

Abstract
Lentiviruses have been adapted as gene delivery vehicles. This article summarized shRNA lentiviral vector methods generally used in research laboratories. The main procedures of shRNA lentiviral vector include that (1) Target sequences screening and shRNA oligonucleotides designing, (2) insert designed oligonucleotides into lentiviral vectors, (3) using packaging cells to produce shRNA lentivirus, and (4) transducing target cells with shRNA lentivirus.
Keywords: Lentiviral vector, laboratories, shRNA, lentivirus, cells, oligonucleotides
Small hairpin RNA (shRNA) and lentiviral vector
RNA interference has been used as a research tool to control the expression of specific genes in numerous experimental organisms and has potential as a therapeutic strategy to reduce the expression of problem genes[1,2]. RNAi can be performed through a synthetic small interfering RNA (siRNA) or a vector-based shRNA. shRNAs, which are stem-loop RNA structures, can be used to silence gene expression via RNA interference after processing by DICER.
As a tool to deliver genetic material into cells (in vivo or in vitro)[1], lentiviral vectors are able to integrate shRNAs which can be used to down regulate specific gene into the genome of both dividing and non-dividing cells make them highly attractive[1]. Key properties of a viral vector are safety (including low toxicity), stability, cell type specificity, and markers. For safety reasons, to produce a lentivirus, packaging plasmids, which encode the virion proteins (the capsid and the reverse transcriptase), are transfected into packaging cell line (i.e., HEK 293). Lentiviral vectors are transcribed to produce the single-stranded RNA viral genome (Lentivirus is a subclass of retroviruses). Ψ (psi) sequence in lentiviral vectors is used to package the genome into the virion.
In this article, we aimed to summarize this useful technique that has been used in our laboratories. shRNA oligonucleotides designing, lentiviral vectors constrcution, o shRNA lentivirus producing, transducing target cells with shRNA lentivirus etc are described in this article in detail.
siRNA sequences selecting and shRNA oligonucleotides designing
Choosing siRNA sequences within target gene is critical[1]. There are some rules can be followed: 1, Avoid GC rich sequences (> 50%). 2, Avoid stretches of 4 or more nucleotide repeats.3, Avoid sequences homology with other related or unrelated genes, etc. However, select such sequence manually is not easy. Fortunately, there are lots of websites provide this service (i.e. http://www.ambion.com/techlib/misc/siRNA_finder.html). Since some siRNAs provide low reduction (<50%), testing more than one siRNA sequence recommended by software for each target gene (i.e. 4-6 sequences) is highly recommended. The hairpin having the strongest inhibitory activity is often determined empirically by testing the shRNA sequence. siRNA with the same nucleotide composition as target siRNA but which lacks significant sequence homology to the genome can be a negative control[2].
The shRNA oligonucleotides should include the following two complementary oligonucleotides (an upper and lower strand) are needed for each shRNA target site (Fig. 1). The oligonucleotide sequences should provide a preferred Pol III transcription start site, a guanine (G) residue should be added upstream of the 5’- end of the shRNA, if the target sequence does not start with a purine:
Fig. 1.
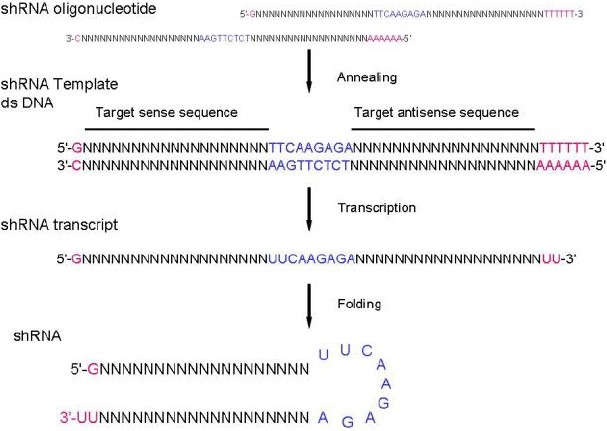
Form oligonucleotides to shRNA harepin
A restriction site overhangs on the upper strand;
The target siRNA sequence;
A nucleotide hairpin loop sequence. Various research groups have reported: TTCAAGAGA[1], AAGTTCTCT (Promega), TTTGTGTAG[1], etc;
The target siRNA antisense sequence;
The other restriction site overhangs on the lower strand. These restriction sites enable directional cloning of the annealed oligonucleotides into the digested Lentiviral vector;
A RNA Pol III terminator sequence (a 5-6 nucleotide poly (T) tract);
Highly recommended: a diagnostic restriction site for convenient restriction digest analysis to confirm the presence of the cloned insert.
Insert shRNA oligonucleotides into lentiviral vectors
A. Annealing the shRNA Oligonucleotides
Option 1
Resuspend both complementary oligonucleotides at a ratio 1:1, using Annealing Buffer (10 mM Tris, pH 7.5–8.0, 50 mM NaCl, 1 mM EDTA). Annealing should perform over a wide range of oligo concentrations. Heat the mixture to 95°C for 3mins to 5 mins to remove secondary structure. This promotes intermolecular annealing. Gradually reduce the heat until the oligonucleotides have reached room temperature. Proceed to a storage temperature of 4 °C.
Option 2
Heat to 95 °C and remain at 95 °C for 2 minutes. Ramp cool to 25 °C over a period of 45 minutes. Proceed to a storage temperature of 4 °C. The annealed oligonucleotides are now ready for ligation into the vector. Alternatively, the annealed oligonucleotides can be stored at –20°C for later use.
B. Preparing the Lentiviral Vector for Cloning
The annealed shRNA oligonucleotides will be inserted between the restriction sites in vector. Digest 1 μg of Vector DNA with restriction enzyme, using the manufacturer's protocol. Purify the digested vector DNA using a spin column from the Extract Kit (using the manufacturer's protocol), or on an agarose gel. Resuspend digested plasmid in 10–20 μl TE buffer (~50 ng/μl). Store the purified vector DNA at –20°C.
C. Ligating the Annealed Oligonucleotides into the Lentiviral Vector
Dilute the annealed oligonucleotides TE buffer to obtain a concentration of 0.5 μM. Otherwise, large excess of oligonucleotides will inhibit ligation. Prepare a ligation reaction for each annealed pair of oligonucleotides. Incubate the reaction mixture according to the ligase kit manufacturer's protocol.
Prepare DNA for transfection
A. Transform Competent Cells
Transform competent E. coli with 2-5 μl of the ligation reaction, using the protocol supplied with competent cells or using standard methods:
Remove cells from the -80 freezer, add 100 μl of cells to an eppendorf containing the vector, and leave eppendorf on ice for 30 minutes. Put agar plates from cold room into the 37°C incubator and set the water bath to 42 °C. Heat shocks the bacteria in the 42°C water bath for 60 sec then IMMEDIATELY put the test tubes in the ice bucket. If using the metal heating block, it may be necessary to heat for 90 sec. Place the eppendorf on ice for 2-5 minutes. Add 900 μl of LB broth to the cell suspension. Tap the tubes lightly with fingers to gently mix the solution. Incubate the tubes at 37°C with gentle shaking for 45-60 minutes.
B. Identify Recombinant Clones
Add 50 -150 μl cells suspension from each transformation on LB agar + ampicillin plates (50–100 μg/ml). Spread cells quickly. Otherwise, cells will be absorbed into the agar as concentrated spots. Place plates at 37°C Incubate overnight. Pick 5-20 well isolated colonies from each transformation and inoculate each into a small-scale (3-5 ml) LB + ampicillin (50–100 μg/ml). Grow overnight at 37°C with shaking (150-200 RPM). Extract plasmid vector using plasmid minipreps kit or standard methods. Identify the target vector by restriction analysis using the diagnostic restriction site within the shRNA oligonucleotide sequence then sequence shRNA insert (Fig. 2). Sequencing more than two clones is highly recommended due to potential synthesis errors. After identified a positive clone, make a large-scale plasmid prep according to the plasmid prep kit manufacturer's protocol, or standard methods.
Fig. 2.
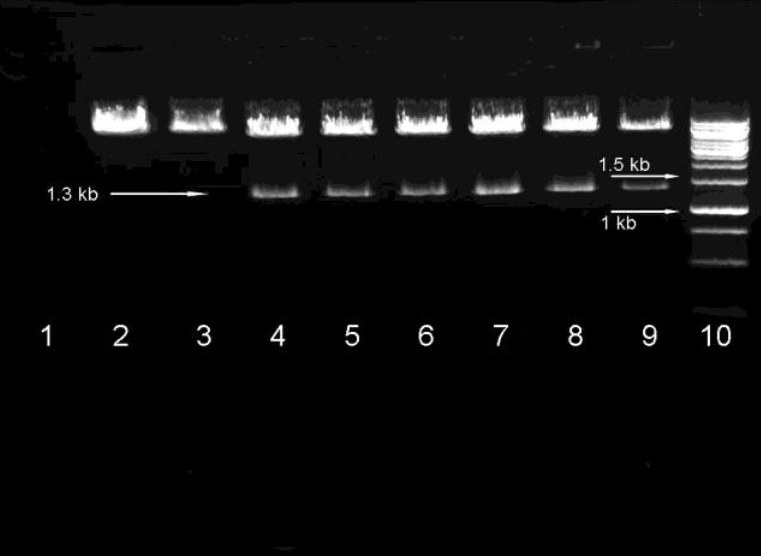
Identify the target vector by restriction analysis using the diagnostic restriction site. 1 negative control, 2 empty plasmid, 3 empty plasmid digested by other enzyme, 4 positive clone 1 digest by selected enzyme which yield 1.3 kb production, 5 positive clone 2, 6 positive clone 3, 7 positive clone 4, 7 positive clone 5, 8 positive clone 6, 9 Marker,
Using packaging plasmids to produce shRNA lentivirus from lentiviral vectors
Normally, HEK 293 cell line will be used to obtain lentivirus from packaging plasmids and lentiviral vector. Key properties of a successful packaging can be culture size and volume, DNA amounts and transfectiongrade quality, and incubation times. Optimize transfection reagents, volumes, and conditions used with vector are very important. Approximately 24 hr before transfection, seed 1-5 × 106 HEK 293 cells into 10 cm tissue culture plates in 10 ml of growth medium. Incubate at 37°C, 5% CO2 overnight. The cells should be 70% confluent at the time of transfection. After transfection 4-8 hours, replace the transfection medium (supernatants may contain infectious lentivirus) with 10 ml fresh complete growth medium and incubate at 37°C for an additional 24–48 hours. Viral titers will generally be highest at 48 hr after the start of transfection (Figs. 3–5). Harvest and filter the lentiviral supernatants through a 0.45 μm low protein binding filter to remove cellular debris. shRNA lentiviruses can be concentrated by ultracentrifugation (2 hours at 50,000 × g) and subsequently purified on a sucrose 20% gradient (2 hours at 46,000 × g)[1] for future use.
Fig. 3.
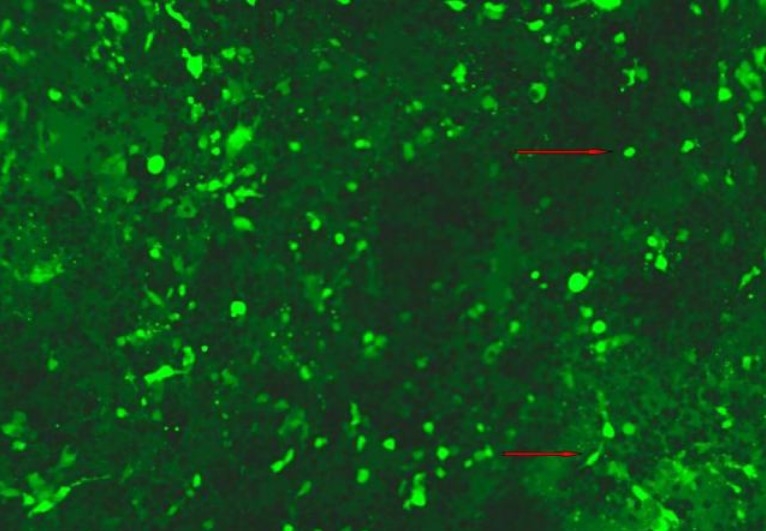
HEK 293 cells after transfection 60 hours. The brightly stained cells are GFP positive cells.
Fig. 5.
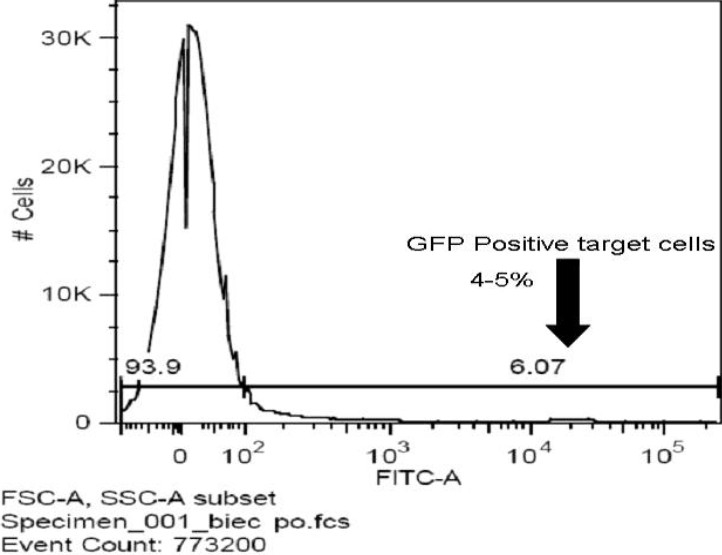
Harvest the cells for proceed with selection using FACS.
Fig. 4.
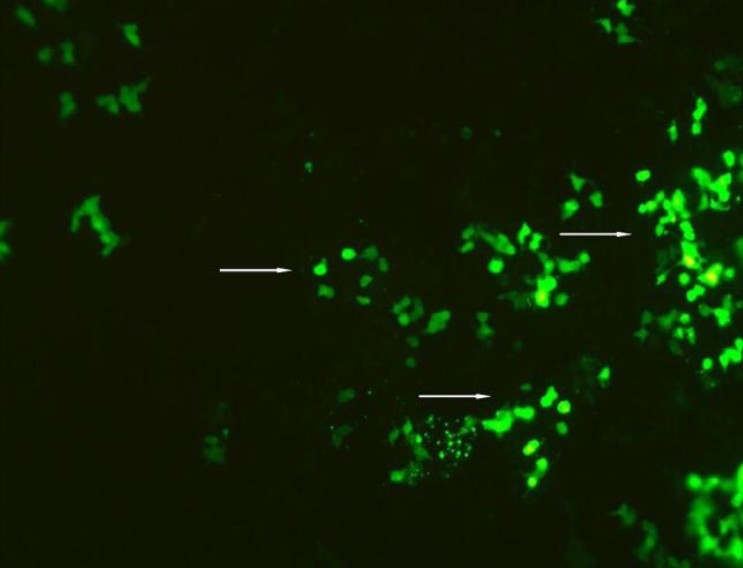
Macrophage cell line after transfection 48 hours. The brightly stained cells are GFP positive cells.
Transducing target cells with shRNA lentivirus
Plate 1-5 × 106 target cells in 10 cm tissue culture dish in 10 ml complete growth medium, Incubate 12-24 hours at 37°C in a humidified incubator in an atmosphere of 5% CO2 before infection. For different cell lines, adjusting the density of target cells and the length of growing time may be necessary. A range of volume or MOI test may be needed before first transducing. 20-150 μl of lentiviral particles per 1-5 × 106 cells or MOIs of 0.5- 5 can be used to determine the optimal transduction efficiency. Dilute the shRNA lentivirus with medium to obtain the desired MOI. Add viral supernatant to the cells and transduce for 8–24 hour. Centrifuge the cultures to improve infection efficiency. To avoid target cells influenced by viral supernatant, limit the infection less than 8 hours. Remove and discard the virus-containing transduction medium and add fresh growth medium. Continue to incubate the cells for 24–48 hours to allow the shRNA to reach its maximum effect. A time course experiment may be necessary in order to determine the optimum time for harvesting the cells. Harvest the cells for analysis or proceed with selection using FACS for GFP or proper antibiotics for resistance gene. Since appropriate concentration of antibiotics for each cell type is different, if the concentration for the target cell type is unknown, a titration test is necessary. If the resistance gene is anti-puromycin, normally, titration can start from 1-10 μg/ml. After first dose, replace media with fresh media containing antibiotics every 2-3 days until resistant colonies can be identified.[9]
References
- 1.Mello CC, Conte D., Jr Revealing the world of RNA interference. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- 2.Gregory J. Hannon. RNA interference, Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 3.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 4.Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA. 2003;100(4):1844–1848. doi: 10.1073/pnas.0437912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumiko Ui-Tei, Yuki Naito, Fumitaka Takahashi, Takeshi Haraguchi, Hiroko Ohki-Hamazaki, Aya Juni, Ryu Ueda, Kaoru Saigo. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32(3):936–948. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whither RNAi? Nat Cell Biol. 2003;5:489–490. doi: 10.1038/ncb0603-490. [DOI] [PubMed] [Google Scholar]
- 7.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2(3):243–724. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 8.Scherr M, Battmer K, Dallmann I, Ganser A, Eder M. Inhibition of GM-CSF receptor function by stable RNA interference in a NOD/SCID mouse hematopoietic stem cell transplantation model. Oligonucleotides. 2003;13(5):353–363. doi: 10.1089/154545703322617032. [DOI] [PubMed] [Google Scholar]
- 9.Naldini l, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93(21):11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]