

ABSTRACT
Precise coordination of meiotic progression is a critical determinant of an egg's capacity to be fertilized successfully, and zinc has emerged as a key regulatory element in this process. An early manifestation of a regulatory role for this transition metal is the significant increase in total intracellular zinc. This accumulation is essential for meiotic progression beyond telophase I and the establishment of meiotic arrest at metaphase II. The subsequent developmental event, fertilization, induces a rapid expulsion of labile zinc that is a hallmark event in meiotic resumption. In the present study, we show that the zinc fluxes work, in part, by altering the activity of the cytostatic factor (CSF), the cellular activity required for the establishment and maintenance of metaphase II arrest in the mature, unfertilized egg. We propose a model in which zinc exerts concentration-dependent regulation of meiosis through the CSF component EMI2, a zinc-binding protein. Together, the data support the conclusion that zinc itself, through its interaction with EMI2, is a central component of the CSF.
Keywords: cytostatic factor, EMI2, meiosis, meiotic arrest, meiotic maturation, metal biology, oocyte, oocyte maturation, zinc
Zinc dynamics in the mammalian oocyte may work through EMI2 to modulate establishment, maintenance, and release of metaphase II arrest.
INTRODUCTION
The necessity of inorganic elements in biological systems is well established. Whereas “bulk” elements, such as sodium, magnesium, potassium, calcium, and phosphorus, have known, concentration-dependent cellular activities, the biological role of so-called “trace” elements, such as iron, copper, and zinc, have largely been confined to that of protein cofactors. In terms of total cellular content, iron and zinc can be as abundant as calcium, on the scale of millimolar concentrations [1], suggesting that “trace element” is a misnomer in a cellular context. Furthermore, recent evidence supports functions for the transition metals in biology akin to their alkali and alkaline earth metal counterparts—namely, regulation of cellular processes by fluxes in intracellular concentration.
Zinc, in particular, has gained attention as key agent in cellular signaling processes. In a biological context, it shares many features with calcium, the ubiquitous inorganic second messenger. Like calcium, zinc is redox inactive and stable at physiological pH as a divalent cation. Calcium works via intracellular oscillations (or transients) resulting from rapid increases and decreases in concentrations of the free or kinetically labile species—that is, calcium that is free or loosely bound within the cell (for review, see [2]). Most notably, calcium oscillations, as detected by calcium-sensitive fluorophores, such as fura-2 [3], have been associated with cell cycle progression [4]. Similarly, evidence indicates the intracellular concentration of labile zinc can also fluctuates depending on whether a cell is at rest, proliferating, or differentiated [5]. To our knowledge, rapid fluxes in free zinc on the time scale of calcium oscillations have not been documented in the literature; however, it has been suggested that zinc can act in concert with the immediate effects of the calcium transients by exerting a slower, prolonged signal [6]. Connections between intracellular calcium and zinc fluxes have also been reported in the endoplasmic reticulum of cultured cells, wherein calcium signaling influences zinc homeostasis, which in turn modulates calcium homeostasis [7].
An additional example of a calcium-zinc biological partnership was recently described in the mammalian egg. In mammals, a characteristic feature of a mature, fertilizable egg is cell cycle arrest at metaphase II (MII) of meiosis. This arrest is relieved upon fertilization, catalyzed by a series of calcium oscillations that activate downstream molecular targets (for review, see [8]). Meiotic resumption, also known as egg activation, can be achieved without sperm using reagents that elevate intracellular levels of calcium [9–12] or, curiously, decrease intracellular levels of zinc [13–15]. Eggs activated with the heavy metal chelator N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN), which is zinc-selective within the context of the egg [14], were robust enough to support live births in mice [15]. A physiological mechanism for labile zinc loss in fertilized eggs was reported soon thereafter; the calcium oscillations induce a series of coordinated exocytosis events, called zinc sparks, that occur rapidly in response to the elevations in intracellular calcium [13].
The zinc sparks represent a cellular activity that regulates the levels of labile zinc in the egg. As it turns out, changes in total intracellular zinc are also critical to the creation of a fertilizable egg. Female mammals are born with all of the germ cells they will ever have, and these are stored as prophase-arrested oocytes within the ovary. During each ovulatory cycle, a select group of these oocytes is stimulated to grow and resume meiosis, progressing from prophase arrest to MII arrest in a process known as meiotic maturation. The total cellular content of zinc increases by more than 50% during this brief, half-day process [14]. This accumulation is necessary for proper MII arrest, because oocytes that mature under zinc-insufficient conditions stall at telophase I (TI) instead [14]. This phenotype is explained, in part, as a defect in regulation of maturation-promoting factor (MPF) activity during the later stages of meiotic maturation [16], though the direct link between the change in total cellular zinc and MPF activity has yet to be discerned.
A wealth of evidence indicates the importance of fluctuations in total zinc in the mammalian egg, and our focus now turns to molecular mechanisms by which zinc exerts these regulatory effects. In the present study, we examine the zinc-dependent regulation of MPF, which drives progression through meiosis, and the role of the cytostatic factor (CSF), a multicomponent cellular activity that maintains MPF activity at MII arrest. The CSF is known to act primarily through inhibition of the anaphase-promoting complex/cyclosome (APC/C) [17, 18], and the zinc-binding APC/C inhibitor EMI2 (official symbol FBXO43) has been shown to be a critical component of the CSF. In this study, we show that zinc and EMI2 play a concerted role in the establishment of, maintenance of, and ultimately, exit from MII arrest. Thus, mammalian EMI2 requires zinc for its proper cellular function, and a physiological reduction in intracellular zinc (via the zinc sparks) is required for egg activation. The correlation between zinc and EMI2 at MII arrest, when intracellular zinc is at its highest and EMI2 activity is required, leads us to present a model in which EMI2 acts as a zinc-responsive switch that responds to dynamic fluxes in zinc over the course of oocyte maturation and early embryo development to drive meiotic progression.
MATERIALS AND METHODS
Reagents and Antibodies
Anti-cyclin 1 (CCNB1) antibody (ab72) was purchased from Abcam. The anti-FBXO43 (EMI2) antibody (EB06061) was from Everest Biotech. Culture medium, fetal bovine serum (FBS), rhodamine-phalloidin (R415), Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin (Ig) G (A11001), horseradish peroxidase (HRP)-conjugated anti-mouse IgG (62–6520), 10% BisTris NuPAGE gels, and 3-(N-morpholino)propanesulfonic acid-SDS running buffer were purchased from Invitrogen. Immobilon-P polyvinylidene fluoride (PVDF) membranes and potassium simplex optimized medium (KSOM) were from Millipore. Peroxidase conjugated anti-goat IgG (PI-9500) and Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) were from Vector Laboratories. HRP-conjugated anti-rabbit IgG (NA934) and ECL Advance detection reagent were purchased from Amersham Biosciences. Equine chorionic gonadotropin (eCG) was purchased from Calbiochem. Anti-α-tubulin (T9026 for immunofluorescence, T6199 for Western blot analysis) and all other chemicals and reagents not specifically noted were purchased from Sigma-Aldrich.
Animals
Mice of the CD1 strain were maintained in accordance with the policies of Northwestern University's Animal Care and Use Committee and the National Research Council publication Guide for Care and Use of Laboratory Animals. Mice were bred and housed within a controlled barrier facility within Northwestern University's Center for Comparative Medicine. They were provided food and water ad libitum and were maintained on food free of soybean or alfalfa meal (2919 Teklad Global), therefore minimizing the impact of phytoestrogens. Humidity, temperature, and photoperiod (14L:10D) were kept constant.
Oocyte and Egg Collection and In Vitro Maturation
For in vitro maturation (IVM) studies, immature female mice (age, 17–21 days) were injected with 5 IU of eCG. After 44–48 h, mice were anesthetized using isoflurane and euthanized by cervical dislocation. Dissected ovaries were placed in Leibovitz L-15 medium containing 1% FBS (L-15/FBS) and 0.2 mM 3-isobutyl-1-methylxanthine (IBMX), and large antral follicles were punctured using 28-gauge needles to release cumulus-oocyte complexes (COCs). COCs were washed through several drops of culture medium consisting of Minimum Essential Medium (MEM)-alpha with GlutaMAX supplemented with 10% FBS, 1.5 IU/ml of human chorionic gonadotropin (hCG), and 5 ng/ml of epidermal growth factor and then placed in culture medium with or without 10 μM TPEN. Previously, 10 μM TPEN was shown to be less than the concentration necessary to cause apparent cell toxicity in oocytes [14]. In some experiments, Z-Leu-Leu-Leu-al (MG132) was added to culture medium at 20 μM. COCs were cultured in drops of pre-equilibrated medium covered with embryo-quality mineral oil at 37°C in 5% CO2 with a humid environment. Ovulated MII eggs were collected from 4- to 6-wk-old mice primed with 5 IU of eCG followed 48 h later by 5 IU of hCG. Oviducts were dissected into L-15/FBS, and eggs were removed and treated with 0.03% (w/v) hyaluronidase to remove cumulus cells. Eggs were washed and cultured in KSOM covered with embryo-quality mineral oil at 37°C in 5% CO2.
SDS-PAGE and Western Blot Analysis
Sample preparation and SDS-PAGE/Western blot analysis of oocytes were performed essentially as previously described [16]. Briefly, 15–25 cumulus-denuded oocytes were collected in a minimal amount of medium and lysed in 8 μl of 1× SDS-PAGE sample buffer [19]. SDS-PAGE gels were run and transferred to PVDF membranes. Blocking and antibody incubations were in 2% (w/v) Amersham ECL Advance blocking reagent in TBS-T (20 mM Tris [pH 7.4], 137 mM NaCl, and 0.1% [v/v] Tween 20). Dilutions were as follows: anti-CCNB1, 1:500; anti-FBXO43, 1:1000; and anti-tubulin and HRP-conjugated secondary antibodies, 1:10 000. Detection was performed using ECL Advance detection reagent. Kodak BioMax MR films were exposed and developed, or an Alpha-Innotech MultiImage II system was used.
Immunofluorescence and Laser-Scanning Confocal Microscopy
Oocytes were fixed in a microtubule stabilizing buffer [20] containing 2% formaldehyde and 1% Triton X-100 for 30 min at 37°C. They were then washed and blocked in PBS containing 0.1 M glycine, 3 mg/ml of bovine serum albumin, 0.01% Tween 20, and 0.01% sodium azide for at least 1 h and then stored in blocking solution at 4°C for up to 2 wk. Oocytes were incubated with anti-tubulin antibody (1:100) for 1 h at 37°C followed by three washes in blocking solution, incubation for 1 h in Alexa Fluor 488-conjugated anti-mouse secondary antibody (5 μg/ml) and rhodamine-phalloidin (2 U/ml), three additional washes in blocking solution, and mounting in Vectashield with DAPI. Microscopy was performed using a Leica SP5 inverted laser-scanning confocal microscope with a 63× oil-immersion objective (Leica Microsystems). Images were processed using Leica LAS-AF software.
Histone H1 and Myelin Basic Protein Dual Kinase Assays
Histone H1 and myelin basic protein (MBP) dual kinase assays were performed as previously described [21]. Dried gels were exposed to Kodak BioMax MR films for 12–72 h at −80°C using intensifying screens; films were developed, scanned, and analyzed using ImageJ software (National Institutes of Health). Assays were performed on single oocytes, and densitometric analysis is expressed relative to levels in control eggs.
Plasmids, In Vitro Transcription, Morpholinos, and siRNA
Empty pIVT vector was kindly provided by Richard Schultz [22], and pRN3-CCNB1(Δ90)-enhanced green fluorescent protein (EGFP) was kindly provided by Karen Schindler [23]. To construct Emi2 cRNA expression constructs, a full-length cDNA clone for mouse Emi2 was obtained from the IMAGE collection (Thermo-Fischer, Open Biosystems), and a missense mutation at codon 393 was corrected using QuickChange site-directed mutagenesis (Agilent). Full-length Emi2 sequence along with an N-terminal T7 tag were cloned into pIVT using XbaI and KpnI sites as a PCR-generated fragment. C573A mutations (TGC to GCC) were produced using Agilent QuickChange and confirmed by sequencing. Plasmids were linearized, and capped RNA was produced using the Ambion mMESSAGE mMACHINE T3 Kit. RNA was purified with Qiagen RNeasy columns and eluted in 10 mM Tris [pH 7.4] and 0.1 mM ethylenediaminetetra-acetic acid at a final concentration of 0.5 μg/μl, and aliquots were stored at −80°C. The sequence of the Emi2 morpholino (MO) used has been previously published and validated [24]. MOs purchased from Gene Tools were dissolved in molecular-grade water at 5 mM, aliquotted and stored at −80°C, and heated to 65°C for 10 min before injection.
Oocyte Microinjection
Microinjection was performed essentially as previously described [16]. Germinal vesicle (GV)-intact oocytes were collected and manually denuded of cumulus cells in L-15/FBS containing 0.2 mM IBMX, then transferred to drops of L-15 medium containing 0.05% (w/v) polyvinyl alcohol and 0.5% (v/v) penicillin-streptomycin (Invitrogen) under light mineral oil on a heated stage set to 34°C for injection. Injection of oocytes at the metaphase I (MI)-MII transition was performed in medium without IBMX and with stage temperature turned down to between 27°C and 30°C; when appropriate, 10 μM TPEN was included in holding medium. From 3 to 10 pl of in vitro-synthesized RNA, MO, or siRNA were injected into the oocyte cytoplasm using an Eppendorf FemtoJet pressure microinjector with Femtotip injection capillaries. GV-stage oocytes were held in MEM-alpha containing IBMX and 1% FBS for 2–6 h (depending on the experiment) before being transferred to IVM medium; MI-MII transition oocytes were returned to culture medium without IBMX. Following culture, oocytes were fixed for spindle staining or collected for Western blot and kinase assays.
Zinc Pyrithione Treatments
Ovulated MII eggs were collected as described above. Eggs were transferred to KSOM medium with or without 10 μM zinc pyrithione (ZnPT; 1:1000 dilution of a 10 mM stock in dimethyl sulfoxide [DMSO]) for exactly 5 min. Eggs were then washed in KSOM and allowed to recover for 10–15 min before activation. For SrCl2 activation, eggs were transferred to drops of calcium-free KSOM containing 10 mM SrCl2 for 2 h followed by an additional 4 h of culture in calcium-containing KSOM without SrCl2. The TPEN activation was performed as previously described [13]; eggs were cultured for 6 h in KSOM containing 10 μM TPEN. Unactivated controls were cultured in KSOM for the same 6-h time period to assess levels of spontaneous parthenogenesis. Before ZnPT treatment, some eggs were incubated in 10 μM FluoZin-3 AM (Invitrogen) in KSOM for 60 min. After 5 min of ZnPT treatment, these eggs were washed and transferred to drops of KSOM under embryo-quality mineral oil in glass-bottom dishes (Bioptechs, Inc.) followed by imaging on a Leica SP5 confocal microscope equipped with Tokai Hit stage-top incubator using a 20× objective. Other groups of eggs were activated by incubation in 10 mM SrCl2 treatment for 2 h followed by 5 min of ZnPT treatment. These eggs were incubated with 10 μg/ml of dithizone (1:1000 dilution of a DMSO stock) in KSOM for 10 min and imaged using a Leica DM-IRB microscope with a 40× objective.
Statistical Analysis
Analysis of kinase assay data was performed by one-way ANOVA using Prism4 software (GraphPad Software) or by two-tailed Student t-tests performed in Excel (Microsoft). The level of statistical significance is indicated in each figure legend.
RESULTS
Zinc Insufficiency Restricted to the MI-MII Transition Disrupts Meiotic Maturation
Previously, we have shown that mouse oocytes cultured under conditions of limited zinc availability (“zinc insufficiency”) due to the presence of the heavy metal chelator TPEN fail to undergo proper asymmetric division, have reduced MPF activity and CCNB1 protein levels, and arrest with a TI-like spindle phenotype following a 14- to 16-h IVM period [14, 16]. While we have shown that this arrest is not due to impairment of the MOS-mitogen-activated protein kinase (MAPK) pathway [16], the exact mechanism through which zinc insufficiency causes this TI arrest remains unknown. To narrow the time frame of meiotic maturation during which zinc is required, we selectively applied TPEN after in vitro-matured COCs had formed MI spindles. Following an initial 7.5-h culture in control medium, oocytes were transferred to medium containing 10 μM TPEN and cultured for an additional 6.5 h. These oocytes failed to progress to MII, mirroring the effect of TPEN treatment during the entire 14-h culture period (Fig. 1 and Supplemental Table S1, all supplemental data are available online at www.biolreprod.org). In contrast, oocytes maintained in control medium for 14 h matured normally and arrested at MII (Fig. 1A and Supplemental Table S1). Of oocytes cultured in TPEN during only the MI-MII transition (i.e., beginning at 7.5 h), 30% had TI-arrested spindles (Fig. 1A, ii), and an additional 51% had chromatin masses rather than discrete chromosomes but without complete retention of a telophase microtubule conformation (Fig. 1A, iii). Most of the remaining oocytes had more than one chromatin mass (Fig. 1A, iv) or had not completed cytokinesis, and none had formed MII spindles. Western blot analysis and histone H1 kinase assays showed that CCNB1 protein levels and MPF activity were low in oocytes cultured with TPEN during the MI-MII transition (Fig. 1, B and C), as has been shown previously for oocytes matured for 12–16 h in the presence of TPEN [16]. Thus, zinc insufficiency limited to the MI-MII transition period causes a meiotic arrest phenotype and failure to reactivate MPF, leading us to conclude that increased availability of intracellular zinc is required for normal MI-MII transition.
FIG. 1. .

Zinc is required for successful MI-MII transition. A) COCs were cultured in control medium for 7.5 h and transferred to TPEN-containing medium at MI for an additional 6.5 h followed by removal of cumulus cells. Immunofluorescence analysis of the spindle shows that rather than forming normal MII spindles of controls (i), 30% of treated oocytes were arrested at TI (ii), whereas 51% had a single chromatin mass without complete retention of midbody microtubules (iii). Others had multiple chromatin masses (iv) or failed to complete cytokinesis. (Refer also to Supplemental Table S1.) Three-dimensional projections of confocal Z stacks with actin (red), tubulin (green), and DAPI (blue) are shown. Bar = 20 μm. B and C) Western blot analysis for CCNB1 and EMI2 and histone H1 kinase activity of oocytes from the same treatments as in A are shown. Western blots were repeated at least three times, showing similar results. Equal numbers of GV stage oocytes, which have low levels of CCNB1 and EMI2 protein, were also included for comparison. Graph presents densitometric analysis for at least seven individual oocytes per group. Error bars represent the SEM, and different letters indicate significant differences according to ANOVA with Bonferroni post-hoc test (P < 0.001).
Restoration of MPF Activity Following the MI-MII Transition Partially Rescues Zinc-Insufficient Oocytes
Zinc-insufficient oocytes fail to increase MPF activity following the first meiotic division and have low CCNB1 protein levels [16]. To determine whether this decrease in CCNB1 is the major cause of the observed meiotic arrest phenotype, we tested whether inhibiting the proteasome at the end of the MI-MII transition, thereby halting CCNB1 degradation and increasing its total protein levels, would be sufficient to rescue MII entry in zinc-insufficient oocytes. Experimental oocytes were cultured for 10 h in TPEN-containing medium and then transferred into medium containing 10 μM TPEN and the proteasome inhibitor MG132 at a 20 μM concentration. MG132 was added after first polar body extrusion, because earlier addition results in MI arrest due to failure to degrade APC/C targets that prevent cell division. Oocytes were cultured for an additional 6 h in the presence of MG132 to allow time for CCNB1 reaccumulation, for a total IVM culture period of 16 h. Proteasome inhibition led to a partial rescue of the zinc insufficiency phenotype. In all, 69% of treated oocytes formed spindle-like structures, and none was in TI arrest, despite being cultured in the presence of TPEN for the entire culture period (Fig. 2A and Supplemental Table S2). MII spindles of proteasome-treated eggs had varying degrees of organization, with 19% showing aligned metaphase plates (Fig. 2A, i), whereas other eggs had slight (31%) to severe (19%) scattering of chromosomes along the length of the spindle (Fig. 2A, ii [≤3 misaligned chromosomes] and iii [>3 misaligned chromosomes]). Notably, eggs that had divided symmetrically during the first meiotic division frequently had two spindles, one in each cell (Fig. 2A, i). Histone H1 kinase assays of MG132-treated oocytes show that MPF activity was increased compared to that in oocytes cultured with TPEN alone (Fig. 2B). CCNB1 protein levels were only slightly increased in MG132-treated oocytes (Supplemental Fig. S1), showing that proteasome inhibition could partially, but not completely, restore CCNB1, corresponding to the observed partial rescue.
FIG. 2. .

Restoration of MPF activity following the MI-MII transition partially rescues zinc-insufficient oocytes. A) COCs were cultured in TPEN-containing medium for 10 h and transferred to medium containing both TPEN and MG132 for an additional 6 h followed by removal of cumulus cells. Spindle stains show that 69% of oocytes had some degree of MII spindle formation, with 19% showing aligned metaphase plates (i), 31% showing three or fewer misaligned chromosomes (ii), and 19% showing more than three misaligned chromosomes (iii). A majority of oocytes cultured in TPEN without MG132 for the entire culture period displayed the TI-arrested spindles associated with zinc insufficiency during IVM (iv). (Refer also to Supplemental Table S2.) Three-dimensional projections of confocal Z stacks with actin (red), tubulin (green), and DAPI (blue) are shown. Bar = 20 μm. B) Graph of histone H1 kinase activity showing densitometric analysis for at least six individual oocytes per group. Error bars represent the SEM, and different letters indicate significant differences according to ANOVA with Bonferroni post-hoc test (P < 0.01). C) Histone H1 kinase activity for oocytes cultured in TPEN containing medium for 10 to 12.5 h followed by injection with CCNB1(Δ90)-EGFP cRNA and transfer back to TPEN containing medium for 3.5 to 6 h is presented as in B.
Because proteasome inhibition was an indirect and somewhat nonspecific way to increase CCNB1, we also tested more directly whether restoring CCNB1 levels could rescue MPF activity by injecting zinc-insufficient oocytes with cRNA coding for EGFP-fused CCNB1(Δ90), which lacks an APC/C interaction domain and, thus, is not subject to APC/C-mediated degradation [25]. Oocytes were cultured in TPEN-containing medium for 10–12.5 h before injection to allow meiosis I progression before increasing CCNB1. Following injection, oocytes were cultured for an additional 3–6 h in the presence of TPEN to allow time for expression of CCNB1(Δ90) protein. We previously reported that expression of CCNB1(Δ90)-EGFP led to a partial rescue of the zinc-insufficient spindle phenotype, with 89% of those oocytes that had completed the first meiotic division by the end of culture displaying some degree of spindle-like structures, although most of these spindles were quite disorganized [16]. In the present study, we also show that MPF activity in injected cells is significantly increased compared to uninjected oocytes cultured in the presence of TPEN for the same period of time (Fig. 2C). MPF levels in injected eggs were, in fact, slightly higher than those in control IVM MII eggs. Taken together, these results show that restoration of MPF activity through re-establishment of CCNB1 after the first meiotic division in zinc-insufficient oocytes results in a partial rescue of meiotic progression to MII, indicating that inappropriate degradation of CCNB1 is a primary cause of the meiotic arrest observed with oocyte zinc insufficiency. Because APC/C is responsible for targeting CCNB1 for degradation, we sought to further investigate the impact of zinc insufficiency on APC/C and its regulators, particularly the zinc-binding APC/C inhibitor EMI2.
EMI2 Zinc-Binding Region Contributes to Progression Through the MI-MII Transition
Previously, EMI2 was shown to be required for progression through the MI-MII transition [24, 26, 27]. Work in Xenopus sp. has shown that the C-terminal zinc-binding region (ZBR) is required for emi2 APC/C inhibitory function in vitro [28], and recent work has shown that a functional EMI2 ZBR contributes to the ability of EMI2 to artificially arrest oocytes at MI, when expressed prematurely in mouse oocytes [29]. Because the first and second meiotic divisions are regulated differentially [30], we examined the importance of the EMI2 ZBR in the more physiological context of MII arrest. GV-stage oocytes were injected with an Emi2 MO targeted against a sequence specific to the Emi2 5′ untranslated region (UTR), as has been previously reported and validated [24], and were held in medium containing 0.2 mM IBMX for 5–6 h. Oocytes were then cultured in IVM medium until the first polar body was produced (7.5–11 h of IVM). Oocytes injected with Emi2 MO underwent accelerated first meiotic divisions, with polar bodies present in a higher proportion of MO-injected oocytes than in uninjected controls by 7 h of IVM. This acceleration of meiotic maturation likely related to the amount of time oocytes were held in IBMX-containing medium, because no such acceleration of first polar body formation was reported with oocytes held for only 2 h in milrinone-containing medium after Emi2 MO injection [24].
Within 2 h of polar body formation, oocytes were injected with either wild-type Emi2 cRNA or Emi2-C573A cRNA containing a mutation in the first of eight putative zinc-binding residues in the EMI2 ZBR [28, 29]. Both constructs used to rescue the EMI2 knockdown lacked the MO-targeted UTR sequence. Following injection, oocytes were returned to culture medium for the remainder of a 15-h total IVM period. The majority (73%) of oocytes injected with only EMI2 MO did not have MII spindles after 15 h of IVM and, instead, had masses of chromatin without discretely visible chromosomes and varying degrees of midbody microtubule retention (Supplemental Fig. S2). Injection of oocytes with Emi2 cRNA after first polar body extrusion restored MII spindle formation in 73% of cells (Fig. 3). Failure to completely rescue spindle formation may have been due to difficulty in timing injections precisely to the MI-MII transition period. Overexpression of EMI2 before first polar body extrusion would likely cause MI arrest, as has been reported for injection of Emi2 cRNA into GV oocytes [24, 29], whereas expression too long after the first meiotic division could fail to rescue spindle formation if CCNB1 levels can no longer be restored. Injection of Emi2-C573A cRNA, which contains a mutation in the putative EMI2 ZBR, restored MII spindle formation in only 41% of cells (with a basal level of 23% of oocytes injected with MO only producing MII spindles) (Fig. 3). Therefore, mutation of a single amino acid in the EMI2-ZBR impairs, but does not completely abrogate, the ability of EMI2 to establish MII arrest. This is consistent with the partially reduced ability of EMI2 ZBR mutants to induce arrest at MI [29] and shows that zinc binding is required for full EMI2 function.
FIG. 3. .

Intact EMI2 ZBR is required for MI-MII transition. A) Cumulus-denuded GV oocytes were injected with Emi2 MO, held in IBMX for 5 h, and transferred to IVM medium. Following first polar body extrusion, groups of oocytes were injected with cRNA coding for T7-tagged wild-type Emi2 or for T7-tagged C573A mutated Emi2. B–D) Following 15 h total IVM, oocytes were fixed; stained for actin (red), tubulin (green), and DNA (blue); imaged by confocal microscopy; and scored for spindle stage. In all, 94% of uninjected eggs had normal MII spindles, whereas 73% of MO-injected oocytes had chromatin masses and partial retention of midbody microtubules. (Refer also to Supplemental Fig. S2.) Wild-type Emi2 cRNA rescued MII spindle formation in 73% of oocytes, whereas ZBR-mutant Emi2 cRNA rescued MII formation in only 41%. Compiled data from three independent experiments are shown; the total number of oocytes scored for each treatment group is shown below the graph in A. Projections of confocal Z stacks are shown for MO injected (B), Emi2 cRNA rescue (C), and Emi2-C573A cRNA rescue (D). Bar = 20 μm.
Previously, knockdown of EMI2 during oocyte maturation has been described using both MO [24] and siRNA [29, 31] approaches; however, those groups did not report an impact of EMI2 depletion on first polar body size. We found that MO-injected oocytes frequently divided to produce large first polar bodies, with up to 22% of cells having a first polar body diameter greater than 50% of the oocyte diameter, depending on the experiment. In addition to failure of MII spindle formation, disruption of asymmetric division as well as slight acceleration of meiosis I are also characteristics of zinc-insufficient oocytes [14, 16], consistent with the hypothesis that zinc insufficiency interferes with EMI2 function. However, some EMI2 depleted oocytes do go on to produce MII spindles and divide to produce second polar bodies (Supplemental Fig. S2) [31]. Because MII spindles are never observed in TPEN-treated oocytes, this suggests either that some endogenous EMI2 expression may persist in MO-injected oocytes, allowing brief MII establishment, or that zinc insufficiency could also impact other pathways involved in MII establishment.
TPEN Interferes with the Ability of EMI2 to Induce Metaphase Arrest
Injection of Emi2 cRNA into GV oocytes before IVM results in MI arrest [24, 29]. Because of the apparent overactivity of APC/C following the first meiotic division of zinc-insufficient oocytes and the requirement for zinc binding to allow proper EMI2 function, we hypothesized that zinc insufficiency interferes with the ability of EMI2 to support the MI-MII transition. We therefore sought to test whether zinc insufficiency could also impair the ability of EMI2 to induce artificial MI arrest when expressed prematurely. GV oocytes were injected with Emi2 cRNA, held in IBMX-containing medium for 2–3 h to allow EMI2 overexpression, and then transferred to IVM medium with or without 10 μM TPEN for 14 h. Consistent with previous reports, 100% of the Emi2 cRNA-injected oocytes matured in control IVM medium arrested at MI. However, only 44% of the Emi2 cRNA-injected oocytes arrested at MI when cultured in TPEN-containing medium (Fig. 4), demonstrating that zinc insufficiency accomplished by TPEN treatment can interfere with the ability of EMI2 to induce metaphase arrest and lending support to our hypothesis.
FIG. 4. .

TPEN inhibits the ability of EMI2 to maintain metaphase arrest in oocytes. A) Cumulus-denuded GV oocytes were injected with Emi2 cRNA, held in IBMX for 2–3 h, and transferred to IVM medium for 14 h using control medium or medium containing 10 μM TPEN. B–D) Oocytes were fixed; stained for actin (red), tubulin (green), and DNA (blue); imaged by confocal microscopy; and scored for spindle stage. In all, 100% of Emi2 cRNA-injected oocytes cultured in control medium arrested at MI, whereas EMI2 expression caused MI arrest in only 44% of oocytes cultured in TPEN-containing medium. This experiment was repeated four times; data from one representative experiment are shown. Projections of confocal Z stacks are shown for Emi2 cRNA-injected oocytes cultured in TPEN containing medium arrested at MI (B), TI (C), and MII (D). Bar = 20 μm.
MPF Activity Declines after TPEN Treatment of MII Eggs
In addition to its role during the MI-MII transition, EMI2 has been identified as a critical component of the CSF, which is necessary for the maintenance of MII arrest in vertebrate eggs [17, 28, 31, 32]. EMI2 inhibits the APC/C during MII arrest but is degraded rapidly upon egg activation, thus releasing inhibition of the APC/C and promoting CCNB1 degradation and meiotic progression [24, 33]. Recent studies have also drawn attention to the role of zinc at MII exit. We recently described a series of zinc exocytosis events (zinc sparks) occurring at fertilization, establishing zinc loss as a hallmark of egg activation [13]. Additionally, sequestration of zinc using TPEN has been shown to induce egg activation and embryo development [13, 15]. Histone H1 and MBP dual kinase assays were performed on individual in vivo-ovulated eggs cultured for 1–8 h in KSOM or in KSOM containing 10 μM TPEN to measure kinase activities of MPF and MAPK, respectively. We found MPF activity is decreased in TPEN-treated eggs, relative to controls, and this reduction becomes statistically significant after 4 h (Fig. 5A), consistent with the previously reported decrease in CCNB1 levels in TPEN-treated eggs [15]. MAPK activity in TPEN-treated eggs remains similar to the level in controls until 6 h after TPEN addition, when MAPK activity becomes significantly lower (Fig. 5B). This is consistent with the timing of pronuclear formation in TPEN-treated eggs [13]. Thus, the kinase activities of MPF and MAPK decrease following zinc sequestration by TPEN, mirroring changes observed in physiological egg activation induced by fertilization.
FIG. 5. .

MPF and MAPK activity decline after TPEN treatment of MII eggs. In vivo-ovulated MII eggs were cultured in KSOM medium with or without 10 μM TPEN for 1–8 h. Graphs of histone H1 kinase activity (A) and MBP kinase activity (B) show densitometric analysis for four to seven individual eggs per data point. Error bars represent the SEM, and asterisks indicate significant differences according to Student t-test comparing TPEN-treated and control groups at individual time points (P < 0.05).
Increasing Intracellular Zinc in MII Eggs Prevents Activation
Egg activation by zinc sequestration demonstrates that decreasing intracellular zinc availability is sufficient to cause meiotic resumption, and the occurrence of zinc sparks at the time of egg activation shows that this is reduction in zinc is also biologically relevant [13, 15]. To further assess the role of zinc in the events of egg activation, we sought to determine whether this decrease in available zinc is also necessary for egg activation. MII eggs were treated with the ionophore ZnPT for 5 min to increase intracellular zinc, as corroborated by increases in both dithizone and FluoZin-3 AM staining (Fig. 6, A–D). Following a 10- to 15-min recovery period, eggs were then treated with an activating agent, either SrCl2 or TPEN. Control MII eggs activated normally, with both SrCl2 and TPEN inducing second polar body formation and formation of pronuclei (PN) or PN-like structures by 6 h postactivation (Fig. 6, E and F). Eggs pretreated with ZnPT formed second polar bodies and PN-like structures when incubated subsequently with TPEN but did not show signs of activation after SrCl2 treatment (Fig. 6, E and F). ZnPT treatment caused spindle abnormalities in unactivated eggs and eggs treated with SrCl2 following 6 h of culture, including severely elongated spindle microtubules, presence of astral microtubules, and scattering of chromatin (Fig. 6F). However, chromosomes remained condensed in these eggs, with no indication of cell cycle progression or interphase entry, and this spindle disorganization did not preclude activation by TPEN (which requires an intact spindle [15, 34]), indicating that this disorganization likely occurred upon extended culture and was not the cause of failed activation by SrCl2. This result also corroborates our previous results showing that elevation of the zinc quota following egg activation causes reformation of a metaphase-like state [13]. The inhibition of egg activation by increased intracellular zinc content and the ability of zinc sequestration to cause activation support the conclusion that a reduction in the availability of zinc is both necessary and sufficient to activate embryonic development.
FIG. 6. .

Increasing intracellular zinc in MII eggs prevents activation by SrCl2 but not by TPEN. In vivo-ovulated MII eggs were treated with 10 μM ZnPT for 5 min to increase intracellular zinc followed by activation with either 10 mM SrCl2 or 10 μM TPEN. A) To demonstrate the intracellular zinc increase, MII-stage eggs were loaded with FluoZin-3 AM for 60 min before ZnPT treatment followed by live imaging by confocal microscopy. B) FluoZin-3 AM-loaded eggs not treated with ZnPT and imaged with identical settings are shown. C and D) As an additional measure, eggs activated using SrCl2 were treated for 5 min with (C) or without (D) ZnPT 2 h after activation, stained with the colorimetric zinc stain dithizone, and imaged by bright-field microscopy. ZnPT dramatically increased staining in both cases. Bar = 50 μm. E and F) By 6 h post-SrCl2 or TPEN treatment, most control oocytes had formed second polar bodies and PN or PN-like structures, but only TPEN caused activation in ZnPT pretreated eggs. Compiled data from three independent experiments are shown; n signifies the total number of oocytes scored for each treatment group. Representative bright-field images and three-dimensional projections of confocal Z stacks with actin (red), tubulin (green), and DAPI (blue) are shown for each treatment group below the graph. Eggs treated with ZnPT displayed varying degrees of MII-spindle disorganization, ranging from elongated spindles with astral microtubules to scattered chromosomes with sparse tubulin staining. Bar = 20 μm.
DISCUSSION
During the final stages of oocyte development, the level of total cellular zinc increases by more than 50% [14]. Preventing this increase during the 12-h period of oocyte maturation results in premature meiotic arrest at TI, failure to increase CCNB1 levels, and failure to establish MII; thus, a tremendous increase in zinc content over a short period of time is required for proper meiotic progression [14, 16]. In the present study, we show that zinc insufficiency initiated at the end of MI is sufficient to cause meiotic arrest, decreased CCNB1, and reduced MPF activity. Previously, we have shown that oocytes matured in the presence of TPEN for as long as 9 h followed by rescue with endogenous zinc form MII spindles [14]. Combined, these data lead us to conclude that the critical window for zinc action in allowing meiotic progression is at the MI-MII transition. Furthermore, we show that increasing CCNB1 activity, either via proteasome inhibition to limit degradation or by expression of nondegradable CCNB1, restores MPF activity and partially rescues MII spindle formation in zinc-insufficient oocytes. Restoration of meiotic progression indicates that the major impact of zinc insufficiency on meiotic cell cycle is due to perturbed regulation of CCNB1. Because continued CCNB1 degradation could be an effect of overactive APC/C and the APC/C inhibitor EMI2 is a zinc-binding protein required for successful MI-MII transition, we hypothesized that the increase in total cellular zinc directly modulates EMI2 activity to initiate MII entry and arrest and that zinc insufficiency disrupts this effect.
While restoration of CCNB1 dynamics in zinc-insufficient oocytes resulted in MII spindle formation in many cases, these spindles were often disorganized, and failed cytokinesis was frequently observed. These effects are likely due, at least in part, to difficulty in pinpointing rescues to the precise time of the MI-MII transition. In addition, rescue of the zinc insufficient phenotype by non-degradable CCNB1 was less complete than rescue by proteasome inhibition. This may indicate that other APC/C substrates, such as securin, are being inappropriately targeted for degradation, contributing to the phenotype of zinc insufficiency. Failure of CCNB1 to fully rescue the zinc insufficiency phenotype may indicate other effects of inappropriate APC/C activity, or could reflect additional pathways being impacted by zinc insufficiency. The role of zinc in these and other pathways is currently under investigation.
EMI2 is a critical component of the CSF that maintains MII arrest until fertilization [28, 31, 32]. EMI2 is required for MI-MII transition [24, 26, 27] and is degraded rapidly upon fertilization, allowing APC/C activation and MII exit [24, 35–37]. Much of the work unraveling pathways regulating emi2 function has been performed in Xenopus oocytes, and though portions of the pathways mediating CSF arrest are conserved in the mouse [24, 31], important differences have also been reported [29, 38]. While mos-mapk pathway signaling is known to impact emi2 stability via p90rsk in Xenopus sp. [39, 40], mouse oocytes lacking any p90rsk isoform are still able to arrest at MII [41], indicating that an alternate pathway may be involved in mammalian oocytes. In addition, emi2 exhibits different localization and cannot substitute for murine EMI2 in mouse oocytes [29], and the camk2- and plk1-mediated pathway that targets emi2 for degradation upon fertilization in Xenopus sp. [35–37] has yet to be clearly demonstrated in a mammalian system. We propose that precise modulation of intracellular zinc in mammalian oocytes may represent an additional layer of regulation of MII arrest (Fig. 7), acting through the known CSF component, EMI2.
FIG. 7. .
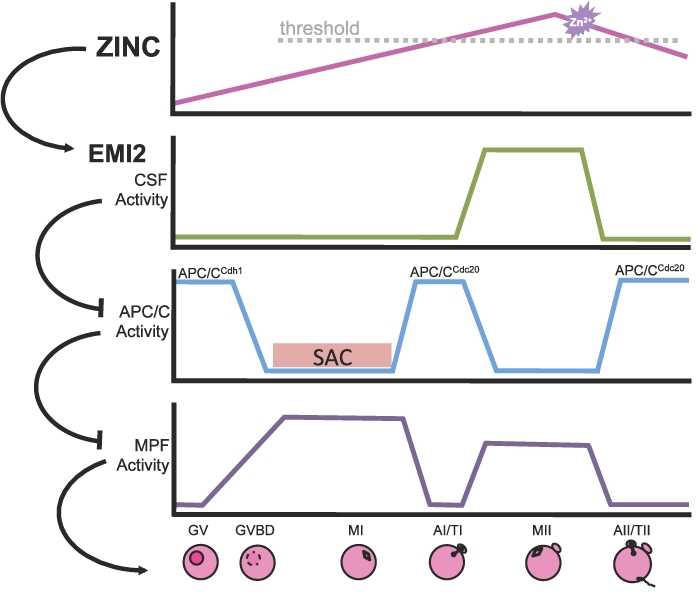
A proposed model for the addition of zinc to the pathways controlling the dynamics of oocyte maturation in which zinc acts as a switch to regulate EMI2 during MII establishment, maintenance, and arrest. The top panel shows approximate relative zinc levels in GV oocytes, MII eggs, and early embryos as determined by x-ray fluorescence microscopy [14]. Subsequent panels represent dynamics of important cellular activities over the course of oocyte maturation and fertilization. In this model, when intracellular zinc is above a threshold level, the APC/C inhibitor EMI2 becomes activated, leading to CSF-mediated MII arrest. After activation, the zinc sparks (indicated by the purple starburst) cause a reduction in cellular zinc and could lead to reduced EMI2 activity upon fertilization, helping to drive the events of egg activation. AI/TI, anaphase I/telophase I; AII/TII, anaphase II/telophase II; GVBD, germinal vesicle breakdown; SAC, spindle assembly checkpoint.
EMI2 contains a ZBR in its C-terminus that is highly conserved among vertebrate species [28, 29]. A functional ZBR has been shown to be required for APC/C inhibitory activity of Xenopus emi2 in cell extracts and in vitro [28], and mutation of putative zinc-binding residues in murine EMI2 reduces its ability to induce artificial arrest at MI [29] or to support MII entry and arrest (present study). We also show in the present study that zinc insufficiency inhibits the ability of exogenously expressed EMI2 to cause MI arrest and that increasing cellular zinc abundance using ZnPT prevents egg activation by SrCl2. These data, along with recent reports of egg activation by zinc sequestration [13, 15] as well as our recent report of a physiological pathway for lowering cellular zinc at the time of fertilization via the zinc sparks [13], establish the reduction in intracellular zinc availability as being both necessary and sufficient to cause activation of the MII-arrested egg. In essence, zinc and EMI2 appear to play a concerted role during two independent stages of oocyte meiotic progression. This idea is further corroborated by similarities between the phenotype of EMI2 knockdown and that of zinc insufficiency during meiotic maturation, as well as the phenotype of EMI2 mutations that alter zinc-binding residues critical for EMI2 function [29]. These results lead us to propose a model in which zinc dynamics control EMI2 activity by altering the metal occupancy of its ZBR. In this model, zinc is a component of the CSF itself. In fact, zinc meets the three criteria described for the CSF [17, 18, 33, 42, 43]—namely, 1) zinc increases over the course of oocyte maturation, 2) zinc is present and necessary during MII arrest, and 3) zinc is lost rapidly at the time of fertilization. Together, these data demonstrate that zinc has a critical role in the ability of EMI2 to support MII and help substantiate a model in which zinc dynamics may act through EMI2 to regulate meiotic progression (Fig. 7). Understanding the biochemistry of mammalian EMI2 and the impact of zinc fluxes could also provide insights regarding differences in EMI2 regulation between mammals and lower vertebrate species.
Utilization of a zinc-responsive system could help oocytes achieve a robust, yet sensitive, system for controlling meiotic progression by using pathways that parallel the effects of the well-characterized calcium oscillations occurring at egg activation, mediated through CaMK2 and other molecules. This model also explains the potential function of the recently described astonishing phenomenon of the zinc sparks [13]. We propose that reduction in cellular zinc achieved through the zinc sparks could ensure rapid and efficient inactivation of EMI2 as a parallel zinc-driven action working in concert with calcium-mediated degradation of EMI2. It remains unclear how fluxes in the total intracellular zinc pool are communicated into changes in zinc occupancy of specific zinc-binding proteins. For example, metallochaperone factors might append the inorganic ion to the target as kinases do with phosphate, or the direct release and uptake of labile and diffusible zinc could be involved. Thus, our model of the zinc-dependent regulation of EMI2 leads to several provocative directions for understanding metalloregulatory switching processes [44].
The regulatory roles of zinc fluxes in mammalian oocyte meiosis have only very recently been recognized. Here, we have presented evidence that the fluxes in intracellular zinc over the course of oocyte meiosis directly mirror the actions of the known CSF component EMI2, which itself requires zinc for proper function. The present study provides compelling support for a model in which zinc dynamics serve as a switch to activate EMI2 during the establishment and maintenance of MII arrest and help to inactivate EMI2 upon egg activation. This model represents a novel cellular function for zinc—as a signal controlling cell cycle progression in a transcription-independent manner—and provides a unified explanation for the physiological changes in zinc over oocyte maturation and egg activation as well as the effects of pharmacological zinc sequestration. Through its key role in the establishment, maintenance, and release of MII arrest, we conclude that zinc is itself a component of the CSF.
Supplementary Material
ACKNOWLEDGMENT
We thank Karen Schindler and Richard Schultz for providing the cRNA expression constructs pRN3-CCNB1(Δ90)-EGFP and pIVT [22, 23]. We also gratefully acknowledge Dragan Mackovic, Sarah Kiesewetter, and Jennifer Jozefik for providing animal care and technical assistance.
Footnotes
Supported by grants from the National Institutes of Health ( P01 HD021921; T32 HD07068) and by the W.M. Keck Foundation Medical Research Award.
REFERENCES
- Outten CE, O'Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 2001; 292: 2488 2492 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003; 4: 517 529 [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985; 260: 3440 3450 [PubMed] [Google Scholar]
- Poenie M, Alderton J, Tsien RY, Steinhardt RA. Changes of free calcium levels with stages of the cell division cycle. Nature 1985; 315: 147 149 [DOI] [PubMed] [Google Scholar]
- Krezel A, Maret W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem 2006; 11: 1049 1062 [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Sakata-Sogawa K, Hasegawa A, Suzuki T, Kabu K, Sato E, Kurosaki T, Yamashita S, Tokunaga M, Nishida K, Hirano T. Zinc is a novel intracellular second messenger. J Cell Biol 2007; 177: 637 645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Dittmer PJ, Park JG, Jansen KB, Palmer AE. Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci U S A 2011; 108: 7351 7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducibella T, Fissore R. The roles of Ca2+, downstream protein kinases, and oscillatory signaling in regulating fertilization and the activation of development. Dev Biol 2008; 315: 257 279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser LR. Strontium supports capacitation and the acrosome reaction in mouse sperm and rapidly activates mouse eggs. Gamete Res 1987; 18: 363 374 [DOI] [PubMed] [Google Scholar]
- Steinhardt RA, Epel D. Activation of sea-urchin eggs by a calcium ionophore. Proc Natl Acad Sci U S A 1974; 71: 1915 1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt RA, Epel D, Carroll EJ, Jr, Yanagimachi R. Is calcium ionophore a universal activator for unfertilized eggs Nature 1974; 252: 41 43 [DOI] [PubMed] [Google Scholar]
- Zhang D, Pan L, Yang LH, He XK, Huang XY, Sun FZ. Strontium promotes calcium oscillations in mouse meiotic oocytes and early embryos through InsP3 receptors, and requires activation of phospholipase and the synergistic action of InsP3. Hum Reprod 2005; 20: 3053 3061 [DOI] [PubMed] [Google Scholar]
- Kim AM, Bernhardt ML, Kong BY, Ahn RW, Vogt S, Woodruff TK, O'Halloran TV. Zinc sparks are triggered by fertilization and facilitate cell cycle resumption in mammalian eggs. ACS Chem Biol 2011; 6: 716 723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AM, Vogt S, O'Halloran TV, Woodruff TK. Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nat Chem Biol 2010; 6: 674 681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Yoshida N, Suzuki E, Okuda E, Perry AC. Full-term mouse development by abolishing Zn2+-dependent metaphase II arrest without Ca2+ release. Development 2010; 137: 2659 2669 [DOI] [PubMed] [Google Scholar]
- Bernhardt ML, Kim AM, O'Halloran TV, Woodruff TK. Zinc requirement during meiosis I-meiosis II transition in mouse oocytes is independent of the MOS-MAPK pathway. Biol Reprod 2010; 84: 526 536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool 1971; 177: 129 145 [DOI] [PubMed] [Google Scholar]
- Tunquist BJ, Maller JL. Under arrest: cytostatic factor (CSF)-mediated metaphase arrest in vertebrate eggs. Genes Dev 2003; 17: 683 710 [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680 685 [DOI] [PubMed] [Google Scholar]
- Ibanez E, Sanfins A, Combelles CM, Overstrom EW, Albertini DF. Genetic strain variations in the metaphase-II phenotype of mouse oocytes matured in vivo or in vitro. Reproduction 2005; 130: 845 855 [DOI] [PubMed] [Google Scholar]
- Svoboda P, Stein P, Hayashi H, Schultz RM. Selective reduction of dormant maternal mRNAs in mouse oocytes by RNA interference. Development 2000; 127: 4147 4156 [DOI] [PubMed] [Google Scholar]
- Igarashi H, Knott JG, Schultz RM, Williams CJ. Alterations of PLCbeta1 in mouse eggs change calcium oscillatory behavior following fertilization. Dev Biol 2007; 312: 321 330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler K, Schultz RM. CDC14B acts through FZR1 (CDH1) to prevent meiotic maturation of mouse oocytes. Biol Reprod 2009; 80: 795 803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madgwick S, Hansen DV, Levasseur M, Jackson PK, Jones KT. Mouse Emi2 is required to enter meiosis II by reestablishing cyclin B1 during interkinesis. J Cell Biol 2006; 174: 791 801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madgwick S, Nixon VL, Chang HY, Herbert M, Levasseur M, Jones KT. Maintenance of sister chromatid attachment in mouse eggs through maturation-promoting factor activity. Dev Biol 2004; 275: 68 81 [DOI] [PubMed] [Google Scholar]
- Liu J, Grimison B, Lewellyn AL, Maller JL. The anaphase-promoting complex/cyclosome inhibitor Emi2 is essential for meiotic but not mitotic cell cycles. J Biol Chem 2006; 281: 34736 34741 [DOI] [PubMed] [Google Scholar]
- Ohe M, Inoue D, Kanemori Y, Sagata N. Erp1/Emi2 is essential for the meiosis I to meiosis II transition in Xenopus oocytes. Dev Biol 2007; 303: 157 164 [DOI] [PubMed] [Google Scholar]
- Schmidt A, Duncan PI, Rauh NR, Sauer G, Fry AM, Nigg EA, Mayer TU. Xenopus polo-like kinase Plx1 regulates XErp1, a novel inhibitor of APC/C activity. Genes Dev 2005; 19: 502 513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Suzuki E, Yoshida N, Kubo A, Li H, Okuda E, Amanai M, Perry AC. Mouse Emi2 as a distinctive regulatory hub in second meiotic metaphase. Development 2010; 137: 3281 3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurumi C, Hoffmann S, Geley S, Graeser R, Polanski Z. The spindle assembly checkpoint is not essential for CSF arrest of mouse oocytes. J Cell Biol 2004; 167: 1037 1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji S, Yoshida N, Amanai M, Ohgishi M, Fukui T, Fujimoto S, Nakano Y, Kajikawa E, Perry AC. Mammalian Emi2 mediates cytostatic arrest and transduces the signal for meiotic exit via Cdc20. EMBO J 2006; 25: 834 845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung JJ, Hansen DV, Ban KH, Loktev AV, Summers MK, Adler JR, III, Jackson PK. A role for the anaphase-promoting complex inhibitor Emi2/XErp1, a homolog of early mitotic inhibitor 1, in cytostatic factor arrest of Xenopus eggs. Proc Natl Acad Sci U S A 2005; 102: 4318 4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Kornbluth S. Across the meiotic divide—CSF activity in the post-Emi2/XErp1 era. J Cell Sci 2008; 121: 3509 3514 [DOI] [PubMed] [Google Scholar]
- Kubiak JZ, Weber M, de Pennart H, Winston NJ, Maro B. The metaphase II arrest in mouse oocytes is controlled through microtubule-dependent destruction of cyclin B in the presence of CSF. EMBO J 1993; 12: 3773 3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Tung JJ, Jackson PK. CaMKII and polo-like kinase 1 sequentially phosphorylate the cytostatic factor Emi2/XErp1 to trigger its destruction and meiotic exit. Proc Natl Acad Sci U S A 2006; 103: 608 613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Maller JL. Calcium elevation at fertilization coordinates phosphorylation of XErp1/Emi2 by Plx1 and CaMK II to release metaphase arrest by cytostatic factor. Curr Biol 2005; 15: 1458 1468 [DOI] [PubMed] [Google Scholar]
- Rauh NR, Schmidt A, Bormann J, Nigg EA, Mayer TU. Calcium triggers exit from meiosis II by targeting the APC/C inhibitor XErp1 for degradation. Nature 2005; 437: 1048 1052 [DOI] [PubMed] [Google Scholar]
- Perry AC, Verlhac MH. Second meiotic arrest and exit in frogs and mice. EMBO Rep 2008; 9: 246 251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Ohe M, Kanemori Y, Nobui T, Sagata N. A direct link of the Mos-MAPK pathway to Erp1/Emi2 in meiotic arrest of Xenopus laevis eggs. Nature 2007; 446: 1100 1104 [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Ohsumi K, Kishimoto T. Phosphorylation of Erp1 by p90rsk is required for cytostatic factor arrest in Xenopus laevis eggs. Nature 2007; 446: 1096 1099 [DOI] [PubMed] [Google Scholar]
- Dumont J, Umbhauer M, Rassinier P, Hanauer A, Verlhac MH. p90Rsk is not involved in cytostatic factor arrest in mouse oocytes. J Cell Biol 2005; 169: 227 231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui Y. From oocyte maturation to the in vitro cell cycle: the history of discoveries of maturation-promoting factor (MPF) and cytostatic factor (CSF). Differentiation 2001; 69: 1 17 [DOI] [PubMed] [Google Scholar]
- Schmidt A, Rauh NR, Nigg EA, Mayer TU. Cytostatic factor: an activity that puts the cell cycle on hold. J Cell Sci 2006; 119: 1213 1218 [DOI] [PubMed] [Google Scholar]
- O'Halloran TV. Transition metals in control of gene expression. Science 1993; 261: 715 725 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.