

During monocyte–macrophage differentiation, C/EBPα transcriptionally activates QKI, which in turn represses CSF1R and thus provides negative feedback to C/EBPα-induced macrophage differentiation. This feedback loop should be important in keeping the balance between cell proliferation and differentiation.
Abstract
Differentiated macrophages are essential for the innate immune system; however, the molecular mechanisms underlying the generation of macrophages remain largely unknown. Here we show that the RNA-binding protein QKI, mainly QKI-5, is transcriptionally activated in the early differentiated monocytic progenitors when CCAAT/enhancer-binding protein (C/EBP) α is expressed. The forced expression of C/EBPα increases the endogenous expression of QKI. Chromatin immunoprecipitation analysis and reporter assays further confirm that C/EBPα activates the transcription of QKI, primarily by binding to the distal C/EBPα-binding site. Blocking the induction of QKI using RNA interference enhances the expression of endogenous CSF1R and facilitates macrophage differentiation. Further study of the mechanism reveals that QKI-5 facilitates the degradation of CSF1R mRNA by interacting with the distal QRE in the 3′ untranslated region. In summary, we show that in committed macrophage progenitors, C/EBPα-activated QKI-5 negatively regulates macrophage differentiation by down-regulating CSF1R expression, forming a negative feedback loop during macrophage differentiation.
INTRODUCTION
Cell lineage specification of hematopoietic progenitors is essential for innate immunity, which is controlled by a complex system composed of interacting transcription factors, microRNAs (miRNAs), and RNA-binding proteins. However, the manner in which these regulators cooperate to control cell differentiation has been poorly understood. In the last two decades, accumulating evidence suggests that lineage-specific transcription factors such as GATA1, CCAAT/enhancer-binding protein (C/EBP) α, and PU.1 dominate in the cell specification process (Clarke and Gordon, 1998). Among them, C/EBPα is well known to be essential for monocyte–macrophage differentiation through transcriptional activation of CSF1R (Stanley, 1986; Zhang et al., 1996; Dahl et al., 2003). CSF1R (also called M-CSFR) is an integral membrane tyrosine kinase encoded by the c-fms proto-oncogene. CSF1R, which is expressed in monocytes/macrophages and their progenitors, is obligatory for macrophage differentiation (Bourette and Rohrschneider, 2000; Pixley and Stanley, 2004). Recently numerous data indicate that C/EBPα is already expressed in hematopoietic stem cells, although at a low level (Hu et al., 1997; Akashi et al., 2003), establishing transcriptional priming. However, the role of C/EBPα in the progenitors remains largely unknown. In particular, how CSF1R is regulated at this stage needs to be clarified.
In recent years, a new network of regulatory circuits that function using miRNAs and RNA-binding proteins at the posttranscriptional level has been discovered. This network is believed to be involved in the regulation of a wide variety of fundamental cellular processes, including cell growth, differentiation, and apoptosis. Multiple posttranscriptional regulators, such as miR-122 and miR-34a, are found to be transcribed by C/EBPα in different cells, adding complexity to the C/EBPα regulatory network (Pulikkan et al., 2010; Zeng et al., 2010) and suggesting that posttranscriptional regulators are involved in the C/EBPα network.
The RNA-binding protein QKI belongs to the evolutionarily conserved signal transduction and activator of RNA (STAR) family and is implicated in embryogenesis and CNS development. The QKI (quaking, Qk) locus encodes a diverse set of proteins created by alternative splicing, and the three well-studied QKI proteins QKI-5, QKI-6, and QKI-7 are constructed with the same 311–amino acid body but with different carboxyl tails (Wu et al., 1999). QKI regulates mRNA stability, nuclear retention, RNA transportation, and translational modulation and functions by dimerizing and binding to QREs located in the untranslated region (UTR) of target mRNAs (Larocque and Richard, 2005). Multiple genes, such as MAG, p53, p27, MBP, and CTNNB1, have been validated as QKI targets (Wu et al., 2002; Larocque et al., 2002; 2005; Schumacher et al., 2005; Yang et al., 2010). The expression of QKI isoforms is developmentally regulated, with QKI5 highly expressed throughout embryogenesis and the neonatal stages and decreasing gradually thereafter (Ebersole et al., 1996). Phylogenetic conservation of QKI proteins and their widespread expression in different cell types (Kondo et al., 1999; Noveroske et al., 2002) underscore the central importance of this gene in the regulation of normal cellular functions. Of interest, QKI is also expressed in the yolk sac endoderm, adjacent to the mesodermal site of developing blood islands, where early hematopoietic and endothelial cells originate. Galarneau and Richard (2005) defined the QKI response element (QRE) as a bipartite consensus sequence NACUAAY-N(1-20)-UAAY and predicted nearly 1430 new putative mRNA targets. CSF1R is included among them, suggesting a potential role of QKI in monocytic differentiation.
In this study, we characterized the expression of QKI, its related targets, and their interactions using two different in vitro macrophage differentiation models. Our data demonstrate for the first time that the transcription of the RNA-binding protein QKI is activated by C/EBPα in early differentiated hematopoietic progenitors. In turn, once transcribed, QKI inhibits the expression of CSF1R and thus delays the terminal differentiation process of hematopoietic progenitor cells toward a macrophage fate. Together these data imply C/EBPα-QKI5-CSF1R negative feedback interaction during macrophage differentiation.
RESULTS
Dynamic expression of QKI during monocyte–macrophage differentiation
To explore the potential role of QKI in the regulation of macrophage differentiation, we first examined the endogenous expression of QKI using Western blot analysis and isoform-specific quantitative real-time (qRT)-PCR during the monocytic differentiation of hematopoietic progenitor cells. The CD34+ subgroup cells, which represent the progenitor population (Li et al., 2011), were enriched and induced toward the macrophage fate in serum-free media supplemented with 1 ng/ml interleukin-6 (IL-6), 100 ng/ml Flt3-ligand, and 50 ng/ml macrophage colony-stimulating factor (M-CSF). As shown in Figure 1A, QKI expression increased at the early stage of differentiation and then declined after 8 d of differentiation, at approximately the same stage that CD14 expression (a widely used marker of differentiation) increased. These data suggest a potential role for QKI in the regulation of monocyte differentiation.
FIGURE 1:

Dynamic expression of QKI during monocyte–macrophage differentiation. (A) The CD34+ cells were induced toward macrophage differentiation, and QKI expression was analyzed by Western blot analysis. CD14 served as an indicator of macrophage differentiation, and tubulin served as an internal control for equal loading. (B) The HL-60 cells grew for the indicated times in the presence of 32 nM of TPA. The expression levels of QKI, CSF1R, C/EBPα, and the differentiation marker CD11b were analyzed by Western blot. (C) The cells were treated in the same manner as described, and the RNA expression of the three QKI isoforms was analyzed by qRT-PCR. GAPDH served as an internal control. The dynamic changes of QKI5 (D), QKI6 (E), and QKI7 (F) during differentiation were calculated by comparing the levels with the ethanol control.
A similar dynamic expression of QKI was also observed during the monocytic differentiation of HL-60 cells. Tetradecanoylphorbol 13-acetate (TPA), also known as phorbol-12-myristate-13-acetate, is a well-known macrophage differentiation inducer of HL-60 cells (Zheng et al., 2002). On TPA induction, the macrophages differentiated, and QKI expression increased at an earlier time point and then declined gradually (Figure 1B). At the terminal differentiation stage, QKI was greatly down-regulated (Figure 1B). The data from the HL-60 cells suggest that this cell line represented the data from the primary culture well. Of note, there was only one detectable band for QKI in these cells. Previously, QKI was found to have at least three isoforms—QKI-5, QKI-6, and QKI-7. To test whether the band represents QKI-5, QKI-6, or QKI-7, the endogenous QKI expression bands were compared with exogenously expressed QKI-5 and QKI-6. As shown in Supplemental Figure S1, the endogenous QKI band was the same size as the exogenously expressed QKI-5. To further confirm that QKI-5 is the main isoform expressed in the hematopoietic progenitors and the differentiated cells, a QKI isoform-specific qRT-PCR was performed. The qRT-PCR analysis revealed that QKI-5 was the dominant isoform that is expressed in these cells, whereas the QKI-6 and QKI-7 expression levels were much lower (Figure 1C). In addition, the expression levels of QKI-6/7 did not change as significantly as that of QKI-5 during differentiation (Figure 1, D–F); however, there was a dynamic change of QKI-6 expression similar to that of QKI-5. For this study, we focused on the role of QKI-5 in monocyte differentiation.
C/EBPα transcriptionally activates the expression of QKI during early monocyte–macrophage differentiation
To determine how QKI is regulated during macrophage differentiation, we performed a bioinformatics study, which revealed that there are multiple C/EBPα-binding sites located in the QKI promoter region (Figure 2A), suggesting potential transcriptional regulation of QKI by C/EBPα. Accordingly, the QKI expression levels showed a similar profile when compared with the expression of C/EBPα during monocyte–macrophage differentiation (Figure 1, A and B). To further test whether C/EBPα can increase the endogenous expression of QKI, C/EBPα was overexpressed in HEK293 cells. As expected, overexpression of C/EBPα increased the endogenous QKI expression both at RNA and protein levels (Figure 2, B and C), further suggesting that C/EBPα activates the transcription of QKI.
FIGURE 2:
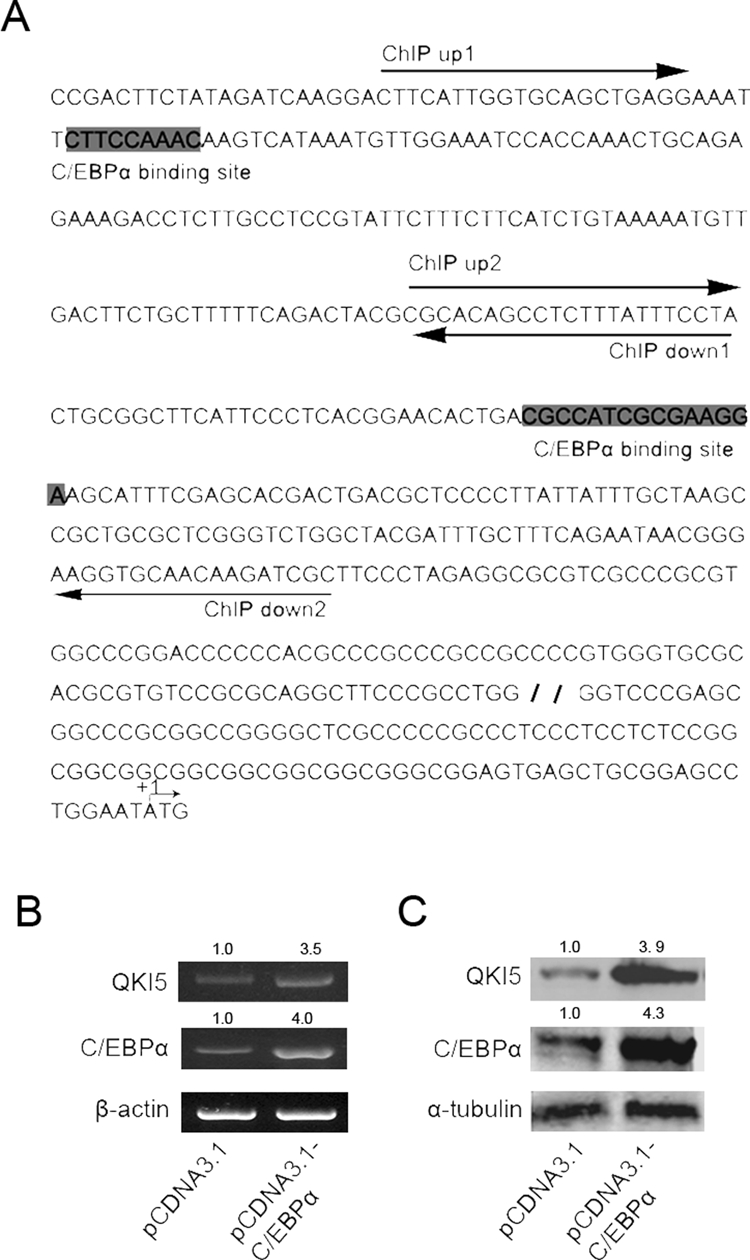
C/EBPα transcriptionally activates the expression of QKI during early monocyte–macrophage differentiation. (A) A bioinformatics study reveals two putative binding sites for C/EBPα, as indicated by the gray background. The sequences of the primers used for the ChIP assay are underlined with arrows showing the direction. (B) The HEK293 cells were transfected with pcDNA3.1-C/EBPα or pcDNA3.1, and QKI expression was analyzed by RT-PCR 48 h after transfection. (C) The cells were treated in the same manner as described, and QKI expression was analyzed by Western blot.
C/EBPα directly activates QKI transcription
On the basis of our data, it is reasonable to deduce that QKI might be directly activated by C/EBPα. To test this interaction, a series of QKI promoter reporter vectors with or without the putative C/EBPα-binding sites and promoters containing mutated C/EBPα-binding sites was constructed. As shown in Figure 3A, C/EBPα overexpression increased the wild-type promoter activity. However, with the proximal C/EBPα-binding site deleted or mutated, the basal promoter activity only weakly responded to the overexpression of C/EBPα. To further confirm the interaction between the QKI promoter and C/EBPα, chromatin immunoprecipitation (ChIP) analysis was performed in control and TPA-treated HL-60 cells. As shown in Figure 3B, after 12 h of TPA treatment, the fragment containing the distal C/EBPα-binding site was significantly enriched in the QKI immunoprecipitated complex, whereas the fragment covering the proximal binding site was less enriched (unpublished data). In contrast, in the ethanol-treated cells or the late-differentiated cells (48 h of TPA treatment) when C/EBPα expression was low, there was a weak enrichment of the QKI promoter by C/EBPα (Figure 3B), further confirming the interaction between the QKI promoter and C/EBPα.
FIGURE 3:

Direct transcriptional activation of QKI by C/EBPα. (A) C/EBPα transcriptionally activates the QKI promoter. In HEK 293 cells, 500 ng of the indicated QKI promoter reporter vectors or the control pGL3 basic plasmids and 50 ng of the internal control pRL-TK were cotransfected for 24 h before luciferase activity was examined. The activity of the luciferase reporter plasmid was normalized against the value of the internal standard pRL-TK. The results were expressed as the means ± SD of at least three different experiments. (B) The in vivo interaction between C/EBPα and the QKI promoter. The HL-60 cells were treated with ethanol or TPA for the indicated time before cross-linking. After DNA/protein cross-linking, the chromatin was immunoprecipitated overnight with 10 μl of anti–C/EBPα antibody. PCR amplifications of the interested genomic regions covering the putative C/EBPα-binding site were performed. The enrichment of the C/EBPα-bound promoter fragment was calculated by normalizing to IgG-immunoprecipitated DNA.
QKI functions as an inhibitor of monocyte–macrophage differentiation
From the foregoing data, we speculate that QKI, specifically QKI-5, might be involved in the fine tuning of macrophage differentiation. To this end, we knocked down QKI in the progenitor cells undergoing differentiation. A stable and efficient knockdown efficiency was confirmed by Western blot (Figure 4A). As shown in Figure 4B, the adhesion of the HL-60 cells induced by TPA occurred much earlier and more obviously in the QKI RNA interference group than in the control group. In addition, when compared with the control, the morphology of the small interference RNA-mediated knockdown of the endogenous QKI (siQKI) cells was more similar to that of monocytes–macrophages. Fluorescence-activated cell sorting (FACS) analysis using the CD14 marker further confirmed that knockdown of QKI facilitated TPA-induced monocyte–macrophage differentiation, whereas QKI had no obvious effects on macrophage differentiation in cells without TPA treatment (Figure 4C).
FIGURE 4:

Knockdown of QKI-facilitated monocyte–macrophage differentiation. HL-60 cells were transfected with 100 nM siRNA against QKI or the NC siRNA and then were induced by 32 nM TPA or ethanol for the indicated time. (A) The knockdown efficiency of QKI during the different periods. (B) The cell morphology was viewed and imaged using a microscope. The siQKI-treated cells displayed a monocyte-macrophage–like appearance much earlier than the control. (C) The CD14+ population was analyzed by FACS. The data presented are representative of three different experiments.
QKI negatively regulates the expression of CSF1R
As an RNA-binding protein, it is highly possible that QKI delays the monocytic differentiation by targeting its downstream mRNAs. CSF1R, an arbiter of macrophage differentiation (Bourette and Rohrschneider, 2000; Pixley and Stanley, 2004), was a potential target of QKI (Figure 5A; Galarneau and Richard, 2005). Bioinformatics analysis and a literature review revealed that CSF1R mRNA might be regulated by miRNAs and the RNA-binding protein HuR (Lewis et al., 2005; Woo et al., 2009), further suggesting that posttranscriptional regulation of CSF1R is of significant importance. To address the regulation of CSF1R, we investigated whether QKI could affect the expression of CSF1R. As expected, siQKI increased the endogenous expression of CSF1R at both mRNA and protein levels in HL-60 cells with 12 h of TPA treatment (Figure 5B), further suggesting that CSF1R is regulated by QKI. In contrast, forced expression of QKI5 in the SKBR3 cells decreased the endogenous expression of CSF1R (Figure 5C). Moreover, expression of QKI in SKBR3 cells decreased the half-life of CSF1R mRNA, suggesting that QKI decreased the expression of CSF1R by destabilizing the CSF1R mRNA (Figure 5D, 5E). Of note, the fold change of CSF1R at the protein level (3.1-fold) was larger than the change at the mRNA level (2.1-fold) under QKI5 knockdown, suggesting that QKI5 might also repress protein expression and destabilize the RNA.
FIGURE 5:

QKI negatively regulates the expression of CSF1R. (A) A schematic representation of the potential posttranscriptional regulation of CSF1R by QKI. The CSF1R mRNA was analyzed by searching a sequence matrix. Potential QREs were indicated by the corresponding binding position in the brackets. (B) HL-60 cells transfected with 100 nM siQKI or the control were cultured in the presence of 32 nM TPA for an additional 12 h. The QKI and CSF1R mRNA abundance and protein expression levels were analyzed by PCR and Western blot analysis. β-Actin and α-tubulin served as internal controls. (C) SKBR3 cells were transfected with either QKI5 or the control. The expression of QKI and CSF1R was analyzed both at mRNA and protein levels by RT-PCR and Western blot 48 h after transfection. (D) QKI reduced the mRNA stability of CSF1R. The cells were treated in the same manner as described. Forty-eight hours after transfection, the cells were treated with actinomycin D for the indicated time, and the mRNA abundances of CSF1R, GAPDH, and β-actin were analyzed by qRT-PCR. (E) The quantification data from D.
The QREs located in the 3′ UTR of CSF1R are highly conserved among multiple species, suggesting a conserved regulation of CSF1R by QKI (Figure 6A). To determine whether CSF1R is regulated by QKI, we constructed serial reporter vectors with wild-type or mutant QREs or without the QREs (Figure 6B). Overexpression of QKI5 decreased the activity of the full-length CSF1R 3′UTR while only weakly changing the activities of the reporters without the distal QRE or with the QRE mutated, suggesting that the distal QRE might be the true responsive QRE (Figure 6B). To further test the possibility of a direct interaction between CSF1R and QKI, we applied an RNA-IP assay in FLAG-QKI5– or FLAG-only–expressing cells (Figure 6C). As expected, the endogenous CSF1R mRNA was pulled down by FLAG-QKI, whereas the unrelated glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was not enriched in the IP complex (Figure 6D). To further confirm whether the QKI5-CSF1R mRNA interaction was dependent on the distal QRE, a CSF1R 3′UTR reporter either with wild-type or mutant QREs was cotransfected with FLAG-only or FLAG-QKI5 into cells. FLAG-QKI5 was found to interact only with the luciferase mRNA harboring the wild-type QRE (Figure 6E).
FIGURE 6:

CSF1R is down-regulated by QKI5 during monocyte–macrophage differentiation. (A) A bioinformatics study revealed that the two QREs in the 3′UTR of CSF1R are conserved across species. (B) Reporter vectors with wild-type or mutant/deleted QREs were constructed. Two hundred nanograms of each indicated CSF1R 3′UTR reporter, or 50 ng of the internal control vector pRL-TK, were cotransfected with the QKI expression vector or the control for 24 h. The fold induction upon QKI expression was calculated and expressed as the means ± SD (n = 3). *p < 0.05. (C) The in vivo interaction between CSF1R and QKI5. pcDNA3.1-3×FLAG-QKI5–transfected SKBR3 cells were immunoprecipitated with an anti-FLAG antibody or the negative control IgG. The immunoprecipitation efficiency is shown. (D) The presence of CSF1R and GAPDH mRNAs in the immunoprecipitation is detected by RT-PCR and visualized by ethidium bromide staining. CSF1R, rather than GAPDH mRNA, was enriched in the pull-down complex. (E) The CSF1R 3′UTR reporter with either wild-type or mutant QRE was cotransfected with FLAG or FLAG-QKI5 into cells. The luciferase and GAPDH mRNAs in the immunoprecipitation were detected by RT-PCR and visualized by ethidium bromide staining. The FLAG-QKI5 was found to interact only with the luciferase mRNA harboring the wild-type QRE.
DISCUSSION
Monocytopoiesis, which produces monocytes–macrophages, is essential for innate immunity and wound healing. Although great advances have been made in the understanding of the myeloid differentiation process, the underlying mechanism is not fully understood, particularly why the primed progenitor cells are maintained in an undifferentiated stage to ensure cell expansion. A feedback loop composed of posttranscriptional regulators is found to play an important role in multiple processes, such as cell cycle and inflammation (Yang et al., 2011; Zhou et al., 2011). Previously, miR-223 was found to function as a fine-tuner of granulocyte production by posttranscriptionally regulating NFI-A and C/EBPα (Fazi et al., 2005; Johnnidis et al., 2008), suggesting that there is a posttranscriptional network in hematopoietic differentiation.
Our study indicates that the RNA-binding protein QKI plays a pivotal role in monocyte–macrophage differentiation. In this study, we found that in the progenitor cells or earlier-differentiated cells, where the differentiation priming transcription factor C/EBPα has been activated, QKI was transcriptionally activated by C/EBPα. Abundant expression of QKI at this stage inhibits the expression of CSF1R, which thus delays the terminal differentiation process of monocytes–macrophages. Here our study showed that in addition to posttranscriptionally regulating NFI-A and C/EBPα to fine-tune the monocytic differentiation process (Johnnidis et al., 2008; Fazi et al., 2005), QKI posttranscriptionally regulates factors downstream of C/EBPα, which adds a novel regulatory layer to this complicated process. In other words, we provided an example in which the undifferentiated progenitor cells simultaneously activate a set of genes priming for differentiation and another set of genes repressing differentiation, thus keeping the progenitors in balance between proliferative and differentiated.
C/EBPα is only one of the important transcription factors activating CSF1R expression and priming the differentiation of monocytes (Zhang et al., 1996; Pan et al., 1999; Feng et al., 2008). Of interest, the regulatory region of the QKI promoter 2 kb upstream of the ATG start codon was found to contain multiple hematopoietic differentiation–related transcriptional factor–binding sites (Cartharius et al., 2005), further suggesting that QKI might be a ubiquitous gatekeeper of the balance between proliferation and differentiation. In light of these findings, we can observe that the switch between differentiation and proliferation is fine-tuned during monocytic differentiation, and altered QKI expression should affect the monocytopoiesis. Accordingly, siQKI influenced the timing of monocytic differentiation. In the future, an in vivo study using conditional transgenic and knockout mice could provide more solid evidence for the role of QKI in monocytic differentiation. Of note, although we focus on the role of QKI-5 in this study, we could not exclude the possibility that QKI-6 and QKI-7 might also be involved in the process, especially when QKI-5 and QKI-6 have similar expression profiles, and the QKI knockdown assay against all the isoforms was considered.
To address how QKI delays the differentiation process and thus ensures that enough mature cells are in the immune system, we determined that CSF1R might be one of the key mediators. It is important to note that although CSF1R is the obligatory regulator of macrophage differentiation (Bourette and Rohrschneider, 2000; Pixley and Stanley, 2004), an experiment is needed to test whether the double knockdown of QKI-5 and CSF1R leads to premature differentiation to show that QKI delays macrophage differentiation by targeting CSF1R.
It has been established that impairment of CSF1R expression is one feature of acute myeloid leukemia (AML; Casas et al., 2003), and thus it is interesting to see that aberrant QKI expression is also involved in this process in a manner other than that of the well-known transcriptional causes. It has been reported that CSF1R is overexpressed in certain breast cancers (Kirma et al., 2004; Kluger et al., 2004; Toy et al., 2005). To this end, in addition to the role in monocytopoiesis, negative regulation of CSF1R by QKI might be aberrant in other cell lineages. In fact, our previous study revealed that QKI might be a tumor suppressor and is down-regulated in some cancer cells (Yang et al., 2010). It is worth testing to determine whether the aberrant QKI expression in cancers causes CSF1R overexpression.
In conclusion, our studies indicate that 1) in committed progenitor cells, the primed C/EBPα activates the expression of CSF1R and QKI, whereas the latter negatively regulates CSF1R expression to maintain the progenitors in an undifferentiated stage; and 2) QKI down-regulation in late monocytopoiesis “unblocks” CSF1R translation, promoting the differentiation and maturation of macrophages (Supplemental Figure S2).
MATERIALS AND METHODS
Reagents
Both anti-CD11b and anti-CD14 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against C/EBPα and CSF1R were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against pan-QKI, α-tubulin, and β-actin were from Sigma-Aldrich (St. Louis, MO). Fluorescein isothiocyanate (FITC)–conjugated monoclonal CD14 or immunoglobulin G (IgG) antibodies (mAbs) were from Immunotech (Marseille, France). The protease inhibitor cocktail was obtained from Calbiochem (La Jolla, CA). The TPA (Sigma-Aldrich) was dissolved in absolute ethanol to a concentration of 1 mM for the stock. Penicillin/streptomycin, trypsin/EDTA, and phosphate-buffered saline (PBS) were obtained from Life Technologies (Carlsbad, CA). Protein A Sepharose was obtained from Amersham Pharmacia Biotech (GE Healthcare Bio-Sciences, Piscataway, NJ). Dithiothreitol was purchased from Invitrogen (Carlsbad, CA). All other reagents were purchased from domestic companies unless specifically stated.
Cell culture
The human cancer HL-60 cell line, originally from the American Type Culture Collection (Manassas, VA), was stored by our laboratory and grown in RPMI 1640 supplemented with 2 mM glutamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, and 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator.
Human cord blood was obtained after informed written consent and processed under the approval of the Fourth Military Medical University Ethics Committee. Isolation of the CD34+ cells from cord blood, unilineage culture, and morphological analysis were performed as previously described (Gabbianelli et al., 2000). Briefly, the CD34+ cells were purified from cord blood by positive selection using the midi-MACS immunomagnetic separation system according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of the CD34+ cells was assessed by flow cytometry using a monoclonal PE (phycoerythrin)-conjugated anti-CD34 antibody and was routinely >90%. CD34+ progenitors were cultured in Iscove's modified Dulbecco's medium supplemented with bovine serum albumin (10 mg/ml), pure human transferrin (700 mg/ml), human low-density lipoprotein (40 mg/ml), insulin (10 mg/ml), sodium pyruvate (10−4 mol/l), l-glutamine (2 × 10−3 mol/l), rare inorganic elements supplemented with iron sulfate (4 × 10−8 mol/l), and nucleosides (10 μg/ml each).
Cell differentiation assay
To induce the monocyte–macrophage differentiation of HL-60 cells, the cells were induced by TPA for the indicated periods at the indicated concentration. Cell morphology was observed by microscope.
To induce CD34+ cord blood human progenitor cell differentiation toward a macrophage fate, the cells were cultured in serum-free medium that was supplemented with 1 ng/ml IL-6 and 100 ng/ml Flt3-ligand combined with saturated-level M-CSF (50 ng/ml).
Plasmid construction, virus packaging, and infection
QP (QKI promoter) and the truncations without the putative C/EBPα-binding sites (ΔQP1, ΔQP2) were constructed as previously described. For the construction of QPm1 (with the distal C/EBPα-binding site mutated), the synthetic two reverse complement DNA sequences harboring the mutated C/EBPα-binding site (synthesized by Sangon, Shanghai, China; Supplemental Table S1) were used and annealed before cloning into the QP1 vector. The QPm2 (with the proximal C/EBPα-binding site mutated) was cloned using a mutagenesis kit according to the manufacturer's instructions (Takara Bio, Otsu, Japan). To construct the CSF1R 3′UTR reporters or truncations without the putative QREs, the fragments were amplified by reverse transcription-PCR from the mRNA using the primers listed in Supplemental Table S1. The PCR products were digested with the indicated restriction enzymes before being ligated into our previously modified pGL3-control vector in which the EcoRI, EcoRV, NdeI, and PstI restriction enzyme sites were inserted downstream the XbaI site. Stealth small interfering RNAs (siRNAs) targeting QKI were synthesized by Invitrogen and dissolved in diethylpyrocarbonate-treated H2O at a concentration of 20 μmol/l as a stock. Construction and infection of the control and QKI overexpression adenovirus were performed as previously described (Yang et al., 2010, 2011).
RNA-IP
RNA-IP was performed as described previously (Yang et al., 2010). The RNA in the immunoprecipitated complex or in the previously saved input fraction was extracted. Specific primers were applied for detection of the target mRNAs (Supplemental Table S1).
Reporter assay
The cells were harvested 24 h after transfection and assayed with the Dual Luciferase Assay Kit (Promega, Madison, WI) according to the manufacturer's instructions. The values were expressed as the means ± SD from at least three independent experiments. Because QKI overexpression might alter the internal control pRL-TK due to alternative splicing, all the fold changes in these vectors by QKI were normalized to the changes in pGL3-SV40. Statistical analyses were performed using Student's t tests. A value of p < 0.05 was considered as a significant difference.
RT-PCR
At the start of each experiment, the cells with the indicated treatment were harvested at the indicated time for RNA extraction, and 2 μg of total RNA was used to prepare the cDNA (TRIzol, Invitrogen; MLV Reverse Transcriptase, Promega). PCR was then performed on 1 μl of cDNA, as described, using specific pairs of primers for the targets (Supplemental Table S1). β-Actin was used as an internal control.
For the mRNA decay assay, the cells were cultured in the indicated conditions and further treated with actinomycin D at time 0, and RNA was extracted at the indicated time to examine the RNA stability.
The qRT-PCR assay was performed using an AB 7500 system. The conditions were as follows: 10 μl of SYBR Green I (Takara Bio), 0.5 μM of each 5′ and 3′ primer, 2 μl of the sample, and H2O to a final volume of 20 μl. The samples were amplified for 45 cycles with a denaturation at 95°C for 5 s, and the annealing and extension were at 60°C for 34 s. SYBR Green fluorescence was measured to determine the amount of double-stranded DNA. To discriminate specific from nonspecific cDNA products, a melting curve was obtained at the end of each run. Relative mRNA levels of different QKI isoforms and CSF1R were normalized to GAPDH levels and compared with the control using the 2−ddCt.
Western blot assay
The levels of QKI, CD11b, CD14, and CSF1R were quantified using the corresponding antibodies listed, with standard procedures for Western blot. The normalization was performed using mouse monoclonal anti–β-actin or anti–α-tubulin antibodies.
Flow-cytometric analyses
HL-60 cells (5 × 105) were incubated for 20 min at 4°C with the FITC-conjugated CD14 mAbs (from Immunotech). As a control, the corresponding cells labeled with a FITC-conjugated isotype IgG were used. After washing with PBS, the cells were analyzed by FACS (Beckman Coulter, Brea, CA).
Statistical analyses
All of the experiments were performed at least in triplicate, and the data are expressed as the means ± SD. A Student's t test was applied for statistical analysis. p < 0.05 was considered as significantly different.
Supplementary Material
Acknowledgments
We thank Achim Leutz for the kind gift of the pcDNA3.1-C/EBPα expression vector. This work was supported by the National Science Foundation of China (30900732, 31100979, 81030046, and 31171112) and the National Key Basic Research and Development Program 2009CB521704.
Abbreviations used:
- AML
acute myeloid leukemia
- C/EBP α
CCAAT/enhancer-binding protein α
- ChIP
chromatin immunoprecipitation
- CSF1R
colony stimulating factor 1 receptor, also known as macrophage colony-stimulating factor receptor
- FACS
fluorescence-activated cell sorting
- NC
negative control
- PE
phycoerythrin
- QRE
QKI response element
- qRT-PCR
quantitative real-time PCR
- RNA-IP
RNA-immunoprecipitation
- RT-PCR
reverse transcription-PCR
- siQKI
small interference RNA–mediated knockdown of the endogenous QKI
- STAR
signal transduction and activator of RNA
- TPA
tetradecanoylphorbol, also known as phorbol-12-myristate-13-acetate
- UTR
untranslated region
- WT
wide type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-05-0412) on March 7, 2012.
REFERENCES
- Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, Zhang J, Haug J, Li L. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood. 2003;101:383–389. doi: 10.1182/blood-2002-06-1780. [DOI] [PubMed] [Google Scholar]
- Bourette RP, Rohrschneider LR. Early events in M-CSF receptor signaling. Growth Factors. 2000;17:155–166. doi: 10.3109/08977190009001065. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Casas S, Nagy B, Elonen E, Aventin A, Larramendy ML, Sierra J, Ruutu T, Knuutila S. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma. 2003;44:1935–1941. doi: 10.1080/1042819031000119299. [DOI] [PubMed] [Google Scholar]
- Clarke S, Gordon S. Myeloid-specific gene expression. J Leukoc Biol. 1998;63:153–168. doi: 10.1002/jlb.63.2.153. [DOI] [PubMed] [Google Scholar]
- Dahl R, Walsh JC, Lancki D, Laslo P, Iyer SR, Singh H, Simon MC. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol. 2003;4:1029–1036. doi: 10.1038/ni973. [DOI] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet. 1996;12:260–265. doi: 10.1038/ng0396-260. [DOI] [PubMed] [Google Scholar]
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, Graf T. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci USA. 2008;105:6057–6062. doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbianelli M, Testa U, Massa A, Pelosi E, Sposi NM, Riccioni R, Luchetti L, Peschle C. Hemoglobin switching in unicellular erythroid culture of sibling erythroid burst-forming units: kit ligand induces a dose-dependent fetal hemoglobin reactivation potentiated by sodium butyrate. Blood. 2000;95:3555–3561. [PubMed] [Google Scholar]
- Galarneau A, Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol. 2005;12:691–698. doi: 10.1038/nsmb963. [DOI] [PubMed] [Google Scholar]
- Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, Enver T. Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 1997;11:774–785. doi: 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- Kirma N, Luthra R, Jones J, Liu YG, Nair HB, Mandava U, Tekmal RR. Overexpression of the colony-stimulating factor (CSF-1) and/or its receptor c-fms in mammary glands of transgenic mice results in hyperplasia and tumor formation. Cancer Res. 2004;64:4162–4170. doi: 10.1158/0008-5472.CAN-03-2971. [DOI] [PubMed] [Google Scholar]
- Kluger HM, Kluger Y, Gilmore-Hebert M, DiVito K, Chang JT, Rodov S, Mironenko O, Kacinski BM, Perkins AS, Sapi E. cDNA microarray analysis of invasive and tumorigenic phenotypes in a breast cancer model. Lab Invest. 2004;84:320–331. doi: 10.1038/labinvest.3700044. [DOI] [PubMed] [Google Scholar]
- Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, Artzt K, Yamamura K, Abe K. Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome. 1999;10:662–669. doi: 10.1007/s003359901068. [DOI] [PubMed] [Google Scholar]
- Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci. 2005;8:27–33. doi: 10.1038/nn1359. [DOI] [PubMed] [Google Scholar]
- Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S. Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron. 2002;36:815–829. doi: 10.1016/s0896-6273(02)01055-3. [DOI] [PubMed] [Google Scholar]
- Larocque D, Richard S. QUAKING KH domain proteins as regulators of glial cell fate and myelination. RNA Biol. 2005;2:37–40. doi: 10.4161/rna.2.2.1603. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li L, et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell. 2011;20(5):591–605. doi: 10.1016/j.ccr.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noveroske JK, Lai L, Gaussin V, Northrop JL, Nakamura H, Hirschi KK, Justice MJ. Quaking is essential for blood vessel development. Genesis. 2002;32:218–230. doi: 10.1002/gene.10060. [DOI] [PubMed] [Google Scholar]
- Pan Z, Hetherington CJ, Zhang DE. CCAAT/enhancer-binding protein activates the CD14 promoter and mediates transforming growth factor beta signaling in monocyte development. J Biol Chem. 1999;274:23242–23248. doi: 10.1074/jbc.274.33.23242. [DOI] [PubMed] [Google Scholar]
- Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628–638. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Pulikkan JA, et al. C/EBPalpha regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood. 2010;116:5638–5649. doi: 10.1182/blood-2010-04-281600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann ER, Hengartner M, Schedl T, Gartner A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell. 2005;120:357–368. doi: 10.1016/j.cell.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Stanley ER. Action of the colony-stimulating factor, CSF-1. Ciba Found Symp. 1986;118:29–41. doi: 10.1002/9780470720998.ch3. [DOI] [PubMed] [Google Scholar]
- Toy EP, Bonafe N, Savlu A, Zeiss C, Zheng W, Flick M, Chambers SK. Correlation of tumor phenotype with c-fms proto-oncogene expression in an in vivo intraperitoneal model for experimental human breast cancer metastasis. Clin Exp Metastasis. 2005;22:1–9. doi: 10.1007/s10585-005-0718-4. [DOI] [PubMed] [Google Scholar]
- Woo HH, et al. Regulation of non-AU-rich element containing c-fms proto-oncogene expression by HuR in breast cancer. Oncogene. 2009;28:1176–1186. doi: 10.1038/onc.2008.469. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem. 1999;274:29202–29210. doi: 10.1074/jbc.274.41.29202. [DOI] [PubMed] [Google Scholar]
- Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci USA. 2002;99:4233–4238. doi: 10.1073/pnas.072090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, et al. RNA-binding protein quaking, a critical regulator of colon epithelial differentiation and a suppressor of colon cancer. Gastroenterology. 2010;138:231–240 e231–235. doi: 10.1053/j.gastro.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Lu X, Wang L, Bian Y, Fu H, Wei M, Pu J, Jin L, Yao L, Lu Z. E2F1 and RNA binding protein QKI comprise a negative feedback in the cell cycle regulation. Cell Cycle. 2011;10:2703–2713. doi: 10.4161/cc.10.16.15928. [DOI] [PubMed] [Google Scholar]
- Zeng C, Wang R, Li D, Lin XJ, Wei QK, Yuan Y, Wang Q, Chen W, Zhuang SM. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology. 2010;52:1702–1712. doi: 10.1002/hep.23875. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Hetherington CJ, Meyers S, Rhoades KL, Larson CJ, Chen HM, Hiebert SW, Tenen DG. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol Cell Biol. 1996;16:1231–1240. doi: 10.1128/mcb.16.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Ravatn R, Lin Y, Shih WC, Rabson A, Strair R, Huberman E, Conney A, Chin K-V. Gene expression of TPA induced differentiation in HL-60 cells by DNA microarray analysis. Nucleic Acids Res. 2002;30:4489–4499. doi: 10.1093/nar/gkf580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci USA. 2011;108:10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.