

Abstract
An efficient formal synthesis of (−)-englerin A (1) is reported. The target molecule is a recently isolated guaiane sesquiterpene that possesses highly potent and selective activity against renal cancer cell lines. The developed strategy proceeds in an enantioselective manner by constructing the BC ring system of 1 via a Rh(II)-catalyzed [4+3] cycloaddition and subsequently attaching the A ring via an intramolecular aldol condensation reaction. As such, this strategy allows the synthesis of truncated englerins. Evaluation of these analogues in A498 renal cancer cell line suggests that the A ring of englerin is crucial to its antiproliferative activity. Moreover, evaluation of these analogues led to the identification of potent growth inhibitors of CEM cells with GI50 values ranging from 1–3 μM.
Keywords: total synthesis, natural product, cycloaddition, renal cancer, leukemia
Introduction
The need to identify new chemical motifs as potential drug leads has spurred the screening of plant extracts used in traditional African, Ayurvedic (Indian) and Chinese medicines.[i] In particular, South Africa has a remarkable botanical diversity with over 30,000 flowering species, from which more than 3,000 are used for medicinal purposes throughout the country.[ii] Among them, plants of the genus Phyllanthus (Euphorbiaceae) are widely distributed and have long been used in African folk medicine to treat kidney and urinary tract infections.[iii] With this in mind, the Beutler laboratory has been screening extracts of the Tanzanian plant Plyllanthus engleri against renal cell carcinoma (RCC) and has recently reported the isolation of two novel bioactive sesquiterpenes, named englerin A (1) and englerin B (2) (Figure 1).[iv]
Figure 1.
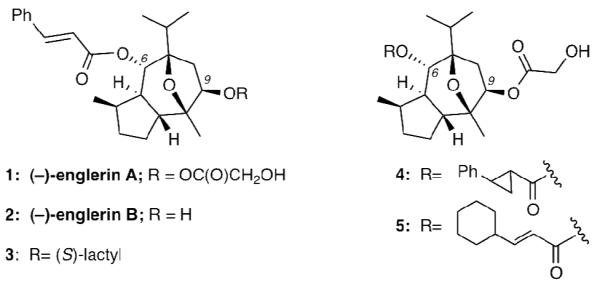
Representative structures of natural and synthetic englerins
Preliminary biological investigations[4] have shown that 1 possesses very potent growth inhibitory activity (GI50 = 1–87 nM) against RCC with approximately 1,000-fold tissue selectivity as compared to other carcinomas. These findings are of particular significance since RCC: (a) is among one of the ten leading cancer types in the US;[v] (b) is characterized by a lack of early warning signs, has diverse clinical manifestations and shows resistance to radiation;[vi] and (c) cannot be effectively treated with current chemotherapeutic agents, leaving surgical procedures as the only treatment option.[vii]
Biogenetically, the englerins belong to the family of guaiane natural products in which a common bicyclic sesquiterpene skeleton has undergone a sequence of oxygenations and oxo-cyclizations.[viii]
Further decoration at the periphery of this core produces the structures of 1 and 2. Due to the combination of uncommon structural architecture, potent and selective cytotoxicity against RCC and promising pharmacology, the englerins have received the attention of the chemical community. Since their isolation in 2009, five total syntheses[ix] and many strategies and studies[x] have been reported. These studies also led to the identification of synthetic englerin analogues, such as 3, 4 and 5, products of various esterifications of the common tricyclic core. Several recent reviews nicely summarize the current status of englerins.[xi] Our continuous interest in exploring natural products from ethnomedicine as medicinal leads[xii] has prompted us to design an enantioselective strategy toward englerins.[xiii] In this account we describe in detail the results of our efforts and the biological evaluation of structurally simplified englerin analogues.
Results and Discussion
Retrosynthetic analysis
Our retrosynthetic strategy is shown in Scheme 1. We anticipated that a sequence of reactions including: inversion of the C6 stereocenter, hydrogenation of the C4-C5 alkene (englerin numbering) and selective esterifications at C6 and C9 hydroxyl groups would form englerin (1) from diol 6. Compound 6 could be derived from enone 7 following a C3 deoxygenation and a regioselective hydroboration of the C8–C9 double bond. We theorized that the cyclopentene moiety of 7 could be constructed from an intramolecular aldol condensation, while an α-hydroxylation would produce the C6 hydroxyl moiety. Disconnection along these lines led us to target the construction of bicyclic motif 8 possessing the BC ring scaffold of englerins. We hypothesized that compound 8 could be formed in enantiomerically pure form by exploring a Rh(II)-catalyzed [4+3] cycloaddition between readily available furan 9, representing the C ring of 1, and chiral pantolactone-derived diazoester 10.
Scheme 1.
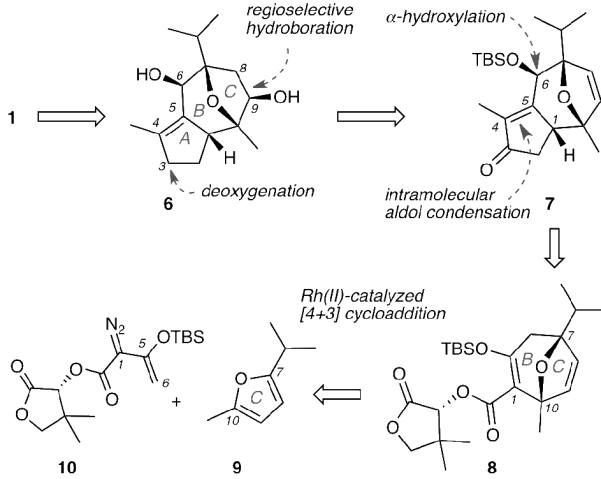
Retrosynthetic analysis of (−)-englerin A (1).
[4+3] Rh-catalyzed cycloadditions
Rhodium-triggered cyclization reactions have been widely applied in synthetic chemistry.[xiv] In 1996, Davies et al. reported an elegant enantioselective Rh(II)-catalyzed [4+3] cycloaddition between furans and chiral diazoesters.[xv] Selection of the appropriate pantolactone enantiomer, as the chiral auxiliary, allows diastereo-control of the produced oxy-bridged adduct. The transfer of chirality, observed in this reaction, can be rationalized by considering that the carbonyl moiety of pantolactone interacts with the Rh-carbenoid as shown in Scheme 2. This interaction blocks one of the two faces of the carbene 10 leading to a selective si-face attack by the 2-methyl-5-isopropyl-furan (9). The stereochemical outcome is consistent with a tandem cyclopropanation/Cope rearrangement (see intermediate [11]). Cyclopropanation of the achiral furan 9 was expected to occur in a regioselective manner at the less substituted and thus less hindered C9-C10 double bond. The potential of this cycloaddition to construct bicyclic structure 8 was of great interest to our strategy, as it provided an efficient, rapid and enantioselective access to the englerin core. Notably, both of starting materials 9[xvi] and 10[15a,xvii] can be readily prepared in more than 50 gram-scale from commercially available materials in 3 steps.
Scheme 2.
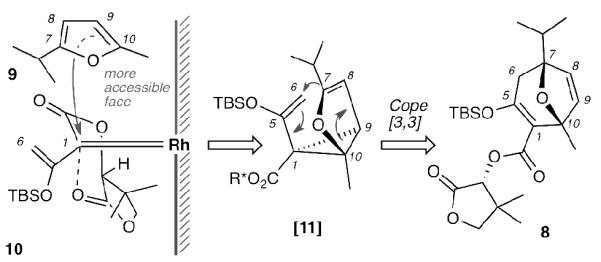
Rationale to stereochemical consequences of the Davies Rh-catalyzed [4+3] cycloaddition.
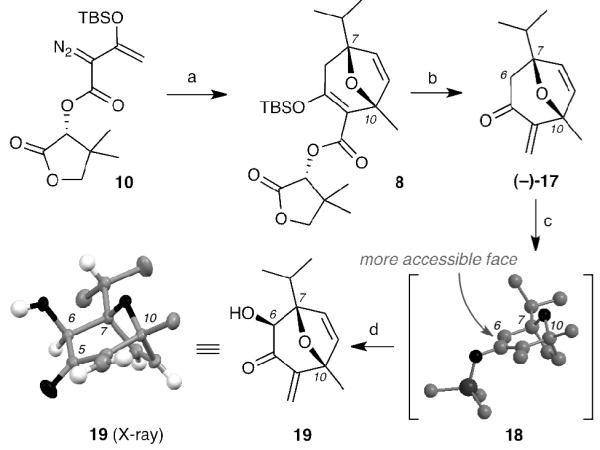
To the best of our knowledge, 2,5-disubstituted furans have not been evaluated as substrates in the Rh(II)-catalyzed [4+3] cycloaddition. With this in mind, we initially evaluated the regioselectivity of this reaction using achiral diazoester 14[xviii] as a model system (Scheme 3). The synthesis of furan 9 is highlighted in Scheme 3. Commercially available 2-methyl furan (12) was treated with n-BuLi and acetone followed by the dehydration (Ac2O/KOAc) to afford alkene 13. A regioselective hydrogenation of 13 proceeded smoothly by controlling the reaction time to 1 hour and the sequential distillation delivered the desired 2,5-disubstituted furan 9. Notably, this 3-step preparation of 9 can be done in more than 50 gram-scale and only one distillation is needed for purification. Refluxing of 9 and diazoester 14 in hexane in the presence of catalytic amounts of rhodium(II) octanoate (2 mol%), afforded oxacyclic product (±)-15 as the only regio-isomer in excellent yields (95%). Cleavage of the ester auxiliary proved to be more challenging than anticipated. In fact, the reported LiAlH4 reductive cleavage led only to decomposition.[15a] However, treatment of ester 15 with DIBAL-H yielded the labile β-hydroxy silyl enol ether 16 that, upon immediate treatment with stoichiometric amounts of BF3•Et2O, underwent rearrangement[xix] to afford exocyclic enone (±)-17 in good yield (81%).
Scheme 3.
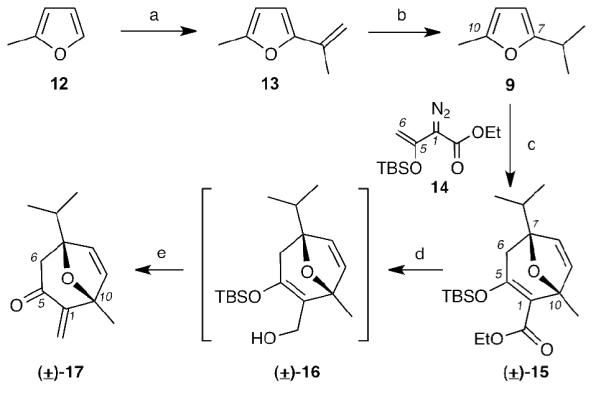
Reagents and conditions: (a) n-BuLi (0.94 equiv), Et2O, −20 °C, reflux; then -20 °C, acetone (1.1 equiv), reflux; then Ac2O (1.5 equiv), KOAc (0.63 equiv), 110 °C; (b) Pd/C (catalytic), H2 (1 bar), 1 h; (c) Rh2(OOct)4 (2 mol%), hexanes, reflux, 95%; (d) DIBAL-H (2.5 equiv), CH2Cl2, −78 °C; (e) BF3•Et2O (1.5 equiv), CH2Cl2, −30°C, 81%.
Encouraged by these results, we synthesized chiral diazoester 10 departing from (R)-pantolactone.[15a,17] The Rh(II)-catalyzed [4+3] cycloaddition of 10 with 9 proceeded efficiently with Rh2(OOct)4 to yield 8 in good yield (90%) albeit in moderate diastereoselectivity (dr 3:1, calculated via 1H-NMR) (Scheme 4). It should be noted that, under the same conditions, use of Rh2(OAc)2 led only to decomposition of the starting materials. Moreover, attempts to increase the d.r. by performing this reaction at lower temperature or by using less catalyst loading led to a significant decrease in the yield without any significant enhancement of diastereoselectivity. Table 1 highlights our efforts to optimize this cycloaddition. The diastereomeric mixture of 8 could be separated via a silica gel column chromatography. Our previously established reductive cleavage conditions were applied to 8 to produce optically active enone (−)-17, although the yield was significantly lowered (59%). This decrease of yield may be due to the presence of the two reactive carbonyl moieties in 8 that require extended reaction times and excess of DIBAL-H (7.5 equiv) as compared to reduction of (±)-15 (2.5 equiv), leading to partial decomposition of the sensitive silyl enol ether moiety.
Scheme 4.
Reagents and conditions: (a) 9 (2.0 equiv), Rh2(OOct)4 (2 mol%), hexanes, reflux, 95% for (±)-16, 90% for (−)-16 (dr 3:1); (b) DIBAL-H (7.5 equiv), CH2Cl2, −78 °C, then BF3•Et2O (1.5 equiv), CH2Cl2, −30 °C, 81% for (±)-17, 59% for (−)-17; (c) LDA (4.6 equiv), TMSCl (4.6 equiv), −78 °C to 0 °C; (d) mCPBA (1.1 equiv), NaHCO3,CH2Cl2, 0 °C, then (COOH)2 (4.6 equiv), 36%; 87% brsm.
Table 1.
Optimization of the key [4+3] cycloaddition reaction between 9 and 10. n.r.= no reaction. ND = not determined
| Furan 9 | Catalyst (mol%) | Temp. | Yield (dr) |
|---|---|---|---|
| 10 equiv | Rh2(OOct)4 (2 mol%) | reflux | 91% (3:1) |
| 2 equiv | Rh2(OOct)4 (2 mol%) | reflux | 90% (3:1) |
| 2 equiv | Rh2(OOct)4 (1 mol%) | reflux | 73% (3:1) |
| 2 equiv | Rh2(OOct)4 (2 mol%) | −78 °C | n.r. |
| 2 equiv | Rh2(OOct)4 (2 mol%) | −40 °C | n.r. |
| 2 equiv | Rh2(OOct)4 (2 mol%) | 0 °C | n.r. |
| 5 equiv | Rh2(OOct)4 (2 mol%) | 30 °C | 31% (ND) |
| 5 equiv | Rh2(OAc)4 (2 mol%) | reflux | n.r. |
The next attempts of installing a hydroxyl group on the C6 carbon of (−)-17 with Davis oxaziridines[xx] proved unsuccessful. However, Rubottom oxidation[xxi] provided the desired hydroxy enone 19 in good yield (36%; 87% brsm), although with the inverse stereochemistry as compared to the englerin C6 hydroxyl moiety. The absolute stereochemistry of hydroxyl enone 19 was established by a single crystal X-ray analysis,[xxii] which simultaneously confirmed the absolute stereochemistry of oxy-bridged ester 8. We were pleased to observe that the Rubottom oxidation proceeded regioselectively at the more electronically rich TMS enol ether without affecting any other alkenes in this molecule. The stereochemical outcome of this reaction was also satisfactory since it proceeded exclusively from the top face of the intermediate TMS-enolate [18] suggesting that this face is less hindered. We predicted that this selectivity would allow us to have complete substrate control during the following steps and invert the C6 stereochemistry at a later stage in our synthesis.
Formation of the A ring via olefin metathesis
The next stage of the synthesis involved constructing the tricyclic core of englerin from compound 19. We envisioned to accomplish this cyclization by the means of a Grubbs ring closing metathesis (RCM)[xxiii] of precursor 21 (Scheme 5) that after oxidation of C6 hydroxyl group would reveal a conjugate system for 1,4-addition of methyl nucleophile at the C4 site. To this end, a Lewis acid-promoted conjugate allylation with allyltrimethylsilane afforded the corresponding ketone 20 in moderate yield (68%) as a single diastereomer. The ensuing olefination of C5, however, proved to be unexpectedly challenging. After attempting many different methods including Wittig reaction, Peterson olefination,[xxiv] Petasis[xxv] and Tebbe olefination[xxvi] etc., only the TiCl4-assisted Nysted olefination[xxvii] gave low yields (15-36%) of trialkene 21. Despite the inefficient formation of 21 we attempted the RCM. We were pleased to see that this reaction proceeded smoothly in almost quantitative yields to give allylic alcohol, which after TPAP/NMO oxidation,[xxviii] formed α,β-unsaturated ketone 22. Unfortunately, all efforts to add methyl nucleophiles to 22 via conjugate addition were unsuccessful. To overcome this issue, we sought to perform the RCM on substrate 24 that contains the challenging C11 methyl group. Consequently, we allylated 19 with methylallyltrimethyl silane. While we were successful in producing 24, olefination of this precursor at C5 resulted in irreproducible results. This prompted us to abandon this approach and seek an alternative strategy for the formation of the 5-membered ring from 19.
Scheme 5.
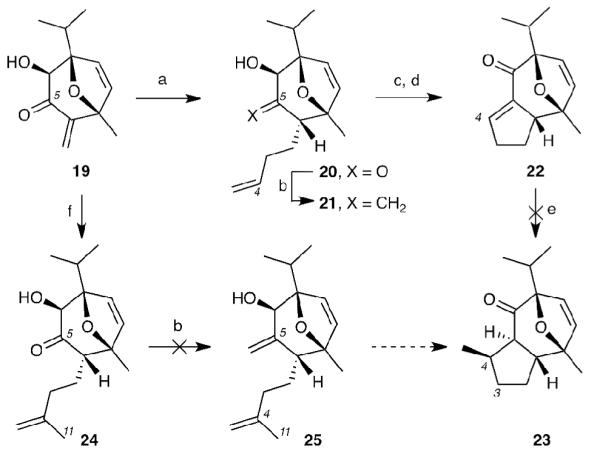
Reagents and Conditions: (a) TiCl4 (1.2 equiv), allyltrimethylsilane (3 equiv), CH2Cl2, −78 °C, 68%; (b) Nysted reagent (10 equiv), TiCl4 (10 equiv), NaHCO3 (7 equiv), −78 °C, 36%, (c) Grubbs 2nd gen. (5 mol%), CH2Cl2, rt, 99%; (d) NMO (3 equiv), TPAP (0.1 equiv), CH2Cl2, rt, 72%; (e) various methyl cuprate reagents; (f) TiCl4 (1.1 equiv), trimethyl(2-methylallyl)silane (5 equiv), CH2Cl2, −78 °C, 71%.
Formation of the 5-membered ring (A-ring) via an intra-molecular aldol condensation and completion of the synthesis
Based on the above results, we pursued an alternative strategy for the construction of the tricyclic core of englerin based on an intramolecular aldol condensation. Initially, we explored the feasibility of this reaction using non-hydroxylated enone 17 as a model system. To this end, treatment of propanal with thiazolium salt 26 and enone 17 under Stetter conditions[xxix] produced diketone 27 (Scheme 6). To our delight, the intramolecular aldol reaction of 27 proceeded under mild conditions (KOH/EtOH, rt, 24 hours) to afford 28 in good yield (76%).
Scheme 6.
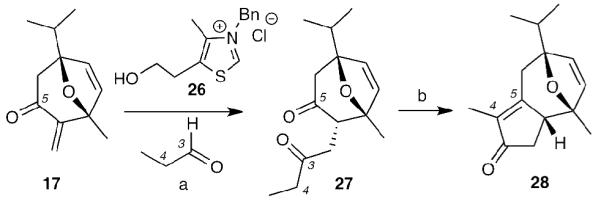
Reagents and conditions: (a) propanal (4 equiv), 26 (20 mol%), Et3N (1.2 equiv), 80 °C, 75%; (b) KOH (5 equiv), EtOH, rt, 76%.
Motivated by this result, we converted 19 to the C6 silyl ether 29 and treated this product with propanal under the previously established Stetter conditions. This reaction proceeded efficiently under basic conditions to furnish diketone 30 in good yield (75% over two steps) as a single diastereomer. However, application of previously successful KOH condensation procedure only resulted in deprotection of the TBS ether with no further reaction. This prompted an extensive investigation on this intramolecular condensation, with different C6 protecting groups (H-, MOM-, TES-, etc.), various bases (t-BuOK, KOH, NaOMe, LDA, etc.) and several reaction conditions. Many failed attempts of the aldol condensation to provide the tricyclic motif proved this reaction to be unexpectedly difficult. Eventually, treatment of diketone 30 with NaHMDS afforded the corresponding aldol addition product, which underwent sequential dehydration process (NaOMe/MeOH, heat) to produce the key tricyclic core 7 in an acceptable yield (36%; 43% brsm). NaBH4 reduction of enone 7 yielded allylic alcohol 31 (99%) as a single diastereomer that, without further purification, was protected as the benzyl ether to furnish 32 in good yield (71%). Treatment of 32 with BH3•THF followed by H2O2 oxidation afforded regio- and stereoselectively alcohol 33 (60%). Silylation of 33 followed by deprotection of the C3 benzyl ether yielded 35 via 34 in 99% overall yield. (Scheme 7). Our efforts to hydrogenate the tetra-substituted alkene of 35 using different catalysts and H2 pressure were unsuccessful presumably due to the steric hindrance of the double bond (C4-C5). This prompted us to deoxygenate the C3 hydroxyl group under Barton-McCombie conditions[xxx] (40% yield). In our efforts to improve this transformation, we discovered that dehydration of 35 with Burgess reagent[xxxi] followed by standard hydrogenation improved significantly the yield to 90% over the two steps. Deprotection of the di-TBS ether 36 with TBAF gave poor results, however, we were pleased to find that a microwave accelerated reaction under similar conditions (TBAF/THF, 80 °C) made diol 6 in a quick and high yielding conversion (45 min, 93%).
Scheme 7.

Reagents and conditions: (a) TBSOTf (2 equiv), NEt3 (5 equiv), CH2Cl2, 0°C to r.t. (b) propanal (4 equiv), 26 (20 mol%), Et3N, 80 °C, 75% over 2 steps; (c) NaHMDS (5 equiv), THF, 0 °C; (d) NaOMe (1.3 equiv), MeOH, 65 °C, 36%; 43% brsm for 2 steps; (e) NaBH4 (5 equiv), MeOH, rt; (f) NaH (4 equiv), BnBr (8 equiv), DMF, 60 °C, 71% for 2 steps; (g) BH3•THF (3 equiv), THF, rt then 3N NaOH/30% H2O2, 60%; (h) TBSOTf (2 equiv), Et3N (4 equiv), CH2Cl2, rt, 97%; (i) H2 (1 atm), Pd(OH)2 (catalytic), MeOH, rt, 99%; (j) Burgess reagent (5 equiv), PhMe, 80 °C, 90%; (k) H2 (1 atm), Pd/C (catalytic), MeOH, rt, 99%; (l) TBAF (3 equiv), THF, 80 °C, microwave, 45 min, 93%.
To access the natural product from diol 6, we would need to achieve stereoselective saturation of the tetra-substituted bond followed by esterification of the C9 hydroxyl group, inversion of C6 stereocenter by oxidation/reduction sequence and finally esterification in the presence of cinnamic acid. An alternative synthesis of (+)-6 was reported by Ma et. al.[9b] Our synthetic route to (+)-6 provides another novel and facile method for the construction of englerin A and related compounds. More significantly, the Rh-catalyzed [4+3] annulation/ intramolecular aldol condensation sequence presents a very useful, practical and general method for the preparation of the guaiane sesquiterpene core present in englerin related compounds.
The intriguing bioactivity of englerin has prompted several groups to perform SAR studies.[9f,10e] These studies have focused exclusively on modifications of the C6 and C9 side chains. An advantage of our strategy is that it can produce in an efficient and stereoselective manner compound 19, which represents the BC ring system of englerins. In other words, this approach could provide information on the biological significance of the A ring of englerin. With this in mind, we synthesized truncated englerins 37, 38 and 39 (Scheme 8). To further expand such SAR studies, we designed a new approach towards compound 46 representing the nor-A-ring englerin A. The synthesis of 46 is highlighted in Scheme 9 and is inspired by Wender’s pioneering [5+2] cycloaddition reactions.[xxxii,9d] 2-Methyl-5-furfural (40) was first alkylated under Grignard conditions and the resulting alcohol was converted to hemiacetal 41 in 75% overall yield. Treatment of 41 with excess acrylonitrile (iPr2NEt/MsCl, 100 °C, 14 h) under gave us the desired bicyclic product 42 in 32% yield. Moreover, we found that this reaction could be accelerated under microwave conditions with improved yield (iPr2NEt/MsCl, 150 °C, 4 h, 45%). To the best of our knowledge, this is the first example of microwave accelerated this type of [5+2] cycloaddition reaction. The Luche reduction[xxxiii] followed by hydrogenation provided compound 43[xxxiv] in almost quantitative yield. The transformation from 43 to 44 was accomplished in a 3-step sequence that included: (a) oxidative cleavage of the cyanide group to the corresponding ketone; (b) esterification with cinnamic acid under Yamaguchi condition;[xxxv] (c) hydride reduction of C9 carbonyl moiety. The last two steps were performed following the reported method[9b] to furnish 46. Moreover, the acetate analogue 45 was readily prepared from acetylation of 44.
Scheme 8.
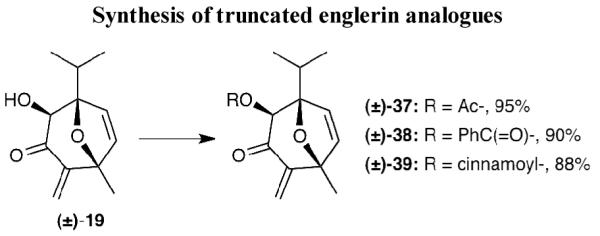
Reagents and conditions: for 37: Ac2O (1.5 equiv), pyridine, 60 °C, 95%; for 38: benzoic acid (2.0 equiv), 2,4,6-trichlorobenzoyl chloride (2.5 equiv), Et3N (3.0 equiv), 4-DMAP (catalytic), toluene, 90%; for 39: cinnamic acid (2.0 equiv), 2,4,6,-trichlorobenzoyl chloride (2.5 equiv), Et3N (3.0 equiv), 4-DMAP (catalytic), toluene,88%.
Scheme 9.
Reagents and conditions: (a) iPr2MgCl (1.5 equiv), Et2O, -10 °C; (b) mCPBA (1.0 equiv), CH2Cl2, 75% over two steps; (c) iPr2NEt (1.2 equiv), MsCl (1.2 equiv), acrylonitrile (80 equiv), 100 °C, microwave, 45%; (d) NaBH4 (1.0 equiv), CeCl3•H2O (1 equiv), MeOH, 0 °C, 98%; (e) H2, Pd/C (catalytic), EtOH, 99%; (f) LDA (2 equiv), THF, -78 °C, then O2, then SnCl2•HCl (30 equiv), 0 °C, 40%; (g) cinnamic acid (2.0 equiv), 2,4,6,-trichlorobenzoyl chloride (2.5 equiv), Et3N (3.0 equiv), 4-DMAP (catalytic), toluene, 95%; (h) NaBH4 (1.0 equiv), MeOH, 0 °C, 100%; (i) Ac2O (1.5 equiv), Et3N (4.0 equiv), 4-DMAP (0.5 equiv), CH2Cl2, r.t., 80%; (j) LiHMDS (1.3 equiv), Im2SO2 (1.3 equiv), THF, 0 °C to r.t., 99%; (k) Cesium hydroxy acetate (5 equiv), 18-crown-6 (5 equiv), toluene, 110 °C, 74%.
Cytotoxicity studies of truncated englerins
The cytotoxicity of the truncated englerins was evaluated in both A498 renal cancer cells and CEM T-cell acute lymphoblastic leukemia (T-ALL) cells using a 3H-thymidine incorporation assay. In our initial study, we compared the growth inhibitory activity of englerin A to that of truncated englerins 44 and 46. We found that englerin A inhibited the growth of A498 renal cancer cells with a GI50 of 45 nM which is in agreement with previous findings (Figure 2).[4,9d,9f,10e] However, compounds 44 and 46 did not have any effect on the growth of A498 cells even at concentrations of 100 nM or greater (Figure 2). These results suggest that the A ring is essential for the growth inhibitory activity of englerin A in renal cancer cells.
Figure 2.
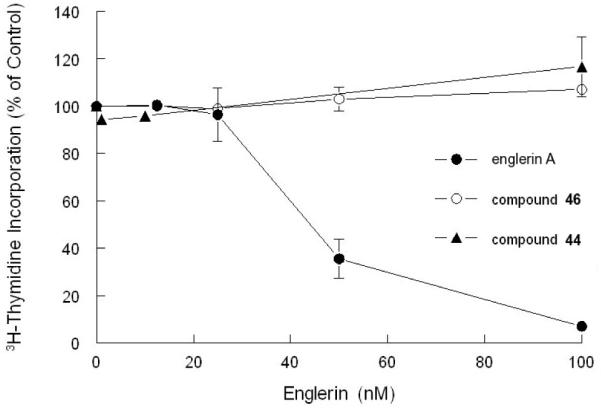
Effect of englerin A and analogues 44, 46 on the proliferation in of A498 renal cancer cells.
We then evaluated the antiproliferative activity of englerin analogues in CEM cells, a cell line in which englerin A has little activity with a reported GI50 of 20.4 μM.[4] Of all analogues tested, 17, (±)-17, and 19 had significant cytotoxicity with GI50 values of 3.3, 1.8, and 2.4 μM, respectively (Figure 3, Table 2). It is likely that these cytotoxicities are due to the exocyclic enone moiety that acts as a conjugate electrophile with bionucleophiles.[xxxvi] In contrast, englerin A and analogues containing an additional ring, such as 7, had little or no cytotoxicity at concentrations as high as 20 μM. These results suggest that the single ring analogues can target leukemia cells effectively and the addition of structural complexity may result in a loss of cytotoxicity to leukemia cells.
Figure 3.
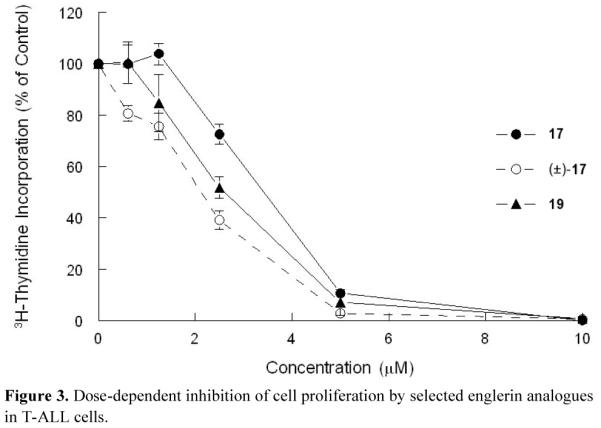
Dose-dependent inhibition of cell proliferation by selected englerin analogues in T-ALL cells.
Table 2.
Inhibition of cell proliferation by (−)-englerin A and analogues in T-ALL cells (ND: not determined)
| Compound | % of Control at 20 μM | GI50 (μM) |
|---|---|---|
| (−)-englerin A | 81.4 ± 2.5 | > 20 |
| 44 | 74.6 ± 2.6 | > 20 |
| 45 | 75.4 ± 4.5 | > 20 |
| 46 | 88.7 ± 4.3 | > 20 |
| 7 | 98.7 ± 4.0 | > 20 |
| 17 | 0.2 ± 0 | 3.3 |
| (±)−17 | ND | 1.8 |
| 19 | 0.4 ± 0.1 | 2.4 |
| 37 | 0.3 ± 0.3 | ND |
| 38 | 0.2 ± 0.1 | ND |
| 39 | 0.1 ± 0 | ND |
Conclusion
We have accomplished an efficient and enantioselective formal synthesis of englerin A (1), a potent and selective growth inhibitor of renal cancer cells. The synthetic approach to intermediate 6 proceeds in 15 steps from readily available compounds 9 and 10 in 5% overall yield. Key to our strategy is the enantioselective formation of the BC ring of 1 via a Rh(II)-induced enantioselective [4+3] cycloaddition, followed by the construction of the A ring via an intramolecular aldol condensation. Inspired by this sequence, we have also synthesized a small family of truncated englerins and have evaluated their growth inhibitory activities against certain renal cancer and leukemia cell lines. These studies suggest that the A-ring of englerin A plays an important role in its bioactivity and tissue selectivity. Interestingly, compounds (−)-17, (±)-17 and (−)-19 have shown significant growth inhibitory activity against CEM cell lines at low micromolar concentrations (GI50 = 1-3 μM). Consequently, these compounds may represent new lead structures for the development of small molecule therapeutics against leukemia.
Experimental Section
(1R,5S)-(R)-4,4-Dimethyl-2-oxotetrahydrofuran-3-yl3-[(tert-butyldimethylsilyl)oxy] -5-isopropyl-1-methyl-8-oxabicyclo[3.2.1]octa-2,6-diene-2-carboxylate (8)
A solution of 10 (11.2 g, 31.6 mmol) in dry hexanes (750 mL) was added dropwise over 5.5 hours to a refluxing solution of 9 (7.85 g, 8.8 mL, 63.2 mmol) and rhodium(II) octanoate dimer (492 mg, 0.63 mmol) in anhydrous hexanes (750 mL). The reaction mixture was stirred for an additional 30 minutes, at which time TLC showed no starting material remaining. The reaction mixture was then allowed to cooled down to room temperature, filtered through a silica plug and concentrated. Crude product purification using flash column chromatography (100:1 to 9:1, slow gradient Hexanes: EtOAc) afforded 8.1 g of bicyclic ester 8 as colorless thick oil (57%). [α]D23 = + 36.40 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 6.38 (dd, J = 5.7, 0.5 Hz, 1H), 5.72 (d, J = 5.7 Hz, 1H), 5.40 (s, 1H), 4.04 (q, J = 8.9 Hz, 2H), 2.41 (d, J = 17.4 Hz, 1H), 1.95 - 1.83 (m, 2H), 1.62 (s, 3H), 1.24 (s, 3H), 1.17 (s, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.92 (s, 9H), 0.19 (s, 3H), 0.18 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.4, 164.2, 157.1, 141.6, 126.8, 117.9, 88.5, 82.8, 76.3, 75.0, 40.2, 37.1, 34.3, 25.8, 23.3, 21.5, 20.6, 18.4, 17.2, 17.1, −3.4, −3.5. HRMS (FAB) m/z: cacld for C24H38O6SiNa(M+Na+) 473.2330, found: 473.2331.
(1R,5S)-5-isopropyl-1-methyl-2-methylene-8-oxabicyclo[3.2.1]oct-6-en-3-one (17)
To a solution of 8 (20.7 g, 45.9 mmol) in dry CH2Cl2 (459 mL) was added quickly dropwise via addition funnel a 1.0 M solution of DIBAL-H in heptanes (344.3 mmol, 344.3 mL) at −78°C. After completion of addition, the reaction was stirred for 15 min at which time TLC showed no starting material. The reaction mixture was quenched with saturated Rochelle salt solution (300 mL), was allowed to reach room temperature and was stirred for 1 hour. The reaction mixture was filtered through Celite and washed with CH2Cl2 until TLC showed no more crude product remaining in the filter cake. The filtered mixture was separated, extracted with CH2Cl2 (2 × 150 mL), washed with brine (300 mL) dried over Na2SO4, filtered, and concentrated under reduced pressure to 500 mL of CH2Cl2. This solution was flushed with Argon and treated directly with BF3•Et2O (68.9 mmol, 8.7 mL) dropwise at −30 °C. 5 min after addition TLC showed no starting material. The reaction mixture was further diluted with CH2Cl2 (250 mL), quenched with saturated NaHCO3 solution (350 mL) and extracted with CH2Cl2 (2 × 200 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated under vacuum. Crude product purification using flash column chromatography afforded 5.2 g of ketone 17 as a yellow oil (59%). [g=a]D23 = + 103.01 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 6.05 (d, J = 5.8 Hz, 1H), 5.96 (s, 1H), 5.92 (d, J = 5.7 Hz, 1H), 5.24 (s, 1H), 2.56 (d, J = 17.7 Hz, 1H), 2.46 (d, J = 17.7 Hz, 1H), 2.00 – 1.89 (m, 1H), 1.61 (s, 3H), 0.99 (d, J = 4.9 Hz, 3H), 0.98 (d, J = 4.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 197.8, 147.3, 136.3, 133.4, 115.3, 89.8, 84.9, 45.9, 33.7, 19.8, 17.3, 17.3. HRMS (FAB) m/z: cacld for C12H16O2Na (M+Na+) 215.1043, found: 215.1045.
(1S,2S,5R)-2-hydroxy-1-isopropyl-5-methyl-4-methylene-8-oxabicyclo[3.2.1]oct-6-en-3-one (19)
n-Butyllithium (77.6 mL, 124.2 mmol, 1.6 M in hexane) was added dropwise to a solution of dry diisopropylamine (19 mL, 135.0 mmol) in dry THF (800 mL) at −78°C. The reaction mixture was stirred for 15 minutes, and then a solution of 17 (5.2 g, 27.0 mmol) in dry THF (500 mL) was added quickly dropwise to the reaction mixture. The reaction mixture temperature was raised from −78 °C to 0 °C over 45 minutes and stirred for 1 hour at 0 °C. The reaction mixture was cooled down to −78 °C and TMSCl (15.8 mL, 124.2 mmol) was added dropwise. The reaction mixture temperature was raised from −78 °C to 0 °C over 45 minutes and stirred for 1 hour at 0°C at which time TLC showed no starting material. The reaction mixture was diluted with hexane (650 mL) quenched with 5% NaHCO3 solution (500 mL), washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated under vacuum. To a solution of the crude enol ether product in CH2Cl2 (250 mL) a solution of NaHCO3 (250 mL, 10%) was added all at once at 0°C. Then a solution of mCPBA (5.2 g, 30.0 mmol) in CH2Cl2 (60 mL) was added slowly while vigorous stirring of the reaction mixture. The reaction was closely monitored by TLC. When traces of enol ether (<5%) was observed the reaction was further diluted in CH2Cl2 (200 mL), quenched with saturated NaHSO3 solution (300 mL), allowed to reach room temperature, extracted with CH2Cl2 (2 × 200 mL), washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated under vacuum to approximately 250 mL. To this solution of crude epoxide product was added a solution of (COOH)2 (15.6 g, 124.2 mmol) in MeOH (150 mL). After 30 min of stirring at room temperature TLC showed no epoxide product remaining. The reaction mixture was slowly quenched with saturated K2CO3 solution until neutral pH was achieved and extracted with CH2Cl2 (2 × 200 mL). The combined organic layers were washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated under vacuum. Crude product purification using flash column chromatography (100:1 to 4:1, hexanes: EtOAc) afforded 17 (2.7 g) and α-hydroxy ketone 19 as a crystalline solid (2.2 g, 36%; 87% b.r.s.m). Recrystallization from hexanes afforded high purity crystals for X-Ray characterization. [g=a]D23 = + 101.00 (c 1.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 6.08 (dd, J = 5.8, 0.9 Hz, 1H), 6.00 (d, J = 5.9 Hz, 1H), 5.97 (s, 1H), 5.28 (s, 1H), 3.81 (d, J = 6.4 Hz, 1H), 2.37 – 2.28 (m, 1H), 1.62 (s, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 199.0, 146.9, 140.2, 130.1, 116.5, 93.4, 85.5, 73.3, 28.4, 19.4, 17.7, 17.0. HRMS (ESI) m/z: cacld for C12H16O3Na (M+Na+) 231.0992, found: 231.0993.
(1S,2S,5R)-1-isopropyl-5-methyl-4-methylene-3-oxo-8-oxabicyclo[3.2.1]oct-6-en-2-yl acetate (37)
To a solution of 19 (5 mg, 0.024 mmol) in pyridine (0.25 mL) was added 4-DMAP (0.6 mg, 0.005 mmol) and Ac2O (0.011 mL, 0.12 mmol). The reaction was heated to 60 °C for 3 hours. The reaction was cooled down to room temperature quenched with saturated NaHCO3 and extracted 3 times with ethyl acetate. The combined organic layers were dried over MgSO4, concentrated and the residue was purified by preparatory plate chromatography to yield 37 as a white foam (5.7 mg, 95%). 1H NMR (400 MHz, CDCl3) δ: 6.16 (d, J = 5.9 Hz, 1 H), 6.02 (d, J = 5.9 Hz, 1 H), 6.00 (s, 1H), 5.30 (s, 1H), 5.27 (s, 1H), 2.16 (s, 3H), 2.16 (m, 1H), 1.65 (s, 3H), 0.95 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 194.1, 170.0, 146.6, 141.2, 129.3, 116.9, 92.6, 85.7, 71.7, 29.9, 28.8, 19.5, 17.4, 17.1. HRMS (ESI) m/z: cacld for C14H18O4 Na (M+Na+) 273.1097, found: 273.1098.
(1S,2S,5R)-1-isopropyl-5-methyl-4-methylene-3-oxo-8-oxabicyclo[3.2.1]oct-6-en-2-yl benzoate (38)
To a stirred mixture of benzoic acid (7.3 mg, 0.06 mol) and 19 (6.5 mg, 0.026 mol) in dry toluene (0.55 mL) were added successively NEt3 (15 μL, 0.11mol) and 2,4,6-trichlorobenzoyl chloride (9 μL, 0.075 mol). The reaction mixture was stirred for 10 min, then catalytic amount of 4-DMAP (1 crystal) was added. After 16 h of stirring at room temperature, the reaction was diluted with ethyl acetate, and the organic phase was washed successively with aqueous HCl solution (1M), saturated sodium bicarbonate solution, and brine. The organic phase was dried over MgSO4 and concentrated in vacuum after filtration. The residue was purified by flash chromatography to afford the benzoic keto-ester 38 (9.1 mg, 90%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.10 (m, 2H), 7.57 (m, 1H), 7.24 (m, 2H), 6.21 (dd, J = 5.8, 0.9 Hz, 1H), 6.09 (d, J = 5.8 Hz, 1H), 6.02 (s, 1H), 5.51 (s, 1H), 5.32 (s, 1H), 2.25 (m, 1H), 1.69 (s, 3H), 0.96 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 193.8, 165.5, 146.7, 141.1, 133.4, 130.1, 129.4, 128.4, 92.8, 85.6, 72.0, 28.9, 19.6, 17.4, 17.1. HRMS (ESI) m/z: cacld for C19H20O4 Na (M+Na+) 335.1254, found: 335.1256.
(1S,2S,5R)-1-isopropyl-5-methyl-4-methylene-3-oxo-8-oxabicyclo[3.2.1]oct-6-en-2-yl cinnamate (39)
To a stirred mixture of cinnamic acid (8 mg, 0.053 mol) and 19 (5.5 mg, 0.026 mol) in dry toluene (0.5 mL) were added successively NEt3 (11 μL, 0.08 mol) and 2,4,6-trichlorobenzoyl chloride (7 μL, 0.07 mol). The reaction mixture was stirred for 10 min, then catalytic amount of 4-DMAP (1 crystal) was added. After 16 h of stirring at room temperature, the reaction was diluted with ethyl acetate, and the organic phase was washed successively with aqueous HCl solution (1M), saturated sodium bicarbonate solution, and brine. The organic phase was dried over MgSO4 and concentrated in vacuum after filtration. The residue was purified by flash chromatography to afford compound 39 (7.8 mg, 88%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 15.9 Hz, 1H), 7.52 (m, 2H), 7.39 (m, 3H), 6.52 (d, J = 16.0 Hz, 1H), 6.19 (d, J = 5.8 Hz, 1H), 6.06 (d, J = 5.8 Hz, 1H), 6.03 (s, 1H), 5.42 (s, 1H), 5.33 (s, 1H), 2.22 (m, 1H), 1.68 (s, 3H), 0.96 (t, J = 6.9 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 194.1, 166.0, 146.5, 141.2, 134.3, 130.7, 129.4, 129.0, 128.4, 117.2, 116.9, 92.8, 85.7, 71.7, 28.8, 19.6, 17.5, 17.1. HRMS (ESI) m/z: cacld for C21H22O4Na(M+Na+) 361.1416, found: 361.1417.
(1R,5S,6S)-1-isopropyl-5-methyl-2-oxo-8-oxabicyclo[3.2.1]oct-3-ene-6-carbonitrile (42)
To a solution of 41 (1.6 g, 11.0 mmol) in acrylonitrile (60 mL) was added diisopropylethylamine (2.3 mL, 13 mmol), followed by methanesulfonyl chloride (1.0 mL, 13 mmol). The resulting solution was microwave heated at 150°C for 4 hours. After cooling to room temperature, the reaction mixture was filtered through a plug of Celite, and the filtrate was concentrated in vacuo. Flash chromatography of the residue (20% ethyl acetate in hexanes) provided the cycloadduct 42 (880 mg, 45%) as colorless oil. 1H NMR (500 MHz, CDCl3) δ: 6.93 (d, J = 9.7 Hz, 1H), 6.01 (d, J = 9.8 Hz, 1H), 3.06 (dd, J = 9.2, 2.9 Hz, 1H), 2.52 (dd, J = 14.3, 2.9 Hz, 1H), 2.30 (p, J = 7.0 Hz, 1H), 2.19 (dd, J = 14.3, 9.3 Hz, 1H), 1.74 (s, 3H), 1.08 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 6.9 Hz, 3H) 13C NMR (126 MHz, CDCl3) δ: 196.7, 151.8, 128.1, 118.8, 100.0, 90.4, 37.4, 35.2, 30.0, 21.6, 17.6, 16.5. HRMS (ESI) m/z: calcd for C12H15NNaO2 (M+Na+) 228.0995, found: 228.0996.
3H-Thymidine incorporation assay
CEM cells were plated at 10-20 × 103 cells/well and A498 cells at 3,500 cells/well in 96-well plates in RPMI supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 u/ml penicillin/streptomycin (complete medium). Englerin A or its analogues were added to the cells at increasing concentrations and 0.1% DMSO was added to control cells. Cells were incubated for 48 hours and then pulsed with 3H-thymidine for 6 hours. Incorporation of 3H-thymidine was determined in a scintillation counter (Beckman Coulter Inc., Fullerton, CA) after cells were washed and deposited onto glass microfiber filters using a cell harvester M-24 (Brandel, Gaithersbur, MD).
Supplementary Material
Acknowledgements
We gratefully acknowledge the National Institutes of Health (NIH) for financial support of this work through Grant No. R01 GM081484. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We also thank Dr. Anthony Mrse (UCSD NMR Facility), Dr. Yongxuan Su (UCSD MS Facility), and Dr. Arnold L. Rheingold and Dr. Curtis E. Moore (UCSD X-ray Facility).
Footnotes
Supporting information for this article, that includes synthesis and spectroscopic/analytical characterization for all compounds, is available on the WWW under http://dx.doi.org/10.1002/asia.200xxxxxx.
References
- [i].a) Gurib-Fakim A. Mol. Aspects. Med. 2006;27:1–93. doi: 10.1016/j.mam.2005.07.008. [DOI] [PubMed] [Google Scholar]; b) Burslem DFRP, Garwood NC, Thomas SC. Science. 2001;291:606–607. doi: 10.1126/science.1055873. [DOI] [PubMed] [Google Scholar]; c) Cragg G, Newmann DJ. Pure Appl. Chem. 2005;77:7–24. [Google Scholar]; d) Eisner T. Issues Sci. Technol. 1989;6:31–34. [Google Scholar]; e) Rout SP, Choudary KA, Kar DM, DAS L, Jain A. Int. J. Pharm. Pharmaceut. Sci. 2009;1:1–23. [Google Scholar]
- [ii].a) Van Wyk BE, Van Oudtshoorn B, Gericke N. Medicinal plants of SouthAfrica. Briza, Pretoria: 1997. pp. 1–304. [Google Scholar]; b) Fabricius C, Koch E, Magome H, Turner S. Rights, resources and rural development: community-based natural resource management in Southern Africa. Earthscan: 2004. p. 98. [Google Scholar]; (c) Street RA, Stirk WA, Van Staden J. J. Ethnopharmacol. 2008;119:705–710. doi: 10.1016/j.jep.2008.06.019. [DOI] [PubMed] [Google Scholar]
- [iii].a) Calixto JB, Santos ARS, Filho VC, Yunes RA. Med. Res. Rev. 1998;18:225–258. doi: 10.1002/(sici)1098-1128(199807)18:4<225::aid-med2>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; b) Bagalkotkar G, Sagineedu SR, Saad MS, Stanslas J. J. Pharm. Pharmacol. 2006;58:1559–1570. doi: 10.1211/jpp.58.12.0001. [DOI] [PubMed] [Google Scholar]
- [iv].a) Ratnayake R, Covell D, Ransom TT, Gustafson KR, Beutler JA. Org.Lett. 2009;11:57–60. doi: 10.1021/ol802339w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Beutler JA, Ratnayake R, Covell D, Johnson TR. WO 2009/088854
- [v].Jemal A, Siegel R, Xu J, Ward E. CA Cancer J. Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- [vi].a) Schrader AJ, Hofmann R. Anticancer Drugs. 2008;19:235–245. doi: 10.1097/cad.0b013e3282f444de. [DOI] [PubMed] [Google Scholar]; b) Larkin JMG, Kipps ELS, Powell CJ, Swanton C. Ther. Adv. Med. Oncol. 2009;1:15–27. doi: 10.1177/1758834009338430. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cohen HT, McGovern FJ. N. Engl. J. Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]; d) Motzer RJ, D M, Bander NH, Nanus DM. N. Engl. J. Med. 1996;335:865–875. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- [vii].Nelson EC, Evans CP, Lara PN., Jr. Cancer Treat. Rev. 2007;33:299–313. doi: 10.1016/j.ctrv.2006.12.005. [DOI] [PubMed] [Google Scholar]
- [viii].Peng G-P, Tian G, Huang X-F, Lou F-C. Phytochemistry. 2003;63:877–881. doi: 10.1016/s0031-9422(03)00222-x. [DOI] [PubMed] [Google Scholar]
- [ix].a) Willot M, Radtke L, Könning D, Fröhlich R, Gessner VH, Strohmann C, Christmann M. Angew. Chem. Int. Ed. 2009;48:9105–9108. doi: 10.1002/anie.200905032. [DOI] [PubMed] [Google Scholar]; b) Zhou Q, Chen X, Ma D. Angew. Chem. Int. Ed. 2010;49:3513–3516. doi: 10.1002/anie.201000888. [DOI] [PubMed] [Google Scholar]; c) Molawi K, Delpont N, Echavarren AM. Angew. Chem. Int. Ed. 2010;49:3517–3519. doi: 10.1002/anie.201000890. [DOI] [PubMed] [Google Scholar]; d) Nicolaou KC, Kang Q, Ng SY, Chen DY-K. J. Am. Chem. Soc. 2010;132:8219–8222. doi: 10.1021/ja102927n. [DOI] [PubMed] [Google Scholar]; e) Li Z-W, Nakashige M, Chain WJ. J. Am. Chem. Soc. 2011;133:6553–6556. doi: 10.1021/ja201921j. [DOI] [PubMed] [Google Scholar]; f) Radtke LR, Willot M, Sun H, Ziegler S, Sauerland S, Strohmann C, Fröhlich R, Habenberger P, Waldmann H, Christmann M. Angew. Chem. Int. Ed. 2011;50:3998–4002. doi: 10.1002/anie.201007790. [DOI] [PubMed] [Google Scholar]
- [x].a) Sun B-F, Wang C-L, Ding R, Xu J-Y, Lin G-Q. Tetrahedron Lett. 2010;52:2155–2158. [Google Scholar]; b) Wang C-L, Sun B-F, Chen S-G, Ding R, Lin G-Q, Xu J-Y, Shang Y-J. Synlett. 2012:263–266. [Google Scholar]; c) Ushakov DB, Navickas V, Ströbele M, Maichle-Mössmer C, Sasse F, Maier ME. Org. Lett. 2011;13:2090–2093. doi: 10.1021/ol200499t. [DOI] [PubMed] [Google Scholar]; d) Navickas V, Ushakov DB, Maier ME, Strobele M, Meyer HJ. Org. Lett. 2010;12:3418–3421. doi: 10.1021/ol1012185. [DOI] [PubMed] [Google Scholar]; e) Chan KP, Chen DY-K. ChemMedChem. 2011;6:420–423. doi: 10.1002/cmdc.201000544. [DOI] [PubMed] [Google Scholar]
- [xi].a) Willot M, Christmann M. Nature Chem. 2010;2:519–520. doi: 10.1038/nchem.715. [DOI] [PubMed] [Google Scholar]; b) Chain WJ. Synlett. 2011;18:2605–2608. [Google Scholar]; c) Pouwer RH, Richard J-A, Tseng C-C, Chen DY-K. Chem.-Asian. J. 2012;7:22–35. doi: 10.1002/asia.201100780. [DOI] [PubMed] [Google Scholar]; d) Lu Y-Y, Yao H-Q, Sun B-F. Chin. J. Org. Chem. DOI: 10.6023/cjoc1110141. [Google Scholar]
- [xii].For selected publications on this topic see: Ling T-T, Kramer BA, Palladino MA, Theodorakis EA. Org. Lett. 2000;2:2073–2076. doi: 10.1021/ol0060578. Ling T-T, Chowdhury C, Kramer BA, Vong BG, Palladino MA, Theodorakis EA. J. Org. Chem. 2001;66:8843–8853. doi: 10.1021/jo0159035. Tisdale EJ, Slobodov I, Theodorakis EA. Proc. Nat. Acad. Sci. USA. 2004;101:12030–12035. doi: 10.1073/pnas.0401932101. Tisdale EJ, Slobodov I, Theodorakis EA. Org. Biomol. Chem. 2003;1:4418–4422. doi: 10.1039/b311833a. Xu J, Trzoss L, Chang WK, Theodorakis EA. Angew. Chem. Int. Ed. 2011;50:3672–3676. doi: 10.1002/anie.201100313. Trzoss L, Xu J, Lacoske MH, Mobley WC, Theodorakis EA. Org. Lett. 2011;13:4554–4557. doi: 10.1021/ol201742j. Chantarasriwong O, Batova A, Chavasiri W, Theodorakis EA. Chem.-Eur. J. 2010;16:9944–9962. doi: 10.1002/chem.201000741.
- [xiii].Xu J, Caro-Diaz EJE, Theodorakis EA. Org. Lett. 2010;12:3708–3711. doi: 10.1021/ol1015652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [xiv].Jeong N, Robinson JE, Wender PA, Gamber GG, Williams TJ, Davies HML, Walji AM. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Wiley-VCH; Weinheim: 2005. pp. 215–340. Chapter 11-14. [Google Scholar]
- [xv].Davies HML, Ahmed G, Churchill MR. J. Am. Chem. Soc. 1996;118:10774–10782. Jackson KL, Henderson JA, Motoyoshi H, Phillips AJ. Angew. Chem. Int. Ed. 2009;48:2346–2350. doi: 10.1002/anie.200806111.
- [xvi].Weyerstahl P, Brendel J. Liebigs Ann. Chem. 1988:1015–1016. [Google Scholar]
- [xvii].Shan R-D, Howlett SE, Knaus EE. J. Med. Chem. 2002;45:955–961. doi: 10.1021/jm010394k. Clemens RJ, Hyatt JA. J. Org. Chem. 1985;50:2431–2435. c) for detailed experimental procedure: see supporting information.
- [xviii].See supporting information.
- [xix].Poirier J-M, Hennequin L. Tetrahedron. 1989;45:4191–4202. [Google Scholar]
- [xx].a) Davis FA, Chattopadhyay S, Towson JC, Lal S, Reddy T. J. Org. Chem. 1988;53:2087–2089. [Google Scholar]; b) Vishwakarma LC, Stringer OD, Davis FA. Org. Synth. 1993;8:546. Coll. [Google Scholar]; c) Davis FA, Chen B-C. Chem. Rev. 1992;92:919–934. [Google Scholar]
- [xxi].Rubottom GM, Vazquez MA, Pelegrina DR. Tetrahedron Lett. 1974;15:4319–4322. [Google Scholar]
- [xxii].CCDC782508 contains the supplementary crystallographic data for compound 19. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/products/csd/request/
- [xxiii].a) Vougioukalakis GC, Grubbs RH. Chem. Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. references herein. [DOI] [PubMed] [Google Scholar]; b) Grubbs RH. Handbook of Metathesis. Wiley-VCH; Weinheim: 2003. [Google Scholar]; c) Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- [xxiv].Peterson DJ. J. Org. Chem. 1968;33:780–784. [Google Scholar]
- [xxv].Petasis NA, Bzowej EI. J. Am. Chem. Soc. 1990;112:6392–6394. [Google Scholar]
- [xxvi].Tebbe FN, Parshall GW, Reddy GS. J. Am. Chem. Soc. 1978;100:3611–3613. [Google Scholar]
- [xxvii].Nysted LN. US Patent 3865848
- [xxviii].a) Griffith WP, Ley SV, Whitcombe GP, White AD. J. Chem. Soc., Chem. Commun. 1987:1625–1627. [Google Scholar]; b) Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- [xxix].a) Stetter H. Angew. Chem. Int. Ed. 1976;15:639–647. [Google Scholar]; b) Stetter H, Kuhlmann H, Haese W. Org. Synth. 1987;65:26–31. [Google Scholar]; Org. Synth. 1993;8:620–624. Coll. [Google Scholar]
- [xxx].Barton DHR, McCombie SW. J. Chem. Soc., Perkin Trans. 1. 1975:1574–1585. [Google Scholar]
- [xxxi].Atkins GM, Burgess EM. J. Am. Chem. Soc. 1968;90:4744–4745. [Google Scholar]
- [xxxii].a) Wender PA, Mascareñas JL. J. Org. Chem. 1991;56:6267–6269. [Google Scholar]; b) Wender PA, Mascareñas JL. Tetrahedron Lett. 1992;33:2115–2118. [Google Scholar]; c) Williams DR, Benbow JW, McNutt JG, Allen EE. J. Org. Chem. 1995;60:833–843. [Google Scholar]; d) Marshall KA, Mapp AK, Heathcock CH. J. Org. Chem. 1996;61:9135–9145. [Google Scholar]
- [xxxiii].Gemal AL, Luche JL. J. Am. Chem. Soc. 1981;103:5454–5459. [Google Scholar]
- [xxxiv].CCDC857553 contains the supplementary crystallographic data for compound 43. This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/products/csd/request/
- [xxxv].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- [xxxvi].For selected examples of natural products that exhibit potent bioactivities due to enone functionalities in their structures see: Siedle B, García-Piñeres AJ, Murillo R, Schulte-Mönting J, Castro V, Rüngeler P, Klaas CA, Da Costa FB, Kisiel W, Merfort I. J. Med. Chem. 2004;47:6042–6054. doi: 10.1021/jm049937r. Kwok BHB, Koh B, Ndubuisi MI, Elofsson M, Crews CM. Chem. & Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. Rizvi SA, Courson DS, keller VA, Rock RS, Kozmin SA. Proc. Nat. Acad. Sci. USA. 2008;105:4088–4092. doi: 10.1073/pnas.0710727105.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.