

ABSTRACT
Fibroblast growth factor (FGF) signaling is essential for the development of the gonadotropin-releasing hormone (GnRH) system. Mice harboring deficiencies in Fgf8 or Fgf receptor 1 (Fgfr1) suffer a significant loss of GnRH neurons, but their reproductive phenotypes have not been examined. This study examined if female mice hypomorphic for Fgf8, Fgfr1, or both (compound hypomorphs) exhibited altered parameters of pubertal onset, estrous cyclicity, and fertility. Further, we examined the number of kisspeptin (KP)-immunoreactive (ir) neurons in the anteroventral periventricular/periventricular nuclei (AVPV/PeV) of these mice to assess if changes in the KP system, which stimulates the GnRH system, could contribute to the reproductive phenotypes. Single hypomorphs (Fgfr1+/− or Fgf8+/−) had normal timing for vaginal opening (VO) but delayed first estrus. However, after achieving the first estrus, they underwent normal expression of estrous cycles. In contrast, the compound hypomorphs underwent early VO and normal first estrus, but had disorganized estrous cycles that subsequently reduced their fertility. KP immunohistochemistry on Postnatal Day 15, 30, and 60 transgenic female mice revealed that female compound hypomorphs had significantly more KP-ir neurons in the AVPV/PeV compared to their wild-type littermates, suggesting increased KP-ir neurons may drive early VO but could not maintain the cyclic changes in GnRH neuronal activity required for female fertility. Overall, these data suggest that Fgf signaling deficiencies differentially alter the parameters of female pubertal onset and cyclicity. Further, these deficiencies led to changes in the AVPV/PeV KP-ir neurons that may have contributed to the accelerated VO in the compound hypomorphs.
Keywords: female infertility, gonadal function, gonadotropin-releasing hormone (GnRH/GnRH receptor), kisspeptin
Single or combined deficiencies in Fgfr1 and Fgf8 differentially alter reproduction and the kisspeptin system in female mice.
INTRODUCTION
Puberty is a critical developmental stage characterized by the initiation and maturation of reproductive capacity. In pubertal female rodents, increased levels of gonadotropin-releasing hormone (GnRH), driven in part by the kisspeptin (KP) system, activate the hypothalamic-pituitary-gonadal (HPG) axis to initiate estrogen-dependent changes such as vaginal opening (VO) and the commencement of estrous cycle [1]. Following pubertal transition, the normal progression of estrous cyclicity continues to require functional GnRH neurons and possibly the KP system. The requirement for the latter was supported by the observations that 1) pubertal ablation of KP-producing neurons lead to the cessation of estrous cycles [2], 2) KP signaling is needed for the tonic release of GnRH during estrous cycle [3], and 3) estrogen receptor α-positive KP neurons are required to maintain estrous cyclicity [4].
It has been shown that fibroblast growth factor (FGF) signaling is critical to the formation of a functional GnRH system in humans [5, 6] and rodents [7]. An interesting convergence between humans and rodents is the dependence of both models on Fgf receptor 1 (Fgfr1) and Fgf8 to support GnRH neuronal development [8–10]. In humans, partial loss-of-function mutations on Fgfr1 or Fgf8 genes lead to highly variable reproductive phenotypes including hypogonadotropic hypogonadism (HH) with or without anosmia, delayed puberty, reversible HH, and hypothalamic amenorrhea [10–16]. The underlying basis of this variability is not clearly understood, but is thought to involve mutations on additional genes [17] or environmental factors [18]. In mice, a hypomorphic Fgfr1 or Fgf8 allele leads to a 30–40% loss of GnRH neurons [8, 10, 19], but the reproductive phenotypes associated with these alleles have not yet been characterized. Such studies would permit a better understanding of specific reproductive defects stemming from Fgf signaling deficiency in a genetically and environmentally defined setting. Further, it is unclear if Fgfr1 and Fgf8 mutations alter the KP system via developmental disruption or postnatal mechanisms. This is an important question, as alterations in the KP system can either exacerbate or compensate for the reproductive deficits.
In this study, we first examine several mouse models deficient in FGF signaling for the disruption of female pubertal onset and reproductive cycle. These models include mice hypomorphic for Fgfr1 [20], Fgf8 [21], and both [19]. The latter is included because compound mutations in both Fgfr1 and Fgf8 have been reported in humans to cause more severe reproductive deficits [10]. Second, we examine these mouse models for the size of the KP neuronal population thought to be responsible for the ovulatory luteinizing hormone (LH) surge, the anteroventral periventricular/periventricular (AVPV/PeV) population [22, 23]. Our results reveal a profound additive effect of FGF signaling deficiencies on female reproductive performance. Further, they suggest FGF signaling deficiencies alter the KP system in a fashion specific to the nature of the FGF deficiency and age.
MATERIALS AND METHODS
Transgenic Animals
Fgf8 (129P2/OlaHsd* CD1) [21] and Fgfr1 (129sv/CD1) [20] hypomorphs were obtained from the Canadian Mutant Mouse Repository (Toronto, ON) and Mouse Regional Resource Centers (Davis, CA), respectively. They were housed in the Department of Integrative Physiology Animal Facility at the University of Colorado, Boulder, under a 12L:12D photoperiod and fed ad libitum. Because of cryptic splice sites in the neomycin resistance gene inserted into the noncoding regions of the Fgf8 or Fgfr1 gene, these hypomorphic mice produce significantly less functional Fgf8 or Fgfr1 transcript. Homozygous Fgf8 hypomorphic mice (Fgf8−/−) have been reported to exhibit about a 54% decrease in the functional Fgf8 mRNA [21], whereas homozygous Fgfr1 hypomorphic mice (Fgfr1−/−) showed a 66–80% decrease in Fgfr1 mRNA [20]. Fgf8 and Fgfr1 transcript levels in the corresponding heterozygous hypomorphs were thought to be midway between wild-type (WT) and homozygotes, as the loss of GnRH neurons observed in these heterozygotes was approximately halfway between WT and homozygotes [8]. Homozygous Fgf8−/− or Fgfr1−/− mice died within 24 h after birth [20, 21]. However, heterozygous Fgf8 (Fgf8+/−) or Fgfr1 (Fgfr1+/−) hypomorphs survived normally with no obvious health problems. For this study, the compound hypomorphs deficient in both Fgfr1 and Fgf8 (Fgfr1+/−/Fgf8+/−) were generated by crossbreeding Fgfr1+/− with Fgf8+/−. In addition to compound hypomorphs, the crossbreeding also generated WT mice and two single hypomorphs (Fgf8+/− or Fgfr1+/−). Thus, compound hypomorphs were analyzed with three other genotypes to test if the effects of Fgf gene deficiency were additive. All comparisons were made among siblings with the same hybrid genetic background to minimize effects due to strain differences. Animals were weighed on Postnatal Day (PND) 30 and PND 60 to gauge their somatic growth and genotyped by PCR of genomic DNA isolated from tail biopsies. All animal procedures complied with protocols approved by the Institutional Animal Care and Use Committee at the University of Colorado.
Examination of VO and Estrous Cycle
VO is an estrogen-dependent phenomenon commonly used to gauge the onset of puberty. VO was checked in female mice of different genotypes from PND 20 until VO was observed. Following VO, daily vaginal smears were performed at 1600 h continuously for 20 days to assess reproductive cyclicity.
Assessment of Fertility in WT and Compound Hypomorphic Mice
Each WT or Fgfr1+/−/Fgf8+/− female was paired with a WT male over 3 mo beginning at PND 45. The cages were checked daily for pup birth. Total number of litters produced by each pair over the 3-mo period and the number of pups within each litter were recorded.
Tissue Collection for KP Immunohistochemistry
Female mice at PND 15, 30, and 60 were lightly anesthetized with isoflurane vapor and killed by decapitation. The brains were dissected and immersion fixed in 5% acrolein for 6 h on ice. Brains were then cryoprotected in 30% sucrose overnight at 4°C and cut at 40-μm thickness using a cryostat. Floating brain sections were stored at 4°C until processed for KP immunohistochemistry (IHC). All PND 60 females used in the study were verified to be in estrus at the time of killing.
KP IHC
Sections were rinsed and incubated in 1% NaBH4 for 15 min to neutralize the residual acrolein, followed by vigorous washing and quenching of endogenous peroxidase activity in 1% H2O2. Sections were then rinsed and incubated with a rabbit anti-KP antibody (1:10 000) generously donated by Dr. Alain Caraty (Université de Tours, Tours, France). This antibody [24] has been used widely for the specific detection of KP in the mouse brain [25, 26]. After 3 days of primary antibody incubation at 4°C, sections were incubated sequentially in a biotinylated donkey anti-rabbit antibody (1:500) and an avidin/biotin-coupled horseradish peroxidase complex (Vector Labs, Burlingame, CA). Sections were reacted with nickel-enhanced diaminobenzidine, mounted on gelatin-coated slides, dehydrated, and coverslipped with Permount (Fisher, Pittsburgh, PA). KP-immunoreactive (ir) neurons were counted in every other section on slides coded to conceal their identity. All sections including the anteroventral periventricular nucleus and periventricular nucleus were scored. Negative controls for the KP IHC included the omission of the primary antibody and preadsorption of the primary antibody with 200 μg/ml of KP (KP10; Genscript, Piscataway, NJ).
Statistical Analysis
Normally distributed samples were analyzed by the one-way ANOVA followed by the Student-Newman-Keuls post hoc test or by the Student t-test. The Kruskal-Wallis test followed by the Dunn test was used when samples failed the homoscedasticity test. Statistical analyses were carried out using Prism or Instat software (GraphPad, La Jolla, CA). Differences were considered significant when P < 0.05. Sample size ranged from 5 to 10 per group for reproductive characterization, from 5 to 14 for mass measurement, and from 4 to 6 for the KP IHC.
RESULTS
FGF Signaling Defects Differentially Impact the Pubertal Onset
To gauge if reductions in GnRH neurons previously seen in these transgenic mice [8, 10, 19] were translated into abnormal pubertal onset, we assessed puberty using two criteria: age of VO and the lag between VO and the first estrus (Fig. 1). Neither of the single hypomorphs, Fgfr1+/− (n = 8) or Fgf8+/− (n = 5), exhibited altered timing of VO compared to WT (n = 6) (Fig. 1A). In contrast, compound hypomorphs (Fgfr1+/−/Fgf8+/−; n = 10) exhibited significantly earlier VO compared to WT or Fgf8+/− (Fig. 1A). Interestingly, although Fgfr1+/− and Fgf8+/− had what appeared to be normal timing for VO, they took longer to reach the first estrus compared to WT (Fig. 1B). This prolonged lag was not observed in the compound hypomorphs (Fig. 1B). The observed differences in pubertal parameters were not the result of altered somatic growth, because no significant differences in the body mass were detected among the genotypes on PND 30 (P = 0.25) or PND 60 (P = 0.81; Table 1).
FIG. 1. .
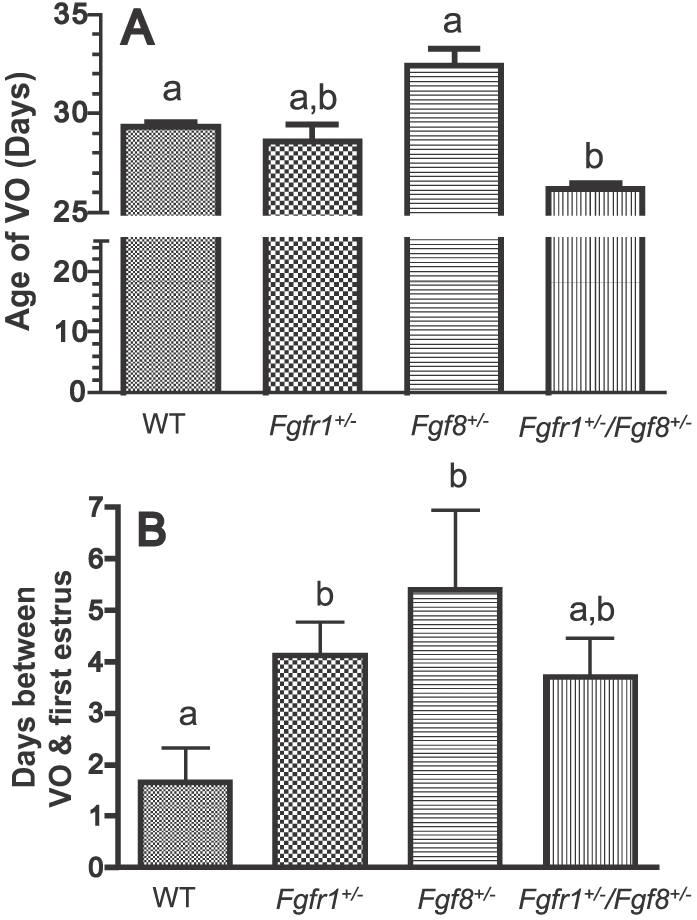
Parameters of pubertal onset in WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− female mice. A) Age of VO. B) Time between VO and first estrus. Different letters represent significant differences (P < 0.05). n = 6, 8, 5, and 10 for WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− mice, respectively.
TABLE 1. .
Body mass (g) of PND 30 and PND 60 WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− hypomorphic female mice.*

No significant differences were observed among genotypes of the same age group. Data are represented as mean ± SEM.
FGF Signaling Defects Differentially Impact Stages of the Estrous Cycle
After VO, female reproductive cyclicity was assessed by continuous vaginal smears for the next 20 days. Compared to WT (n = 6), compound hypomorphs (n = 10) spent a greater fraction of their time during estrous cycle in diestrus, and less in proestrus and metestrus (Fig. 2). In contrast, neither of the single hypomorphs, Fgfr1+/− (n = 8) or Fgf8+/− (n = 5), exhibited altered estrous cycle compared to WT.
FIG. 2. .

Percentage of time spent in diestrus (A), proestrus (B), estrus (C), or metestrus (D) during the estrus cycle of WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− female mice. Data were derived from continuous vaginal smears over a 20-day period. Different letters represent significant differences (P < 0.05). n = 6, 8, 5, and 10 for WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− mice, respectively.
Compound Hypomorphs Exhibited Fewer Complete Estrous Cycles and Reduced Fertility
Because compound hypomorphs showed abnormal progression of estrous cycles, we hypothesized that these mice also underwent fewer complete estrous cycles and produced fewer litters. An estrous cycle was identified as complete only if the four stages occurred in the right sequence within no more than 2 days of each other. The number of complete estrous cycles in Fgfr1+/− (n = 8) and Fgf8+/− (n = 5) was not significantly different from the number in WT (Fig. 3A). As expected, over the 3-mo study period, compound hypomorphs (n = 10) exhibited significantly fewer complete cycles compared to WT (n = 5) (Fig. 3A). Consistent with this observation, the total number of litters produced by compound hypomorphs (n = 5) was reduced by 50% compared to WT (n = 5) (Fig. 3B). The number of pups per litter, however, was not significantly different (P = 0.13) between WT (10.4 ± 2.1 pups; n = 5) and compound hypomorphs (5.6 ± 1.9 pups; n = 5).
FIG. 3. .
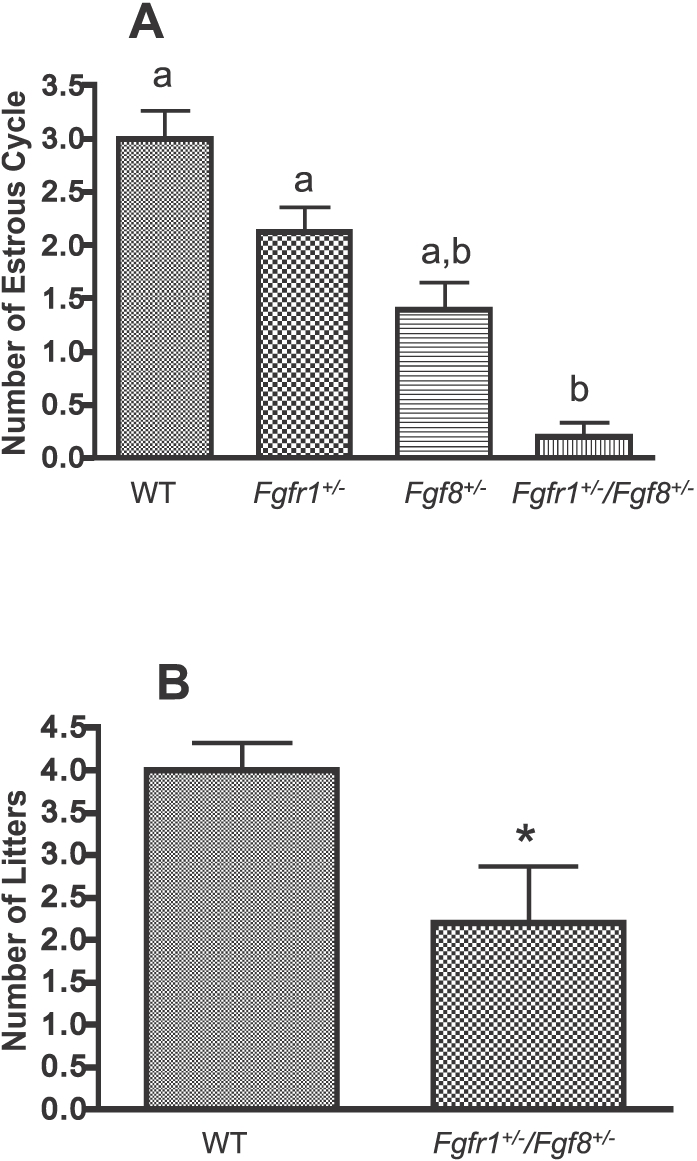
A) Number of complete estrous cycles over a 20-day period in WT (n = 5), Fgfr1+/− (n = 8), Fgf8+/− (n = 5), and Fgfr1+/−/Fgf8+/− (n = 10) female mice. Different letters represent significant differences (P < 0.05). B) Number of litters produced over a 3-mo period in WT (n = 5) and Fgfr1+/−/Fgf8+/− (n = 5) female mice. *Significant difference (P < 0.05).
FGF Signaling Defects Differentially Impact the KP System
All hypomorphic mice, including the single and compound hypomorphs, had significantly reduced GnRH neuronal populations [8, 10, 19]. In particular, the compound hypomorphs showed an exacerbated reduction of GnRH neurons above that of the single hypomorphs [19]. The unexpected early VO of compound hypomorphs prompted us to examine if compensatory mechanisms exist at the level of the KP system to allow them to overcome the loss of GnRH neurons and undergo early VO. All mice (n = 4–6 per genotype), including WT and hypomorphs, underwent marked increases in AVPV/PeV KP-ir neurons from PND 15 to PND 60 (Fig. 4, A–C). In all age groups examined, the compound hypomorphs consistently had more KP-ir neurons compared to WT (Fig. 4, A–C). Fgfr1+/− mice had significantly more KP-ir neurons on PND 15 (Fig. 4A), but this difference disappeared at later ages (Fig. 4, B and C). In contrast, Fgf8+/− mice had significantly fewer KP-ir neurons on PND 30 (Fig. 4B) but not at other ages (Fig. 4, A and C). On PND 30, in the midst of pubertal transition, the differences in the number of AVPV/PeV KP-ir neurons among mice of different genotypes could be clearly visualized (Fig. 4, D–G). Omission of the primary antibody (not shown) or preadsorption of the KP antibody with KP10 (Fig. 4, H and I) abolished the staining.
FIG. 4. .

Total AVPV/PeV KP-ir neurons in WT, Fgfr1+/−, Fgf8+/−, and Fgfr1+/−/Fgf8+/− mice on PND 15 (A), PND 30 (B), and PND 60 (C). Different letters represent significant differences (P < 0.05). n = 4–6 for all age groups. D–G) Representative photomicrographs of KP-ir neurons in PND 30 WT (D), Fgfr1+/;− (E), Fgf8+/− (F), and Fgfr1+/−/Fgf8+/− (G) female mice. H, I) Representative images of adjacent sections incubated with KP antibody that has not (H) or has (I) been preadsorbed with 200 μg/ml KP10. Bar in D applies to D–G; bar in H applies to H and I. Bars = 50 μm.
DISCUSSION
Several key observations emerge from the current study. First, transgenic female mice with compound hypomorphic alleles in both Fgf8+/− and Fgfr1+/− undergo early VO but have disorganized estrous cycles that subsequently reduce their fertility. Second, females with a single hypomorphic allele in either Fgf8 or Fgfr1 suffer delayed first estrus but have normal estrous cycles once estrous cycles are initiated. Lastly, the AVPV/PeV KP system is differentially altered in the hypomorphic mice, which may, in part, account for the varied reproductive phenotypes.
Human studies show a proband with digenic mutations on both Fgf8 and Fgfr1 exhibits absent puberty compared to the proband's father, who harbors only an Fgfr1 mutation and exhibits only delayed puberty [10]. Consistent with this, mice hypomorphic for both Fgf8 and Fgfr1 have the greatest reduction in GnRH neuronal population (∼65% reduction) compared to either of the single hypomorphs [19]. As such, one might predict a significant delay or absence of pubertal onset in the compound hypomorphs. Unexpectedly, these animals undergo early VO and have seemingly normal timing for their first estrus. Also unexpectedly, these mice have more KP-ir neurons in their AVPV/PeV throughout all ages examined, including during the period approximating pubertal onset (PND 30). In female rodents, elevated numbers of AVPV/PeV KP-ir neurons correlate with pubertal onset [25] and the ability to generate the ovulatory LH surge [27]. Thus, the increased number of KP-ir neurons in compound hypomorphs could drive early VO. However, once reproductively mature, compound hypomorphs experience difficulty in producing normal estrous cycles, which is manifested in prolonged diestrus and shortened proestrus and metestrus. This implies that the mice have a short follicular phase with difficult entry into, and termination of, the luteal phase, potentially indicating a dysregulation of LH and FSH secretory patterns [28, 29]. This, in turn, may be the result of abnormal GnRH and/or KP output, or local dysregulation directly at the level of the ovary. The latter is important to keep in mind, as Fgfr1 and Fgf8 are expressed in the adult oocytes and granulosa cells and may mediate local ovarian functions [30, 31]. Overall, although increased KP-ir neurons may drive early VO in the compound hypomorphs, they are unable to rescue defects in estrous cycle and fertility that may have stemmed, in part, from the reduced GnRH neuronal number.
In contrast to the more deleterious reproductive defects seen in compound hypomorphs, Fgf8+/− or Fgfr1+/− mice exhibit only a modest delay in the first estrus, and begin to cycle normally after the first estrus. This suggests a 30–40% reduction in GnRH neurons impacts predominantly the initiation of the first estrus cycle but not the cyclicity once it commences. This is consistent with numerous studies reporting that only a few GnRH neurons are needed to support female puberty and ongoing fertility [32–34]. These results also bear similarities to studies reporting some humans with monogenic mutations on Fgfr1 or Fgf8 experienced only delayed puberty with no further reproductive consequence [10, 15, 16].
At present, mechanisms responsible for the altered KP-ir neuron numbers in single and compound hypomorphs are unclear. Two possible explanations are 1) the AVPV/PeV KP neuronal population is altered developmentally by these mutations and 2) the production of KP is altered indirectly by downstream changes within the HPG axis. We believe both are possible. Supporting the former, the persistent presence of 75–100 additional KP-ir neurons in compound hypomorphs of all ages (Fig. 4, A–C) suggests there may be a developmental surplus of KP-ir neurons in the compound hypomorphs. Fgf-signaling genes, including Fgfr1 and Fgf8, are expressed in the presumptive rodent hypothalamus and thus positioned for the developmental regulation of diverse hypothalamic neurons [35–37]. Supporting this, mice hypomorphic for Fgf8 exhibited significantly reduced neuroendocrine oxytocin neurons in the paraventricular and supraoptic nuclei [38]. Of further interest, mice null for Bax, a proapoptotic gene, exhibit a modest but significant increase in KiSS-1 neurons in the AVPV/PeV, although the sexually dimorphic prevalence of KiSS-1 neurons in females remained unaltered [39]. Currently we have no direct evidence to suggest FGF signaling deficiency attenuates the apoptotic pathway, but it is certainly one mechanism by which the size of KP neuronal population can be enhanced in the compound hypomorphs. Further verification of this notion will involve a detailed examination of FGF signaling in the differentiating AVPV/PeV. On the other hand, the alterations of KP-ir neurons in the single hypomorphs are transient and disappeared on PND 60, suggesting the differences may have arisen from temporary changes in the HPG axis, such as gonadal steroid levels [40], as they matured. Of note, our IHC studies quantified the number of neurons with detectable levels of KP peptide, not the actual number of KP neurons. Because dynamic postnatal changes in KP levels of KP-ir neurons may be responsible for the observed differences in KP-ir neuronal number, future lineage-tracing studies or the use of markers alternative to KP are required to resolve this issue.
Together, the current study shows that reproductive disruption in FGF signaling-deficient female mice is complex and occurs differentially at the level of both pubertal onset and subsequent reproductive cyclicity. Because humans with FGF signaling deficiencies have never been reported to undergo early puberty, one might question the appropriateness of extrapolating data from the rodent model to humans. It has been known for some time that different mechanisms drive pubertal onset in rodents and primates [41]. However, there are also features of the reproductive neuroendocrine systems shared by both rodents and primates, such as the origin of GnRH neurons [42–44], the dependence of GnRH neurons on Fgf signaling for proper development [8, 9], and the reliance on KP signaling for normal puberty [45, 46]. In our study, despite the unexpected finding that the compound hypomorphs undergo early VO, there is still information to be gained with regard to how humans with FGF signaling deficiency could overcome some reproductive deficits through other compensatory mechanisms. According to our observations, the activation of the KP system may be one of the mechanisms. Further, our results highlight the possible involvement of the KP system in the manifestation of the reproductive phenotype in FGF signaling-deficient animals. The variable reproductive deficits in our hypomorphic mice speak to the difficulty in predicting the reproductive outcome based solely on size of the GnRH system.
ACKNOWLEDGMENT
Dr. Alain Caraty (Université de Tours, Tours, France) generously donated the rabbit anti-KP antibody.
Footnotes
Supported by National Institutes of Health grants HD042634 and HD058044 and National Science Foundation grant IOS 0743818.
REFERENCES
- Millar RP, Roseweir AK, Tello JA, Anderson RA, George JT, Morgan K, Pawson AJ. Kisspeptin antagonists: unraveling the role of kisspeptin in reproductive physiology. Brain Res 2010; 1364: 81 89 [DOI] [PubMed] [Google Scholar]
- Mayer C, Boehm U. Female reproductive maturation in the absence of kisspeptin/GPR54 signaling. Nat Neurosci 2011; 14: 704 710 [DOI] [PubMed] [Google Scholar]
- Dungan HM, Gottsch ML, Zeng H, Gragerov A, Bergmann JE, Vassilatis DK, Clifton DK, Steiner RA. The role of kisspeptin-GPR54 signaling in the tonic regulation and surge release of gonadotropin-releasing hormone/luteinizing hormone. J Neurosci 2007; 27: 12088 12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, Levine JE. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc Natl Acad Sci U S A 2010; 107: 22693 22698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF., Jr Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology 2010; 92: 81 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraoui H, Dwyer A, Pitteloud N. Role of fibroblast growth factor (FGF) signaling in the neuroendocrine control of human reproduction. Mol Cell Endocrinol 2011; 346: 37 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PS, Brooks LR, Rochester JR, Kavanaugh SI, Chung WC. Fibroblast growth factor signaling in the developing neuroendocrine hypothalamus. Front Neuroendocrinol 2011; 32: 95 107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through FGF receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology 2008; 149: 4997 5003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pêcheux C, Le Tessier D, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003; 33: 463 465 [DOI] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer AA, Quinton R, Na S, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 2008; 118: 2822 2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, Avbelj M, Dhruvakumar S, Plummer L, Hughes VA, Seminara SB, Boepple PA, et al. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med 2011; 364: 215 225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing A, Elinseekova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A 2006; 103: 6281 6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ND, Seminara SB, Welt CK, Au MG, Plummer L, Hughes VA, Dwyer AA, Martin KA, Quinton R, Mericq V, Merino PM, Gusella JF, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab 2011; 96: E566 E576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF., Jr Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab 2005; 90: 1317 1322 [DOI] [PubMed] [Google Scholar]
- Trarbach EB, Abreu AP, Silveira LF, Garmes HM, Baptista MT, Teles MG, Costa EM, Mohammadi M, Pitteloud N, Mendonca BB, Latronico AC. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J Clin Endocrinol Metab 2010; 95: 3491 3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Meysing A, Quinton R, Acierno JS, Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol 2006; 254–255: 60 69 [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, Li WP, Maccoll G, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 2007; 117: 457 463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab 2011; 22: 249 258 [DOI] [PubMed] [Google Scholar]
- Chung WC, Matthews TA, Tata BK, Tsai PS. Compound deficiencies in fibroblast growth factor signalling components differentially impact the murine gonadotrophin-releasing hormone system. J Neuroendocrinol 2010; 22: 944 950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev 1998; 12: 2332 2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp- mediated recombination. Nat Genet 1998; 18: 136 141 [DOI] [PubMed] [Google Scholar]
- Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of female mouse. Endocrinology 2005; 146: 3686 3692 [DOI] [PubMed] [Google Scholar]
- Smith JT, Popa SM, Clifton DK, Hoffman GE, Steiner RA. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci 2006; 26: 6687 6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini I, Lomet D, Cateau M, Delsol G, Tillet Y, Caraty A. Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor alpha. Neurosci Lett 2006; 401: 225 230 [DOI] [PubMed] [Google Scholar]
- Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 2006; 147: 5817 5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J. d'Anglemont de Tassigny X, Colledge WH, Caraty A, Herbison AE. Distribution of kisspeptin neurones in the adult female mouse brain. J Neuroendocrinol 2009; 21: 673 682 [DOI] [PubMed] [Google Scholar]
- Kauffmann AS. Gonadal and nongonadal regulation of sex differences in hypothalamic Kiss1 neurones. J Neuroendocrinol 2010; 22: 682 691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone NA, Kaiser UB. The biology of gonadotroph regulation. Curr Opin Endocrinol Diabetes Obes 2009; 16: 321 327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R, Webster NJ. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocr J 2009; 56: 729 737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Blasio AM, Vigano P, Cremonesi L, Carniti C, Ferrari M, Ferrari A. Expression of the genes encoding basic fibroblast growth factor and its receptor in human granulosa cells. Mol Cell Endocrinol 1993; 96: R7 11 [DOI] [PubMed] [Google Scholar]
- Valve E, Penttilä TL, Paranko J, Härkönen P. FGF-8 is expressed during specific phases of rodent oocyte and spermatogonium development. Biochem Biophys Res Commun 1997; 232: 173 177 [DOI] [PubMed] [Google Scholar]
- Gibson MJ, Charlton HM, Perlow MJ, Zimmerman EA, Davies TF, Krieger DT. Preoptic area brain grafts in hypogonadal (hpg) female mice abolish effects of congenital hypothalamic gonadotropin-releasing hormone (GnRH) deficiency. Endocrinology 1984; 114: 1938 1940 [DOI] [PubMed] [Google Scholar]
- Gibson MJ, Krieger DT, Charlton HM, Zimmerman EA, Silverman AJ, Perlow MJ. Mating and pregnancy can occur in genetically hypogonadal mice with preoptic area brain grafts. Science 1984; 225: 949 951 [DOI] [PubMed] [Google Scholar]
- Herbison AE, Porteous R, Pape JR, Mora JM, Hurst PR. Gonadotropin-releasing hormone neuron requirements for puberty, ovulation, and fertility. Endocrinology 2008; 149: 597 604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfralpha, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci 2003; 25: 83 95 [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 1991; 113: 1419 1434 [DOI] [PubMed] [Google Scholar]
- Wanaka A, Johnson EM, Jr, Milbrandt J. Localization of FGF receptor mRNA in the adult rat central nervous system by in situ hybridization. Neuron 1990; 5: 267 281 [DOI] [PubMed] [Google Scholar]
- Brooks LR, Chung WC, Tsai PS. Abnormal hypothalamic oxytocin system in fibroblast growth factor 8-deficient mice. Endocrine 2010; 38: 174 180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semaan SJ, Murray EK, Poling MC, Dhamija S, Forger NG, Kauffman AS. BAX-dependent and BAX-independent regulation of Kiss1 neuron development in mice. Endocrinology 2010; 151: 5807 5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM, Tena-Sempere M. Kisspeptins and the neuroendocrine control of reproduction. Front Biosci (Schol Ed) 2011; 3: 267 275 [DOI] [PubMed] [Google Scholar]
- Plant TM. Hypothalamic control of the pituitary-gonadal axis in higher primates: key advances over the last two decades. J Neuroendocrinol 2008; 20: 719 726 [DOI] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature 1989; 338: 161 164 [DOI] [PubMed] [Google Scholar]
- Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci U S A 1989; 86: 8132 8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res 1989; 6: 311 326 [DOI] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, et al. The GPR54 gene as a regulator of puberty. N Engl J Med 2003; 349: 1614 1627 [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A 2003; 100: 10972 10976 [DOI] [PMC free article] [PubMed] [Google Scholar]