pontine neurons of Machado-Joseph disease brains. Exp Neurol. 2002;178(1):124–128. doi: 10.1006/exnr.2002.8028. [DOI] [PubMed] [Google Scholar]
- 52.Chou AH, Yeh TH, Kuo YL, Kao YC, Jou MJ, Hsu CY, Tsai SR, Kakizuka A, Wang HL. Polyglutamine-expanded ataxin-3 activates mitochondrial apoptotic pathway by upregulating Bax and downregulating Bcl-xl. Neurobiol Dis. 2006;21:333–345. doi: 10.1016/j.nbd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Yu YC, Kuo CL, Cheng WL, Liu CS, Hsieh M. Decreased antioxidant enzyme activity an increased mitochondrial DNA damage in cellular models of Machado-Joseph disease. J Neurosci Res. 2009;87:1884–1891. doi: 10.1002/jnr.22011. [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez E, Rivera I, Astorga S, Mendoza E, García F, Hernández-Echeagaray E. Uncoupling oxidative/energy metabolism with low sub chronic doses of 3-mitropropionic acid or iodoacetate in vivo produces striatal cell damage. Int J Biol Sci. 2010;22:199–212. doi: 10.7150/ijbs.6.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sandhir R, Mehtora A, Kamboj SS. Lycopene prevents 3-nitropropionic acid-induced mitochondrial oxidative stress and dysfunctions in nervous system. Neurochem Int. 2010;57:579–587. doi: 10.1016/j.neuint.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion. 2010;10:393–401. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fukui H, Moraes CT. Extended polyglutamine repeats trigger a feedback loop involving the mitochondrial complex III, the proteasome and huntingtin aggregates. Hum Mol Genet. 2007;16(7):783–797. doi: 10.1093/hmg/ddm023. [DOI] [PubMed] [Google Scholar]
- 58.Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13(5):567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruan Q, Lesort M, MacDonald ME, Johnson GV. Striatal cells from huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death trough a non-apoptotic pathway. Hum Mol Genet. 2004;13(7):669–681. doi: 10.1093/hmg/ddh082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1- Cell susceptibility to antimycin A is not altered by human expanded ataxin-3 expression. Untransfected HEK293 cells and HEK293 cells transfected with EGFP-ataxin-3 (Q28) or (Q84) were exposed to increasing concentrations of antimycin A (10 nM – 30 μM) for 24 hours, before cell viability assessment through MTT (0.5 μg/ml) assay. Graph summarizes the mean ± SEM of the percentage of control for each cell type of 4 independent experiments, run in duplicates.
Figure S2- Protein levels of mitochondrial complex II subunits are not changed in MJD lymphoblastic cell lines. (A) 40 μg of protein from mitochondrial extracts of normal and MJD (JMMA, JMJW) lymphoblastic cell lines were run in SDS-PAGE and mitochondrial complex II (succinate dehydrogenase) subunits (SdhA and SdhB) were detected through immunoblotting with anti-SdhA and anti-SdhB antibodies. Quantification of SdhA (B) and SdhB (C) protein levels in MJD lymphoblastic cell lines relatively to the control line in 3 independent experiments, run in triplicates.