

Abstract
Nontypeable Haemophilus influenzae (NTHi) is an important bacterial pathogen associated with lower respiratory tract colonization and with acute exacerbations and disease progression in chronic obstructive pulmonary disease (COPD). Why the immune system fails to eliminate NTHi and the exact contribution of the organism to COPD progression are not well understood, in part because we lack an animal model that mimics all aspects of COPD. For this study, we used an established murine model that exhibits typical features of COPD. Elastase/LPS-exposed mice infected with NTHi showed persistence of bacteria up to 5 days after infection, whereas mice exposed to elastase, LPS, or PBS cleared all bacteria by 3 days. Elastase/LPS-exposed mice also showed sustained lung neutrophilic inflammation, goblet cell metaplasia, airway hyperresponsiveness, and progression of emphysema at 15 days after infection. Alveolar macrophages isolated from elastase/LPS-exposed mice showed impaired bacterial phagocytosis, reduced expression of MARCO and of mannose receptor, and absent expression of scavenger receptor-A (SR-A). Neutralization of SR-A significantly decreased phagocytosis of NTHi by normal alveolar macrophages. Our results suggest that elastase/LPS-exposed mice show impaired bacterial clearance and sustained lung inflammation. Lack of SR-A expression may, in part, be responsible for impaired phagocytosis of bacteria by alveolar macrophages of elastase/LPS-exposed mice. These data validate the suitability of elastase/LPS model for investigating NTHi pathogenesis and progression of disease in COPD.
Chronic obstructive pulmonary disease (COPD) is a progressive disorder of the lung parenchyma and airways that became the third-leading cause of death in the United States in 2008.1,2 Acute exacerbations are associated with disease progression and are also a major cause of morbidity and mortality in COPD patients.3,4 Approximately two thirds of all of the infectious exacerbations are associated with bacterial infection, alone or in combination with respiratory viruses.5,6 Acute exacerbations are associated with the acquisition of new strains of nontypeable Haemophilus influenzae (NTHi), Moraxella catarrhalis, and Streptococcus pneumoniae and, in patients with severely reduced lung function, Enterobacteriaceae and Pseudomonas species.7 The lower airways of some COPD patients are also chronically colonized with bacteria readily detectable by conventional culture-dependent methods; these patients show elevated markers of inflammation, compared with individuals who do not show bacterial colonization by this methodology.8,9 NTHi is the most frequently isolated bacterial pathogen from sputum of COPD patients under stable conditions.10 Impaired innate immune defense mechanisms are thought to contribute to the bacterial infection and persistence of bacteria in these patients.
Alveolar macrophages are professional phagocytes that play a critical role in the clearance of infecting bacteria while preventing excessive pulmonary inflammation. Alveolar macrophages express a variety of cell surface receptors that are crucial for recognition and phagocytosis of pathogens and stimulating the appropriate immune response to infection. Compared with alveolar macrophages from healthy nonsmokers, alveolar and monocyte-derived macrophages from COPD patients are deficient in phagocytosis of common respiratory bacterial pathogens, including NTHi and S. pneumoniae,11,12 and of apoptotic cells.13,14 Although the importance of bacterial clearance seems evident, impaired efferocytosis (ie, clearance of apoptotic cells) has also been linked to COPD progression.2,15 The precise mechanisms for the observed defects in COPD are unclear. Defining the molecular basis for defects in alveolar macrophage phagocytic function could lead to novel therapies to improve morbidity and mortality in COPD, which typically progresses even after smoking cessation.
One family of receptors that plays a role in the host innate defense against inhaled particles and pathogens is the class A scavenger receptors, which include macrophage receptor with collagenous structure (MARCO) and scavenger receptor A (SR-A), which is expressed as SR-A I and SR-A II in humans because of a splice variant and as a single form (SR-A I/II) in mice.16–18 Both receptors were shown to be required for efficient clearance of pneumococci from the lungs of infected mice16,17 and also for protection against inhaled oxidants.18 Importantly, the risk of developing COPD is increased in humans with mutations in the macrophage scavenger receptor 1 gene, MSR1, which encodes SR-A types I and II; these mutations result in expression of a receptor lacking the distal collagen-like domain, which is essential in ligand recognition.19–21 Reduced efferocytosis by alveolar macrophages from COPD patients was attributed in part to decreased expression of surface receptors, including CD31, CD91 and CD44.14 However, these studies did not examine expression of SR-A, which we previously showed is essential for optimal efferocytosis by murine peritoneal macrophages and macrophage cell lines.22 Collectively, these findings indicate the importance of defining the possible role of alveolar macrophage scavenger receptors in lung host defense and homeostasis.
To this end, we used an established experimental model in which mice show structural and functional features typical of human COPD, including pulmonary emphysema, loss of lung elastic recoil, hyperinflation, goblet cell metaplasia, markedly increased numbers of neutrophils, T and B lymphocytes, monocytes and macrophages in the airways and alveoli, and a deficiency in clearing rhinovirus infection.23,24 Our results highlight the importance of alveolar macrophage class A scavenger receptors, especially SR-A, for clearance of NTHi.
Materials and Methods
Animals and Treatment
Normal 8- to 10-week-old C57BL/6 mice (Charles River Laboratories International, Wilmington, MA) maintained in a specific pathogen-free environment were exposed to elastase and LPS for four consecutive weeks, as described previously.23,24 At 1 week after the last exposure to LPS, mice were examined for susceptibility to bacterial infection. Mice treated with PBS instead of elastase/LPS were used as controls in all experiments. In selected experiments, mice were exposed to elastase alone or LPS alone once a week for four consecutive weeks. All experiments were approved by the Animal Care and Use Committee of the University of Michigan.
Bacteria and Growth Conditions
NTHi (isolate 6P5H), isolated from a COPD patient at the time of acute exacerbation, was kindly provided by T.F. Murphy (University of Buffalo) and was maintained as glycerol stock at −80°C. Bacteria were subcultured and grown on chocolate agar, as described previously.25 In some experiments, bacteria were labeled with either fluorescein isothiocyanate (FITC) (Pierce; Thermo Fisher Scientific, Rockford, IL) or Alexa Fluor 555 (Invitrogen, Carlsbad, CA), as described previously.26
Infection of Mice
After the 4 weeks of exposure to elastase/LPS, elastase, LPS, or PBS, mice were anesthetized by intraperitoneal injection of xylazine (1 mg/kg body weight) and ketamine (50 mg/kg body weight) and infected with 50 μL of NTHi (5 × 107 CFU) or with the same volume of PBS (sham infection) by the intratracheal route. Mice were euthanized humanely at 1, 3, 5, or 15 days after infection for use in a variety of assays; not all mice were used in each assay.
Lung Function Measurements
Mice were anesthetized and a steel cannula was inserted into the trachea and connected to a miniature computerized FlexiVent ventilator (Scireq, Montreal, QC, Canada). To determine elastic recoil, lungs were gradually inflated to 30 cm H2O; pressure and lung volume were then measured continuously during inflation and deflation of the lungs. Airway responsiveness to methacholine was measured as described previously.23
Lung Histology
Lungs were inflation-fixed with 10% buffer formalin and embedded in paraffin. Sagittal sections, 5 μm thick, were stained with H&E or PAS reagent.
Bacterial Persistence and Measurement of Cytokines
Lungs were harvested from mice under aseptic conditions and homogenized in 2 mL of PBS. Lung homogenates were serially diluted and plated on chocolate agar plates to determine the bacterial load. Homogenate supernatants were used to determine protein levels of proinflammatory cytokines by enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN).
Measurement of Muc5AC and Muc5B Expression
After RNA extraction, Muc5AC and Muc5B mRNA levels were measured by quantitative real-time PCR using specific primers and probes.
Bronchoalveolar Lavage
Mice were euthanized and lungs were lavaged with PBS containing 20 mmol/L EDTA; cells were collected by centrifugation. Total and differential cell counts in bronchoalveolar lavage fluid (BALF) were determined as described previously.23 The cells were immunolabeled with antibodies against CD45, CD11c, CD11b (eBiosciences, San Diego, CA), SR-A, MARCO, mannose receptor, CD80, and CD36 (AbD Serotec, Raleigh, NC) and analyzed on an LSR II flow cytometer (BD Bioscience, San Jose, CA) equipped with 488-nm blue, 405-nm violet, and 633-nm red lasers, as described previously.23
Phagocytosis Assay
BALF cells from PBS or elastase/LPS-exposed mice were centrifuged, suspended in RPMI 1640 medium containing 10% fetal bovine serum, seeded in two-chamber coverslipped glass slides (Invitrogen, Carlsbad, CA), and incubated for 2 hours. The chamber slides were rinsed briefly with medium to remove unattached cells and infected with Alexa Fluor 555-labeled NTHi. Phagocytosis of bacteria in real time was recorded for 30 minutes using a DeltaVision RT-live cell imaging system (Applied Precision, Issaquah, WA).
In some experiments, resident alveolar macrophages plated in chamber slides were infected with FITC-labeled NTHi and extracellular fluorescence was quenched with Trypan Blue, as described previously.26 The number of cells positive for intracellular FITC-labeled bacteria was counted under a fluorescence microscope equipped with phase contrast to determine phagocytosis index.
The capacity of immortalized MH-S murine alveolar macrophages (CRL-2019; ATCC, Manassas, VA) to phagocytose NTHi was quantified by flow cytometry, as described previously.26 Briefly, macrophages were seeded in six-well plates and incubated with FITC-labeled NTHi at 10 MOI (multiplicity of infection) for 1 hour. Cells were washed, extracellular fluorescence was quenched with Trypan Blue, and then cells were analyzed by flow cytometry. In some experiments, macrophages were pretreated with neutralizing antibody to SR-A (Cell Sciences, Canton, MA) or isotype control for 1 hour at 37°C, infected with FITC-labeled NTHi, and analyzed by flow cytometry as above.
Statistical Analysis
Results are presented as means ± SEM. Data were analyzed using SigmaStat statistical software (Systat Software, San Jose, CA). One-way analysis of variance with Tukey-Kramer post hoc analysis was performed to compare more than two groups. To compare two groups, an unpaired t-test with Welch's correction was used. A P value <0.05 was considered significant.
Results
Elastase/LPS-Exposed Mice Show Persistent Pulmonary Bacterial Load and Lung Inflammation
Mice exposed to PBS or to elastase/LPS were infected intratracheally with NTHi and euthanized at 1, 3, and 5 days after infection. Lung bacterial load was determined. Although both PBS-exposed and elastase/LPS-exposed mice showed bacteria in the lungs 24 hours after infection, only elastase/LPS-exposed mice showed persistence of bacteria up to 5 days after infection (Figure 1A).
Figure 1.

Persistence of NTHi and lung inflammation. PBS-exposed or elastase/LPS-exposed mice were either sham-infected with PBS and euthanized 1 day after infection or infected with NTHi and euthanized at 1, 3, or 5 days after infection. A: Lung homogenates from NTHi-infected mice were serially diluted and plated to determine the bacterial load in the lungs. B and C: Bronchoalveolar lavage was performed and numbers of total cells and differential cell counts were determined. D: Myeloperoxidase (MPO) activity was assessed in total lung homogenates. n = 5 to 9 mice per group. *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance.
Bronchoalveolar lavage was performed to determine the total and differential cell counts (Figure 1, B and C). Both PBS-exposed and elastase/LPS-exposed mice showed a 1-log increase in total cell counts 1 day after NTHi infection, compared with their respective sham-infected controls, and neutrophils accounted for 85% to 87% of the cells in both groups. At 3 days after infection, total cells decreased slightly in both groups of mice. Neutrophilic inflammation persisted in both groups, but was significantly higher in the elastase/LPS-exposed group. By 5 days after infection, total and neutrophil cell counts returned to baseline in the PBS group and were similar to those of sham-infected controls. The elastase/LPS-exposed mice, however, showed slightly increased total cell counts and sustained neutrophilic inflammation. Total and differential cell counts in sham-infected animals on days 3 and 5 were similar to those observed at 1 day after infection (data not shown). Myeloperoxidase activity significantly increased in both groups at 1 day after infection, compared with the respective sham-infected animals (Figure 1D). Although myeloperoxidase levels returned to normal levels in the PBS group, the elastase/LPS group showed persistently increased myeloperoxidase levels, consistent with the observed increase in BALF neutrophils.
H&E-stained lung sections from sham-infected animals were histologically similar to those from the respective uninfected mice. At 1 day after infection with NTHi, both PBS-exposed and elastase/LPS-treated mice showed an inflammatory infiltrate, with neutrophils predominating (Figure 2, A, B, and D–F). However, compared with the PBS group, elastase/LPS-exposed mice showed more inflammation (Figure 2, D–G). Whereas PBS-exposed mice showed localized accumulation of inflammatory cells in the peribronchiolar and perivascular areas, elastase/LPS-exposed mice showed widespread neutrophilic inflammation, including in the airway lumina and alveoli (Figure 2, E and F). At 5 days after infection, the inflammation in PBS-exposed mice resolved completely (Figure 2C). In contrast, elastase/LPS-exposed mice showed persistent inflammation up to 5 days after infection (Figure 2G). Consistent with this observation, we observed sustained increases in the proinflammatory cytokines TNF-α, IL-1β, IL-6, KC, MIP-2, and CCL2 up to 5 days after NTHi challenge in elastase/LPS-exposed but not PBS-exposed mice (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 2.

Histology of mice infected with NTHi. Paraffin-embedded lung sections from PBS-or elastase/LPS-exposed mice infected with NTHi were stained with H&E. A: PBS-exposed mouse, 1 day after infection. B: Inflammatory cells seen at higher magnification; the image corresponds to the area outlined in panel A. C: PBS-treated mouse, 5 days after infection. D–F: Elastase/LPS-exposed mice at 1 day after infection. E: Neutrophils in the airway lumen seen at higher magnification; the image corresponds to the area outlined in panel D. F: Neutrophils in the alveolar space. G: Elastase/LPS-exposed mice exhibit moderate lung inflammation, 5 days after infection. Arrows indicate neutrophils; arrowheads indicate macrophages. Images are representative of 3 mice from each group. Scale bars: 50 μm (B, E, and F); 200 μm (A, C, D, and G).
Elastase/LPS-exposed mice with sham infection showed more PAS-positive material than the PBS-exposed mice in both large and small airways, as observed previously (Figure 3, A and B).23,24 PAS-positive cells further increased after bacterial infection in elastase/LPS-exposed mice (Figure 3, C and D), but not in similarly infected PBS-exposed mice (Figure 3, E and F). RT-PCR analysis indicated a significant increase in Muc5AC expression at 1, 3, and 5 days after infection in elastase/LPS-exposed mice, but only 1 day after infection in PBS-exposed mice (Figure 3G). There were no changes in Muc5B expression in either group of mice.
Figure 3.

Mucin expression in NTHi-infected mice. Paraffin-embedded lung sections from PBS-exposed or elastase/LPS-exposed mice infected with NTHi were stained with PAS. A and B: Lung sections from sham-infected elastase/LPS-exposed mice show PAS-positive cells in large (A) and small (B) airways. C and D: Lung sections from elastase/LPS-exposed mice at 5 days after infection show increased PAS-positive cells and PAS-positive material in large (C) and small (D) airways. E and F: Lung sections from PBS-exposed mice at 5 days after infection show few PAS-positive cells in the large airway (E), but not in the small airway (F). Arrows indicate PAS-positive cells and arrowheads indicate PAS-positive material in airway lumen. Images are representative of three animals per group. G: Total RNA was extracted from sham-infected or NTHi-infected mice at 1, 3, or 5 days after infection. Muc5AC and Muc5B gene expression determined by real-time PCR was normalized to expression of β-actin and expressed as fold increase relative to sham-infected PBS-exposed mice. Data are presented as means ± SEM (n = 5 mice/per group). *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance. Scale bars: 50 μm.
NTHi Infection Affects Lung Function in Elastase/LPS-Exposed Mice
To examine whether overt inflammation caused by bacterial infection was associated with altered lung function, we examined pressure-volume relationships and airway cholinergic responsiveness in mice 1 and 5 days after infection. Pressure-volume loops did not change significantly in infected animals and were similar to those of uninfected mice (data not shown). We also measured airway responsiveness to increasing doses of nebulized methacholine. NTHi-infected, PBS-exposed mice showed no significant increase in airway responsiveness, compared with sham-infected mice at either 1 or 5 days after infection. In contrast, elastase/LPS-exposed mice showed significant increases in airway responsiveness at both 1 and 5 days after infection, relative to sham-infected controls, and also relative to similarly treated PBS-exposed mice (see Supplemental Figure S2 http://ajp.amjpathol.org).
Elastase/LPS-Exposed Mice Infected with NTHi Show Progression of Emphysema
To examine the long-term effects of NTHi infection on progression of lung disease, elastase/LPS-exposed mice were infected with NTHi and sacrificed at 15 days after infection. Selected parameters were measured, including bacterial load. None of the groups showed bacteria in their lungs (data not shown). However, compared with sham-infected mice, elastase/LPS-exposed mice infected with NTHi showed small but significantly increased levels of CXCL-1/KC protein (Figure 4A) and Muc5AC mRNA (Figure 4B), but no difference in the expression of Muc5B mRNA (data not shown). In contrast, PBS-exposed mice infected with NTHi showed increases in neither CXCL-1/KC nor Muc5AC levels. Histological evaluation revealed persistence of mild inflammation in both sham-infected and NTHi-infected elastase/LPS-exposed mice, but not in similarly infected PBS-exposed mice (Figure 4, C–E). We also observed sustained goblet cell metaplasia in the small airways of elastase/LPS-exposed mice, which appears to be slightly increased in NTHi-infected mice (Figure 4, F–H). In contrast, mice exposed to PBS did not show PAS-positive cells in the small airways. Notably, NTHi-infected mice in both elastase/LPS and PBS-exposed mice showed slightly leftward and upward shift in pressure-volume curves compared with the respective sham-infected control group (Figure 5A). This shift was more pronounced in NTHi-infected elastase/LPS-exposed mice, suggesting further loss of lung elastic recoil in these mice. Consistent with this observation, NTHi-infected elastase/LPS-exposed mice showed decreased elastance and increased compliance, compared with similarly exposed sham-infected mice. (Figure 5, B and C). Taken together, these results indicate that changes caused by acute NTHi infection may lead to progression of lung disease in elastase/LPS-exposed mice in the absence of detectable bacteria at late phase of infection.
Figure 4.

Lung inflammation and mucus cell metaplasia at 15 days after NTHi infection in elastase/LPS-exposed mice. PBS-exposed or elastase/LPS-exposed mice were sham-infected or NTHi-infected and euthanized 15 days later. A: Lung homogenate supernatant was analyzed by enzyme-linked immunosorbent assay to determine the CXCL-1/KC protein levels. B: mRNA levels of Muc5AC was determined by quantitative PCR using total RNA isolated from lungs. Data are presented as means ± SEM, calculated from triplicate experiments (n = 5 to 8 mice total). *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance. C: H&E-stained lung sections from PBS-exposed NTHi-infected mice show no lung inflammation. D and E: H&E-stained lung section from sham-infected (D) or NTHi-infected elastase/LPS-exposed mice (E) show peribronchiolar inflammation and emphysema. F: PAS-stained lung sections from NTHi-infected PBS-exposed mice show no PAS-positive cells. G and H: PAS-stained lung sections from sham-infected (G) or NTHi-infected elastase/LPS-exposed mice (H) show PAS-positive cells in the airway. Arrows indicate PAS-positive cells. Images are representative of three animals per group. Scale bars: 50 μm (F–H); 200 μm (C–E).
Figure 5.

Lung function in elastase/LPS-exposed mice at 15 days after NTHi infection. PBS-exposed or elastase/LPS-exposed mice were sham-infected or NTHi-infected; mice were anesthetized 15 days later, and pressure-volume relationships (A), compliance (B), and elastance (C) were measured with a FlexiVent system. A: Each curve is representative of pressure-volume curves of three mice/group. B and C: Data are presented as means ± SEM, calculated from triplicate experiments (n = 3). *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance.
Elastase/LPS-Exposed Mice Are Deficient in Phagocytosis
To examine whether the persistence of bacteria in elastase/LPS-exposed mice is caused by a deficiency in phagocytosis of bacteria by alveolar macrophages, we performed ex vivo phagocytosis assays. BALF resident alveolar macrophages obtained from PBS-exposed or elastase/LPS-exposed mice were infected with Alexa Fluor 594-labeled NTHi, and real-time phagocytosis was recorded under time-lapse fluorescence microscopy. Macrophages from elastase/LPS-exposed mice showed decreased capacity to phagocytose bacteria, compared with macrophages from PBS-exposed mice (see Supplemental Movies S1 and S2 at http://ajp.amjpathol.org). Quantification of phagocytosis under microscopy revealed that 78% of macrophages from PBS-exposed mice were positive for intracellular bacteria, whereas only 42% of macrophages from elastase/LPS-exposed mice were positive (Figure 6A). It should be noted that, although elastase/LPS-exposed mice have more macrophages and monocytes put together, the number of resident alveolar macrophages is significantly lower than in the PBS-exposed mice (see below), and therefore it is possible that fewer macrophages will be positive for bacteria in elastase/LPS-exposed mice. Moreover, confocal imaging of macrophages from PBS-treated mice 2 hours after infection revealed degraded bacterial particles (Figure 6B), indicative of efficient killing of phagocytosed bacteria. In contrast, macrophages from elastase/LPS-exposed mice showed intact bacteria on the cell surface and occasionally inside the cells. These data indicate that macrophages from elastase/LPS-exposed mice are not only deficient in bacterial phagocytosis but also appear to be deficient in killing of ingested bacteria.
Figure 6.

Phagocytosis of bacteria by alveolar macrophages. Resident alveolar macrophages were isolated from mice exposed to PBS or elastase/LPS for 4 weeks, seeded in coverslipped chamber slides, and infected with FITC-labeled NTHi bacteria. A: After 30 minutes, extracellular fluorescence was quenched with Trypan Blue and numbers of intracellular bacteria were quantified under fluorescence microscopy (n = 3). Data are presented as means ± SEM. *P ≤ 0.05 versus PBS-treated mice, unpaired t-test with Welch's correction. B: In some experiments, alveolar macrophages were infected with Alexa Fluor 555-labeled NTHi, incubated for 2 hours, fixed, counterstained with DAPI, and visualized under a confocal microscope. Images are representative of three independent experiments. Original magnification, ×1000.
Elastase/LPS-Exposed Mice Have Decreased Levels of Class A Scavenger Receptors
Class A scavenger receptors play a role in optimal phagocytosis of bacteria by macrophages. We therefore assessed the expression of SR-A and MARCO in macrophages from PBS and elastase/LPS-exposed mice. We also examined expression of the class B scavenger receptor CD36, mannose receptor, and the costimulatory receptor CD80. As observed previously, elastase/LPS-exposed mice showed three times more total cells, compared with PBS-exposed mice.23 In PBS-exposed mice, the majority of CD45+ BALF cells were also positive for CD11c (Figure 7, A and B). In contrast, only 10% of CD45+ BALF cells from elastase/LPS-exposed mice were CD11c single-positive and the rest were either CD11b single-positive (recently recruited monocytes) or double positive for both CD11c and CD11b (a population termed exudate macrophages, which would also include any dendritic cells). Because CD45 and CD11c single-positive cells represent resident alveolar macrophages, we compared the expression of scavenger receptors in this cell population. Compared with macrophages from PBS-exposed mice, macrophages from elastase/LPS-exposed mice showed decreased expression of MARCO and of mannose receptor, increased expression of CD36, and absent expression of SR-A (Figure 7C). There was no difference in the expression of CD80 between PBS and elastase/LPS-exposed mice. These results suggest that SR-A may be at least in part responsible for the observed defect in bacterial phagocytosis in these mice.
Figure 7.

Analysis of macrophages and expression of scavenger receptors. BALF cells from PBS-exposed or elastase/LPS-exposed mice were immunostained with a combination of labeled antibodies to CD45, CD11b, CD11c, SR-A, MARCO, CD36, mannose receptor, and CD80 and were analyzed by flow cytometry. A and B: CD45+ cells were analyzed for the expression of CD11b and CD11c. Flow scatter images (A) are representative of three independent experiments. C: CD45 and CD11c+ cells were analyzed for expression of SR-A, MARCO, CD36, mannose receptor, and CD80. Data in B and C are presented as means ± SEM, calculated from two independent experiments (n = 5 mice per experiment). *P ≤ 0.05 versus PBS-treated mice, unpaired t-test with Welch's correction.
Mice Exposed to Elastase or LPS Alone Show Normal Clearance of Bacteria
Previously, we have shown that exposure to both elastase and LPS is required for inducing COPD phenotype including susceptibility to rhinovirus infection in mice,23 but the contribution of elastase or LPS alone to defective bacterial clearance is not known. We therefore exposed mice to elastase or LPS alone and determined their capacity to clear bacteria from their lungs after NTHi challenge. We also determined phagocytosis of bacteria by resident alveolar macrophages ex vivo and expression of MARCO and SR-A on CD45+ and CD11c+ BALF cells. Unlike elastase/LPS-exposed mice, mice exposed to elastase or to LPS cleared all bacteria from their lungs by 3 days, similar to mice exposed to PBS (data not shown). In addition, resident macrophages isolated from mice exposed to elastase or LPS alone did not show deficiency in bacterial phagocytosis (Figure 8A). Analysis of BALF cells revealed the presence of 1 log more total cells in both elastase and LPS-exposed mice, compared with mice exposed to PBS, and 0.6 to 0.7 log fewer cells than in mice exposed to elastase/LPS, as observed previously23 (Figure 8B). Mice exposed to LPS alone or elastase alone showed a decrease in number of CD11c single-positive cells, compared with mice exposed to PBS, but the number of CD11c single-positive cells was still higher than in mice exposed to the elastase/LPS combination. Similar to mice exposed to elastase/LPS, mice exposed to LPS alone or elastase alone also showed increase in number of CD11b single-positive cells and cells positive for both CD11b and CD11c, compared with PBS-exposed mice. However, unlike mice exposed to elastase/LPS, mice exposed to elastase alone or LPS alone did not show significant reduction in the expression of either MARCO or SR-A on CD45+ and CD11c+ cells, compared with mice exposed to PBS (Figure 8C). Taken together, these results suggest that exposure to elastase alone or LPS alone does not affect expression of MARCO or SR-A, nor does it affect the capacity of mice to clear bacteria. In addition, these results further support the notion that absent expression of SR-A in elastase/LPS-exposed mice may contribute to defective bacterial phagocytosis.
Figure 8.
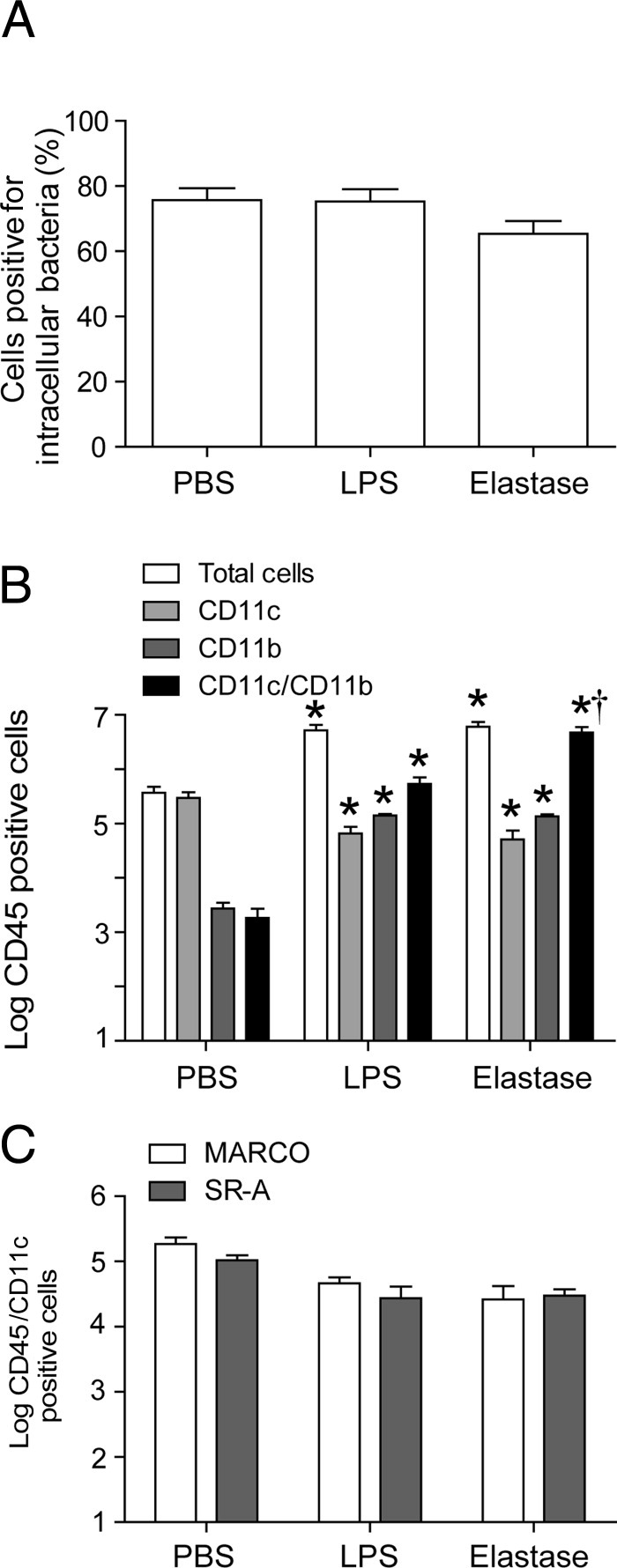
Bacterial phagocytosis and expression of MARCO and SR-A in mice exposed to elastase or LPS alone. BALF cells were isolated from mice exposed to elastase or LPS alone for four consecutive weeks. A: Resident alveolar macrophages were seeded in coverslipped chamber slides and incubated with FITC-labeled NTHi for 30 minutes; numbers of intracellular bacteria were quantified under fluorescence microscopy. B and C: BALF cells were immunostained with a combination of labeled antibodies to CD45, CD11b, CD11c, SR-A, and MARCO and were analyzed by flow cytometry. Data are presented as means ± SEM, calculated from two independent experiments (n = 3 to 6). *P ≤ 0.05 versus PBS-treated mice, unpaired t-test with Welch's correction.
Neutralization Antibody to SR-A Decreases Phagocytosis of NTHi by MH-S Alveolar Macrophages
To determine the requirement of SR-A in NTHi phagocytosis by alveolar macrophages, we used the MH-S mouse alveolar macrophage cell line, which we have shown previously to be efficient in bacterial phagocytosis.26 Initially, we examined the expression of SR-A in MH-S cells by flow cytometry and found that these cells expressed SR-A on the cell surface (Figure 9, A and B). To determine the contribution of SR-A in phagocytosis of NTHi, MH-S cells were preincubated with neutralizing antibody to SR-A or nonspecific IgG isotype control and then infected with FITC-labeled NTHi. Cells were analyzed by flow cytometry, after quenching of extracellular fluorescence. MH-S cells phagocytosed bacteria efficiently, and this was not affected by preincubation with normal IgG (the isotype control for SR-A antibody). In contrast, MH-S cells preincubated with neutralizing antibody to SR-A showed significantly fewer intracellular bacteria than the cells incubated with isotype IgG control or NTHi alone (Figure 9, C and D). These results indicate that SR-A is crucially involved in phagocytosis of NTHi by murine alveolar macrophages. Taken together, these results suggest that absent expression of SR-A in macrophages from elastase/LPS-exposed mice may be in part responsible for deficient bacterial phagocytosis, leading to the persistence of bacteria in these mice.
Figure 9.

Role of SR-A in phagocytosis of bacteria by alveolar macrophages. A and B: Immortalized murine alveolar macrophage cells of the MH-S cell line were incubated with antibody to SR-A or isotype control and were analyzed by flow cytometry to determine the expression of SR-A. A: Representative histograms show expression of SR-A. B: Quantification of SR-A. C and D: MH-S cells were preincubated with neutralizing antibody to SR-A or isotype control or medium and then were incubated with FITC-labeled NTHi for 1 hour. Extracellular fluorescence was quenched and cells were analyzed by flow cytometry to determine intracellular bacteria. C: Representative histograms show intracellular bacteria. D: Quantification of intracellular bacteria in MH-S cells preincubated with anti-SR-A or isotype control (n = 4). *P ≤ 0.05 versus PBS-treated mice; †P ≤ 0.05 versus isotype control, analysis of variance. MFI, mean fluorescence intensity.
Discussion
The principal findings of the present study demonstrate exaggerated inflammation and impaired pulmonary clearance of NTHi, a bacterial pathogen centrally implicated in COPD pathogenesis, in a murine model that exhibits typical histological and functional features of COPD, including emphysema, chronic bronchitis, airway remodeling, goblet cell metaplasia, and reduced elastic recoil.23,24 After NTHi infection, elastase/LPS-exposed mice (but not mice exposed to elastase alone or LPS alone) show delayed bacterial clearance, defective phagocytosis, decreased expression of mannose receptor and MARCO, and absent expression of SR-A on resident alveolar macrophages. Elastase/LPS-exposed mice also showed increased levels of chemokines and proinflammatory cytokines, and neutrophilic lung inflammation up to 5 days after infection. Further, we found that inhibition of SR-A by neutralizing antibody decreases phagocytosis of bacteria in MH-S mouse alveolar macrophages. Thus, these data indicate the importance of alveolar macrophage scavenger receptors, especially SR-A, in defense against NTHi. Finally, elastase/LPS-exposed mice infected with NTHi also showed progression of emphysema at 15 days after infection. Together with our previous demonstration that this model shows delayed viral clearance and increased inflammation during rhinovirus infection,23 and can be ameliorated by the antioxidant quercitin,24 these observations demonstrate important advantages of elastase/LPS-exposed mice as a practical in vivo model of established COPD.
COPD is a heterogeneous syndrome resulting from an abnormal reaction to inhaled oxidant stress. The overwhelming majority of cases in the developed world result from tobacco smoking, but only a minority of smokers develop COPD. Furthermore, fixed airflow obstruction and a phenotype indistinguishable from COPD can also develop in individuals with longstanding asthma who have never smoked,27 and population-based surveys have shown that chronic airflow limitation occurs in ∼10% of never-smokers ≥40 years old.28,29 Moreover, in the developing world, where COPD incidence is increasing even most rapidly, indoor air pollution from biomass fuels may be a more common cause than smoking. These epidemiological considerations point to the need for a variety of experimental approaches to define mechanisms relevant to COPD pathogenesis.
In this regard, we believe that our present murine model importantly complements experimental models that use cigarette smoke exposure, models that have significant limitations. Short-term exposure models (<3 weeks) predominately reveal acute and even toxic effects. Chronic models have been successful in modeling emphysema development, but even after prolonged exposures these models show minimal peribronchial inflammation and fail to develop mucus hypersecretion or goblet cell hypertrophy and airway remodeling. Moreover, the findings from rodent smoke-exposure models sometimes contradict those for the human disease. For example, maturation of lung dendritic cells was suppressed in cigarette smoke-exposed mice, resulting in diminished antigen-specific T-cell proliferation in their regional lymph nodes30; by contrast, lung dendritic cells in humans show progressive increases in their expression of maturation receptors from smokers with preserved lung function through worsening spirometrically defined COPD severity.31 Whether the elastase/LPS model will mimic this and other features of COPD in humans requires further investigation.
As an example of the utility of comparing data from several sources, the deficiency we found in alveolar macrophages from elastase/LPS-exposed mice to phagocytose bacteria both in vivo and ex vivo is in accord with and extends findings observed in alveolar macrophages isolated from COPD patients11,12,14 and from mice exposed to cigarette smoke.32 Because no portion of our in vivo model involves cigarette smoke exposure, these data imply that the alveolar macrophage phenotype can be modified by the chronically inflamed lung environment independently of any toxic effect of tobacco smoke. Thus, this murine model system appears to be well suited for investigation of the molecular mechanisms acting to sustain the pathological changes in COPD that persist after smoking cessation or that occur in never-smokers. Other significant advantages of the elastase/LPS model are the absence of a requirement for specialized exposure equipment and the relatively short duration required to produce robust structural and functional changes.
The findings that the expression by resident alveolar macrophages of the class A scavengers was decreased (MARCO) or absent (SR-A) in elastase/LPS-exposed mice infected with NTHi is significant based on the multiple actions of these receptors in lung host defense.16,17,33,34 Both SR-A and MARCO have been shown to bind Gram-negative and Gram-positive bacteria and to increase their uptake by macrophages.35–37 Collectively, our data extend those studies by demonstrating a key role for SR-A in macrophage ingestion of NTHi. Although we cannot rule out a contribution of MARCO or of mannose receptor, a C-type lectin, in decreased bacterial phagocytosis by alveolar macrophages from elastase/LPS-exposed mice, we think that the smaller magnitude of the difference in expression of these receptors makes it less likely that they are essential. Our data are in accord with accumulating evidence for reduced expression of MARCO in alveolar macrophages from COPD patients and in macrophages exposed to cigarette smoke.14,38 Similarly, the slight increase in expression of SR-B (CD36) in alveolar macrophages of elastase/LPS-exposed mice parallels findings in alveolar macrophages isolated from COPD patients.14 Importantly, both SR-A and MARCO have also been shown to interact with LPS, lipoteichoic acid, and other bacterial cell-surface proteins.35,39,40 Not only do interactions of SR-A with bacterial products or bacteria fail to activate macrophages,35 but they actually dampen their proinflammatory response.41,42 Thus, together with the role we previously showed for this molecule in optimal efferocytosis,22 the present results suggest the potential benefit of therapies to increase alveolar macrophage expression of class A scavenger receptors in COPD patients. However, we are unaware of any published report of SR-A protein expression in alveolar macrophages in COPD, an important future research direction.
The increased goblet cell metaplasia and expression of Muc5AC in both large and peripheral airways in elastase/LPS-exposed mice at 5 days after infection, relative to infected PBS-exposed mice, is noteworthy, given the clinical importance of the chronic bronchitic phenotype. Chronic mucus hypersecretion in COPD patients has been associated with impaired quality of life, accelerated loss of lung function, greater health care utilization, and increased mortality, relative to those without chronic sputum production.43–45 These changes in our murine model may be the direct effect of NTHi-induced TLR2 signaling in bronchial epithelial cells,46 an indirect effect of cytokines such as TNF-α and IL-13 secreted by macrophages in response to infection, or both. Although the TNF-α response of elastase/LPS-exposed mice to NTHi infection was lower at 1 day after infection, it persisted up to 5 days after challenge (see Supplemental Figure S1 at http://ajp.amjpathol.org). IL-13 was also marginally increased in NTHi-infected elastase/LPS-exposed mice at 3 and 5 days after infection. NTHi is particularly associated with risk of exacerbation and chronic sputum production,47 which provides opportunity for considerable additional investigation in this and other model systems.
IL-13 and TNF-α not only increase mucin secretion, but also increase airway responsiveness to methacholine challenge. Previously, we have shown that TNF-α expressed in response to rhinovirus infection increases airway hyperresponsiveness and that blocking neutrophil-elaborated TNF-α partially inhibits rhinovirus-induced airway hyperresponsiveness.48 TNF-α also synergizes with IL-13 to increase airway smooth muscle response to cholinergic compounds.49 It is therefore conceivable that increased IL-13 and TNF-α may be in part responsible for the observed increase in airway hyperresponsiveness to methacholine challenge in NTHi-infected elastase/LPS-exposed mice.
Acute exacerbations in COPD patients are often associated with impairment of respiratory function for prolonged periods of time and sometimes with progression of lung disease and increased mortality.50 These prolonged changes may be due to persistence of inflammatory changes caused by the infecting organism. Consistent with this notion, although elastase/LPS-exposed mice cleared bacteria and partially resolved inflammation by 15 days after NTHi infection, they showed further loss of elastic recoil, indicating progression of emphysema. This is a significant finding not previously shown in an animal model of COPD.
In contrast to our present findings, and to previous reports demonstrating impaired clearance of bacteria in cigarette smoke-exposed mice after bacterial challenge,32,51 Gaschler et al52 found that mice exposed to cigarette smoke for a short duration cleared NTHi more rapidly after challenge, compared with room air-exposed mice. This result was attributed to increased titers of NTHi-specific IgA.53 One explanation for the observed discrepancy may be that mice exposed to cigarette smoke for short durations do not up-regulate MMP-12, as is seen in the elastase/LPS-exposed mice,24 in mice with long-term exposure to cigarette smoke (6 months),54,55 and in sputum of COPD patients.56 Such increases in MMP-12 in the airways can degrade IgA, which is involved in NTHi clearance.53 Whether reduced airway IgA resulting from the increased expression and activity of MMP-12 that we previously observed in elastase/LPS-exposed mice also contributes to the impaired bacterial clearance reported here is beyond the scope of the present study.
In conclusion, we have shown that during NTHi pulmonary infection elastase/LPS-exposed mice, which develop typical features of COPD within 4 weeks, clear the organism more slowly and show sustained lung inflammation and increased mucus production and progression of emphysema. Lack of SR-A expression in alveolar macrophages may contribute to the observed defective clearance of bacteria in these mice. These findings provide a murine model that complements smoke-exposure models to investigate features common in the COPD phenotype.
Acknowledgment
We thank Marisa Linn for her help with histology.
Footnotes
Supported by an NIH grant (AT004793 to U.S.S.) and by a Research Enhancement Award from the Biomedical Laboratory Research & Development Service, Department of Veterans Affairs (J.L.C.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.09.029.
Supplementary data
Cytokine levels in lungs. Sham-infected and NTHi-infected mice were sacrificed at 1, 3, or 5 days after infection. Cytokine levels in the lung homogenates were determined by bioplex enzyme-linked immunosorbent assay (n = 5). *P ≤ 0.05 versus PBS-exposed mice, analysis of variance; †P ≤ 0.05 versus the sham-infected mice, analysis of variance.
Measurement of airway responsiveness to methacholine challenge. Mice were anesthetized and endotracheally intubated; changes in respiratory system resistance to nebulized methacholine were measured using a FlexiVent system. Data are reported as means ± SEM (n = 3). *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance.
Real-time phagocytosis of bacteria by alveolar macrophages. BALF cells collected from PBS-exposed mice were plated onto coverslipped two-chamber slides, infected with Alexa Fluor 594-labeled NTHi, and monitored under time-lapse video microscopy. The video clip is representative of at least 10 alveolar macrophages recorded.
Real-time phagocytosis of bacteria by alveolar macrophages. BALF cells collected from elastase/LPS-exposed mice were plated onto coverslipped two-chamber slides, infected with Alexa Fluor 594-labeled NTHi, and monitored under time-lapse video microscopy. The video clip is representative of at least 10 alveolar macrophages recorded.
References
- 1.Miniño A.M., Xu J., Kochanek K.D. Deaths: Preliminary Data for 2008. Natl Vital Stat Rep. 2010;59(2):1–52. [PubMed] [Google Scholar]
- 2.Tuder R.M., Yoshida T., Arap W., Pasqualini R., Petrache I. State of the art: Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc. 2006;3:503–510. doi: 10.1513/pats.200603-054MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wedzicha J.A. Mechanisms of exacerbations. Novartis Found Symp. 2001;234:84–93. doi: 10.1002/0470868678.ch6. discussion 93–103. [DOI] [PubMed] [Google Scholar]
- 4.Wedzicha J.A. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:115–120. doi: 10.1513/pats.2306030. [DOI] [PubMed] [Google Scholar]
- 5.Sethi S., Evans N., Grant B.J., Murphy T.F. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 6.Sapey E., Stockley R.A. COPD exacerbations. 2: aetiology. Thorax. 2006;61:250–258. doi: 10.1136/thx.2005.041822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsen M.V., Janner J.H., Nielsen S.D., Friis-Møller A., Ringbaek T., Lange P. Bacteriology in acute exacerbation of chronic obstructive pulmonary disease in patients admitted to hospital. Scand J Infect Dis. 2009;41:26–32. doi: 10.1080/00365540802484828. [DOI] [PubMed] [Google Scholar]
- 8.Hill A.T., Campbell E.J., Hill S.L., Bayley D.L., Stockley R.A. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000;109:288–295. doi: 10.1016/s0002-9343(00)00507-6. [DOI] [PubMed] [Google Scholar]
- 9.Soler N., Ewig S., Torres A., Filella X., Gonzalez J., Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J. 1999;14:1015–1022. doi: 10.1183/09031936.99.14510159. [DOI] [PubMed] [Google Scholar]
- 10.Murphy T.F., Brauer A.L., Schiffmacher A.T., Sethi S. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:266–272. doi: 10.1164/rccm.200403-354OC. [DOI] [PubMed] [Google Scholar]
- 11.Berenson C.S., Garlipp M.A., Grove L.J., Maloney J., Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194:1375–1384. doi: 10.1086/508428. [DOI] [PubMed] [Google Scholar]
- 12.Taylor A.E., Finney-Hayward T.K., Quint J.K., Thomas C.M., Tudhope S.J., Wedzicha J.A., Barnes P.J., Donnelly L.E. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35:1039–1047. doi: 10.1183/09031936.00036709. [DOI] [PubMed] [Google Scholar]
- 13.Hodge S., Hodge G., Scicchitano R., Reynolds P.N., Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–296. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [Erratum appeared in Immunol Cell Biol 2003, 81:499] [DOI] [PubMed] [Google Scholar]
- 14.Hodge S., Hodge G., Ahern J., Jersmann H., Holmes M., Reynolds P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37:748–755. doi: 10.1165/rcmb.2007-0025OC. [DOI] [PubMed] [Google Scholar]
- 15.Henson P.M., Cosgrove G.P., Vandivier R.W. State of the art: Apoptosis and cell homeostasis in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:512–516. doi: 10.1513/pats.200603-072MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arredouani M.S., Yang Z., Imrich A., Ning Y., Qin G., Kobzik L. The macrophage scavenger receptor SR-AI/II and lung defense against pneumococci and particles. Am J Respir Cell Mol Biol. 2006;35:474–478. doi: 10.1165/rcmb.2006-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arredouani M., Yang Z., Ning Y., Qin G., Soininen R., Tryggvason K., Kobzik L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med. 2004;200:267–272. doi: 10.1084/jem.20040731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl M., Bauer A.K., Arredouani M., Soininen R., Tryggvason K., Kleeberger S.R., Kobzik L. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest. 2007;117:757–764. doi: 10.1172/JCI29968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hersh C.P., DeMeo D.L., Raby B.A., Litonjua A.A., Sylvia J.S., Sparrow D., Reilly J.J., Silverman E.K. Genetic linkage and association analysis of COPD-related traits on chromosome 8p. COPD. 2006;3:189–194. doi: 10.1080/15412550601009321. [DOI] [PubMed] [Google Scholar]
- 20.Ohar J.A., Hamilton R.F., Jr, Zheng S., Sadeghnejad A., Sterling D.A., Xu J., Meyers D.A., Bleecker E.R., Holian A. COPD is associated with a macrophage scavenger receptor-1 gene sequence variation. Chest. 2010;137:1098–1107. doi: 10.1378/chest.09-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomsen M., Nordestgaard B.G., Tybjaerg-Hansen A., Dahl M. Scavenger receptor AI/II truncation, lung function and COPD: a large population-based study. J Intern Med. 2011;269:340–348. doi: 10.1111/j.1365-2796.2010.02308.x. [DOI] [PubMed] [Google Scholar]
- 22.Todt J.C., Hu B., Curtis J.L. The scavenger receptor SR-A I/II (CD204) signals via the receptor tyrosine kinase Mertk during apoptotic cell uptake by murine macrophages. J Leukoc Biol. 2008;84:510–518. doi: 10.1189/jlb.0307135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sajjan U., Ganesan S., Comstock A.T., Shim J., Wang Q., Nagarkar D.R., Zhao Y., Goldsmith A.M., Sonstein J., Linn M.J., Curtis J.L., Hershenson M.B. Elastase- and LPS-exposed mice display altered responses to rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2009;297:L931–L944. doi: 10.1152/ajplung.00150.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganesan S., Faris A.N., Comstock A.T., Chattoraj S.S., Chattoraj A., Burgess J.R., Curtis J.L., Martinez F.J., Zick S., Hershenson M.B., Sajjan U.S. Quercetin prevents progression of disease in elastase/LPS-exposed mice by negatively regulating MMP expression. Respir Res. 2010;11:131. doi: 10.1186/1465-9921-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sajjan U., Wang Q., Zhao Y., Gruenert D.C., Hershenson M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178:1271–1281. doi: 10.1164/rccm.200801-136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chattoraj S.S., Murthy R., Ganesan S., Goldberg J.B., Zhao Y., Hershenson M.B., Sajjan U.S. Pseudomonas aeruginosa alginate promotes Burkholderia cenocepacia persistence in cystic fibrosis transmembrane conductance regulator knockout mice. Infect Immun. 2010;78:984–993. doi: 10.1128/IAI.01192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange P., Parner J., Vestbo J., Schnohr P., Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med. 1998;339:1194–1200. doi: 10.1056/NEJM199810223391703. [DOI] [PubMed] [Google Scholar]
- 28.Menezes A.M., Perez-Padilla R., Jardim J.R., Muiño A., Lopez M.V., Valdivia G., Montes de Oca M., Talamo C., Hallal P.C., Victora C.G., PLATINO Team Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence study. Lancet. 2005;366:1875–1881. doi: 10.1016/S0140-6736(05)67632-5. [DOI] [PubMed] [Google Scholar]
- 29.Mannino D.M., Homa D.M., Akinbami L.J., Ford E.S., Redd S.C. Chronic obstructive pulmonary disease surveillance–United States, 1971–2000. MMWR Surveill Summ. 2002;51(6):1–16. [PubMed] [Google Scholar]
- 30.Robbins C.S., Franco F., Mouded M., Cernadas M., Shapiro S.D. Cigarette smoke exposure impairs dendritic cell maturation and T cell proliferation in thoracic lymph nodes of mice. J Immunol. 2008;180:6623–6628. doi: 10.4049/jimmunol.180.10.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman C.M., Martinez F.J., Han M.K., Ames T.M., Chensue S.W., Todt J.C., Arenberg D.A., Meldrum C.A., Getty C., McCloskey L., Curtis J.L. Lung dendritic cell expression of maturation molecules increases with worsening chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180:1179–1188. doi: 10.1164/rccm.200904-0552OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phipps J.C., Aronoff D.M., Curtis J.L., Goel D., O'Brien E., Mancuso P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun. 2010;78:1214–1220. doi: 10.1128/IAI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Areschoug T., Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. 2009;11:1160–1169. doi: 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- 34.Areschoug T., Waldemarsson J., Gordon S. Evasion of macrophage scavenger receptor A-mediated recognition by pathogenic Streptococci. Eur J Immunol. 2008;38:3068–3079. doi: 10.1002/eji.200838457. [DOI] [PubMed] [Google Scholar]
- 35.Hampton R.Y., Golenbock D.T., Penman M., Krieger M., Raetz C.R. Recognition and plasma clearance of endotoxin by scavenger receptors. Nature. 1991;352:342–344. doi: 10.1038/352342a0. [DOI] [PubMed] [Google Scholar]
- 36.Elomaa O., Kangas M., Sahlberg C., Tuukkanen J., Sormunen R., Liakka A., Thesleff I., Kraal G., Tryggvason K. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell. 1995;80:603–609. doi: 10.1016/0092-8674(95)90514-6. [DOI] [PubMed] [Google Scholar]
- 37.Sankala M., Brännstrm̈ A., Schulthess T., Bergmann U., Morgunova E., Engel J., Tryggvason K., Pikkarainen T. Characterization of recombinant soluble macrophage scavenger receptor MARCO. J Biol Chem. 2002;277:33378–33385. doi: 10.1074/jbc.M204494200. [DOI] [PubMed] [Google Scholar]
- 38.Baqir M., Chen C.Z., Martin R.J., Thaikoottathil J., Case S.R., Minor M.N., Bowler R., Chu H.W. Cigarette smoke decreases MARCO expression in macrophages: implication in Mycoplasma pneumoniae infection. Respir Med. 2008;102:1604–1610. doi: 10.1016/j.rmed.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Plüddemann A., Hoe J.C., Makepeace K., Moxon E.R., Gordon S. The macrophage scavenger receptor A is host-protective in experimental meningococcal septicaemia. PLoS Pathog. 2009;5:e1000297. doi: 10.1371/journal.ppat.1000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peiser L., Makepeace K., Plüddemann A., Savino S., Wright J.C., Pizza M., Rappuoli R., Moxon E.R., Gordon S. Identification of Neisseria meningitidis nonlipopolysaccharide ligands for class A macrophage scavenger receptor by using a novel assay. Infect Immun. 2006;74:5191–5199. doi: 10.1128/IAI.00124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozeki Y., Tsutsui H., Kawada N., Suzuki H., Kataoka M., Kodama T., Yano I., Kaneda K., Kobayashi K. Macrophage scavenger receptor down-regulates mycobacterial cord factor-induced proinflammatory cytokine production by alveolar and hepatic macrophages. Microb Pathog. 2006;40:171–176. doi: 10.1016/j.micpath.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Haworth R., Platt N., Keshav S., Hughes D., Darley E., Suzuki H., Kurihara Y., Kodama T., Gordon S. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J Exp Med. 1997;186:1431–1439. doi: 10.1084/jem.186.9.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vestbo J., Prescott E., Lange P., Copenhagen City Heart Study Group Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Am J Respir Crit Care Med. 1996;153:1530–1535. doi: 10.1164/ajrccm.153.5.8630597. [DOI] [PubMed] [Google Scholar]
- 44.Pelkonen M., Notkola I.L., Nissinen A., Tukiainen H., Koskela H. Thirty-year cumulative incidence of chronic bronchitis and COPD in relation to 30-year pulmonary function and 40-year mortality: a follow-up in middle-aged rural men. Chest. 2006;130:1129–1137. doi: 10.1378/chest.130.4.1129. [DOI] [PubMed] [Google Scholar]
- 45.Burgel P.R., Nesme-Meyer P., Chanez P., Caillaud D., Carré P., Perez T., Roche N., Initiatives Bronchopneumopathie Chronique Obstructive Scientific Committee Cough and sputum production are associated with frequent exacerbations and hospitalizations in COPD subjects. Chest. 2009;135:975–982. doi: 10.1378/chest.08-2062. [DOI] [PubMed] [Google Scholar]
- 46.Chen R., Lim J.H., Jono H., Gu X.X., Kim Y.S., Basbaum C.B., Murphy T.F., Li J.D. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2004;324:1087–1094. doi: 10.1016/j.bbrc.2004.09.157. [DOI] [PubMed] [Google Scholar]
- 47.Eldika N., Sethi S. Role of nontypeable Haemophilus influenzae in exacerbations and progression of chronic obstructive pulmonary disease. Curr Opin Pulmon Med. 2006;12:118–124. doi: 10.1097/01.mcp.0000208451.50231.8f. [DOI] [PubMed] [Google Scholar]
- 48.Nagarkar D.R., Wang Q., Shim J., Zhao Y., Tsai W.C., Lukacs N.W., Sajjan U., Hershenson M.B. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human rhinovirus infection. J Immunol. 2009;183:6698–6707. doi: 10.4049/jimmunol.0900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deshpande D.A., Dogan S., Walseth T.F., Miller S.M., Amrani Y., Panettieri R.A., Kannan M.S. Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: role of CD38/cyclic adenosine diphosphate ribose pathway. Am J Respir Cell Mol Biol. 2004;31:36–42. doi: 10.1165/rcmb.2003-0313OC. [DOI] [PubMed] [Google Scholar]
- 50.Anzueto A. Impact of exacerbations on COPD. Eur Respir Rev. 2010;19:113–118. doi: 10.1183/09059180.00002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drannik A.G., Pouladi M.A., Robbins C.S., Goncharova S.I., Kianpour S., Stämpfli M.R. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2004;170:1164–1171. doi: 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- 52.Gaschler G.J., Skrtic M., Zavitz C.C., Lindahl M., Onnervik P.O., Murphy T.F., Sethi S., Stampfli M.R. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med. 2009;179:666–675. doi: 10.1164/rccm.200808-1306OC. [DOI] [PubMed] [Google Scholar]
- 53.Gaschler G.J., Zavitz C.C., Bauer C.M., Stämpfli M.R. Mechanisms of clearance of nontypeable Haemophilus influenzae from cigarette smoke-exposed mouse lungs. Eur Respir J. 2010;36:1131–1142. doi: 10.1183/09031936.00113909. [DOI] [PubMed] [Google Scholar]
- 54.Hautamaki R.D., Kobayashi D.K., Senior R.M., Shapiro S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 55.Cawston T., Carrere S., Catterall J., Duggleby R., Elliott S., Shingleton B., Rowan A. Matrix metalloproteinases and TIMPs: properties and implications for the treatment of chronic obstructive pulmonary disease. Novartis Found Symp. 2001;234:205–218. doi: 10.1002/0470868678.ch13. discussion 218–228. [DOI] [PubMed] [Google Scholar]
- 56.Demedts I.K., Morel-Montero A., Lebecque S., Pacheco Y., Cataldo D., Joos G.F., Pauwels R.A., Brusselle G.G. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61:196–201. doi: 10.1136/thx.2005.042432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cytokine levels in lungs. Sham-infected and NTHi-infected mice were sacrificed at 1, 3, or 5 days after infection. Cytokine levels in the lung homogenates were determined by bioplex enzyme-linked immunosorbent assay (n = 5). *P ≤ 0.05 versus PBS-exposed mice, analysis of variance; †P ≤ 0.05 versus the sham-infected mice, analysis of variance.
Measurement of airway responsiveness to methacholine challenge. Mice were anesthetized and endotracheally intubated; changes in respiratory system resistance to nebulized methacholine were measured using a FlexiVent system. Data are reported as means ± SEM (n = 3). *P ≤ 0.05 versus PBS-treated mice, analysis of variance; †P ≤ 0.05 versus sham-infected mice, analysis of variance.
Real-time phagocytosis of bacteria by alveolar macrophages. BALF cells collected from PBS-exposed mice were plated onto coverslipped two-chamber slides, infected with Alexa Fluor 594-labeled NTHi, and monitored under time-lapse video microscopy. The video clip is representative of at least 10 alveolar macrophages recorded.
Real-time phagocytosis of bacteria by alveolar macrophages. BALF cells collected from elastase/LPS-exposed mice were plated onto coverslipped two-chamber slides, infected with Alexa Fluor 594-labeled NTHi, and monitored under time-lapse video microscopy. The video clip is representative of at least 10 alveolar macrophages recorded.