

Abstract
CR1642D, an endophytic isolate of Penicillium sp. collected from a Costa Rican rainforest, was identified through a high-throughput approach to identify natural products with enhanced anti-tumor activity in the context of tumor-stromal interactions. Bioassay-guided separation led to the identification of five xanthones (1-5) from CR1642D. The structures of the xanthone dimer penexanthone A (1) and monomer penexanthone B (2) were elucidated on the basis of spectroscopic analyses, including 2D NMR experiments. All of the compounds were tested against a panel of tumor cell lines in the presence and absence of bone marrow stromal cells. Compound 3 was the most active, with IC50 values of 1~17 μM, and its activity was enhanced two-fold against tumor cell line RPMI8226 in the presence of stromal cells (IC50 1.2 μM, but 2.4 μM without stromal cells).
Identification of agents for the treatment of cancer typically involves target-based screens or phenotypic screens against isolated cancer cell lines. However, tumor cells in patients interact with diverse types of non-malignant cells, which can stimulate tumor cell survival, proliferation and resistance to diverse classes of established anticancer drugs and investigational agents. Recently, we have also shown that the presence of non-malignant accessory cells, such as bone marrow stromal cells (BMSCs), can sensitize tumor cells to other novel agents.1 Therefore, in vitro screening for novel anticancer agents has to be performed in cocultures of tumor cells with non-malignant accessory cells, in order to detect whether an agent is subject to microenvironment-dependent drug resistance or sensitization. In our continuing search for biologically active natural products from Costa Rican tropical rainforests as part of an International Cooperative Biodiversity Group (ICBG) program,2 we screened endophytic fungal extracts from Costa Rica at Harvard Medical School’s high throughput screening facility (ICCB-L) against a panel of cancer cell lines with and without BMSCs (Scheme 1). One extract, designed as CR1642D and identified as being from Penicillium sp., had enhanced anti-tumor activity against MM.1S with 40.2% viability in the presence of BMSCs, but 66.8% without BMSCs at 1 μg/mL. Bioassay-guided fractionation using C18 open column and phenyl-hexyl HPLC yielded five xanthones (1-5). Herein we report the structure elucidation of compounds 1 and 2 and the anti-tumor properties of compounds 1-5.
Scheme 1.

High-throughput screening to identify compounds active against tumor cells interacting in vitro with bone marrow stromal cells.
Compound 1 had the molecular formula C36H36O15 by HRESIMS. The UV spectrum of 1 was very similar to that of phomoxanthone B, a xanthone dimer.3 The IR spectrum of 1 supported the presence of carbonyl groups (1744 and 1605 cm−1). The proton NMR spectrum of 1 showed two aromatic AB spin systems, six protons bound to oxygenated carbons, two methines, two methylenes, three acetyl methyl groups, and two chelated phenolic hydroxyl groups. In ring A of monomer I, H-4 exhibited 3J HMBC correlations to C-2 and C-9a. Peri to a carbonyl group, the C-1 hydroxyl group correlated to C-1, C-2 and C-9a. H-3 was ortho coupled with H-4, and showed 3J HMBC correlations to C-1, C-4a, and C-4′. Therefore ring A was determined as a 2,3,6-trisubstituted phenol (2,4a,9a-trisubstituted phenol). H-2′ and H-3′ showed 3J HMBC correlations to C-4′ and C-9a’, and C-1′ and C-4a’, respectively, hence ring A’ in monomer II was determined as a 2,3,4-trisubstituted phenol (4′,4a’,9a’-trisubstituted phenol), and rings A and A’ were connected through the C-2-C-4′ bond. Since 1-OH was chelated and C-4a was oxygenated, it could be deduced that A/A’ joined C/C’ to form a 4-chromanone.
The remaining parts of the molecule to be determined are rings B and B’. The methyl group at δH 1.07 (H3-11) showed HMBC correlations to C-5 (δC 72.1), C-6 (28.0), and C-7 (33.7). The oxygenated methylene at δH 3.94 and 3.75 (H2-12) correlated to C-10a (δC 80.7), C-5, and C-8a (101.5), while the oxygenated methine at δH 5.55 (H-5) exhibited correlations to C-10a, C-8a, C-6, C-7, C-12 (δC 65.6), C-11 (17.9), and 5-OCOCH3 (170.9). Therefore, this ring was determined to be 4-acetoxyl-3-hydroxymethyl-5-methyl cyclohexenol with the other two substituents at 2,3-positions of this fragment, which could be ring B or ring B’. Similarly, the last ring was determined to 4-acetoxyl-3-acetoxymethyl-5-methyl cyclohexenol with the other two substituents at 2,3-positions of this fragment, which could be ring B’ or B. The only difference between ring B and ring B’ is the substituent at the oxygenated methylene - one is a hydroxyl while another is an acetoxy. A ROESY experiment showed that the oxygenated methylene (12-position) was on one face of the hemi-chair ring while both the methyl (11-position) and the acetoxyl group (5-OAc) were on the other face (Figure 2). Molecular modeling with Chem3D Ultra (9.0) suggested that the substituent at the 10a-position was far away (> 5 Å) from the aromatic protons of ring A. In the NOESY spectrum of compound 1, both 1–OH (δH 11.62) and H-3 (δH 7.16) correlated with the signal at δH 1.83 (12′-OCOCH3), and H-4 (δH 6.43) showed correlation with the signal at δH 2.11 (5-OCOCH3). Since the signals at δH 1.83 (12′-OCOCH3) and 2.05 (5′-OCOCH3) were in the same ring system, the signal at δH 2.11 must be the 5-acetoxyl methyl. Hence, the structure of compound 1 including its relative stereochemistry was determined as shown. Both monomers were assigned the relative stereochemistry (R*,R*,R*) based on the assumption that they share the same biosynthetic pathway. All the 13C NMR chemical shifts except C-9 and C-9′ were obtained from the HSQC and HMBC spectra due to insufficient sample for a good 13C NMR spectrum. The planar structure of 1 was reported in a Korean patent, but with an unclean 1H NMR spectrum, which most likely resulted from an impure sample.5
Figure 2.
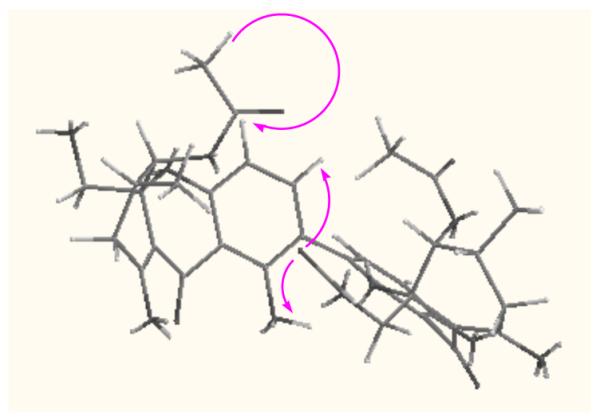
key NOSEY correlations of 1
The molecular weight of compound 2 was 376.1158 as suggested by HRESIMS, which was 1 unit more than that of monomer II of compound 1. The proton NMR data of compound 2 were similar to those of monomer II of compound 1, but with an extra aromatic proton signal. The planar structure and relative stereochemistry of compound 2 were determined by 2D NMR experiments as shown. A structure identical to compound 2 was used for CD calculation, but never isolated.6
Compounds 3-5 were identified as dicerandrols B, A, and C, respectively, by comparison of physical and spectroscopic data (UV, IR, 1H NMR, [α]D, and MS) with literature values.4i These were previously purified from the endophytic fungus Phomopsis longicolla isolated from an endangered mint from Florida.4i Compounds 4 and 5 are homodimers with a 2—2′ connectivity. The heterodimer 3 has the same two monomers as 1, but they are dimerized through a 2—2′ linkage, while the two different monomeric units of 1 are coupled through the 2—4′ positions. Many such xanthone dimers with a methyl at the 6-position and an oxygenated methylene at the 10a-position have been reported, but most of them are symmetrical homodimers as in 4 and 5.4 Usually, the unsymmetrical xanthone heterodimers are different only at either ring A/A’ like phomoxanthone B,3 or ring C/C’ as in 3. Curiously the monomeric units in these xanthone dimers (1, and 3-5) had not been previously isolated from natural sources.
All five pure compounds were tested against a panel of tumor cell lines (from myeloma, lymphoma, leukemia, as well as breast and prostate cancer) in the presence and absence of BMSCs. As expected, the activity was either decreased or enhanced in the context of tumor-stromal interactions. As can be seen in Table 2, the cytotoxic activity of these compounds increases upon dimerization and acetylation of 12′-OH (4) to 12′-OAc (3). The C-12 and C-12′ diacetate (5) exhibits less activity than 3, and the 2-4′ linked monoacetate (1) is much less active than the 2-2′ linked monoacetate (3). The most active compound, 3, exhibits moderate activity against Dox40, Farage, H929, HT, OPM2 and RPMI8226 in the presence of stromal cells with IC50 values of 2.3, 1.3, 3.4, 1.3, 1.5, and 1.2 μM, respectively. The activity of 3 against cancer cell lines RPMI8226 and H929 is doubled or tripled in the presence of stromal cells - results that would not have been identified through a traditional cell based screen. On the other hand, compound 3 is much less toxic against human immortalized non-malignant cells, such as HS-5 bone marrow stromal cells, HOBIT osteoblast-like cells, THLE-3 hepatocytes, and SVGp12 astrocytes, with IC50 values of 13.0, 9.2, 10.0, and 13.7 μM, respectively (Figure 2S). The relative selectivity of these compounds, especially compound 3, warrants further investigation.
Table 2.
Assay results of compounds l~5a
| 1 | 2 | 3 | 4 | 5 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
bIC50 (μM) |
cIC50 (μM) |
dDiff |
bIC50 (μM) |
cIC50 (μM) |
dDiff |
bIC50 (μM) |
cIC50 (μM) |
dDiff |
bIC50 (μM) |
cIC50 (μM) |
dDiff |
bIC50 (μM) |
cIC50 (μM) |
dDiff | |
| Dox40e | 15 | 21.6 | 6.7 | 11.6 | 24.2 | 12.6 | 2.2 | 2.3 | 0.1 | 22.8 | 36.9 | 14.1 | 10 | 13.9 | 3.9 |
| - | |||||||||||||||
| Faragef | 12.4 | 10.5 | −1.9 | 199.8 | 38.7 | 161.1 | 1.6 | 1.3 | −0.3 | 15.6 | 14.8 | −0.8 | 3.5 | 3.3 | −0.3 |
| H929e | 54.5 | 35.2 | −19.3 | 187.5 | 94.8 | −92.7 | 10.2 | 3.4 | −6.8 | 42.5 | 22.9 | −19.6 | 10.8 | 5.4 | −5.4 |
| HTf | 10.8 | 9.9 | −0.9 | 100.0 | 34.9 | −65.2 | 1.2 | 1.3 | 0.1 | ND | ND | 2.1 | 4.0 | 1.9 | |
| KMS34e | 22.6 | 55.6 | 33 | 152.9 | 199.1 | 46.1 | 5.5 | 9.3 | 3.8 | ND | ND | ND | ND | ||
| KU812Fg | 14.6 | 24.2 | 9.5 | 44.9 | 95.9 | 51.0 | 2.7 | 3.6 | 0.9 | 24.6 | 46.6 | 22 | 7.1 | 14.3 | 7.2 |
| - | |||||||||||||||
| L363e | 66.0 | 79.4 | 13.4 | 464.2 | 198.6 | 265.6 | 16.7 | 9.4 | −7.3 | ND | ND | ND | ND | ||
| MDA-MB- 231h |
43.4 | 34.2 | −9.3 | 163.9 | 118.5 | −45.4 | 8.5 | 5.5 | −2.9 | 43.3 | 38.4 | −4.9 | 18.1 | 13.3 | −4.8 |
| MM1Se | 37.1 | 47.1 | 10.0 | 168.5 | 185.6 | 17.1 | 7.3 | 8.0 | 0.7 | 28.9 | 33.3 | 4.4 | 14.7 | 20.8 | 6.1 |
| OCILY17Rf | 31.1 | 47.4 | 16.3 | 1616 | 293.4 | −1323 | 4.6 | 5.2 | 0.6 | ND | ND | ND | ND | ||
| OCIMY5e | 18.8 | 47.7 | 28.8 | 158.0 | 383.9 | 225.8 | 2.8 | 5.7 | 2.9 | ND | ND | ND | ND | ||
| OPM2e | 12.7 | 10.5 | −2.2 | 42.8 | 44.8 | 1.9 | 1.9 | 1.5 | −0.3 | ND | ND | 4.8 | 7.5 | 2.6 | |
| - | |||||||||||||||
| PC3i | 122.3 | 66.3 | −56.0 | 1382. | 216.0 | 1166. | 34 | 14.1 | −19.9 | 100. | 71.7 | −28.4 | 121 | 45.2 | −75.4 |
| RPMI8226e | 12.9 | 8.9 | −4 | 71.6 | 34 | −37.6 | 2.4 | 1.2 | −1.2 | ND | ND | 4.8 | 2.8 | −2.0 |
Activity of five compounds from the PeniciUium expansion extract across various tumor types in the presence and absence of bone marrow stromal cells. Tumor cells of various types were cultured in the presence and absence of HS-5 stromal cells treated with increasing concentrations of compounds. IC50 was calculated both in the presence and absence of stroma.
No stroma.
Plus Stroma.
Difference in IC50 values.
Myeloma.
Lymphoma.
Leukemia.
Breast.
Prostate.
EXPERIMENTAL SECTION
General Experimental Procedures
All NMR experiments were carried out on a Varian INOVA 600 MHz spectrometer. IR spectra were measured on a Bruker Alpha-P spectrometer and UV spectra on an Amersham Biosciences Ultrospec 5300 pro spectrophotometer. All the compounds (1-5) were purified from CR1642D on an Agilent 1100 series HPLC (Agilent Technologies) using a semi-preparative Phenomenex Luna Phenyl-hexyl column (Luna, 25 cm × 10 mm, 5 μm particle size) and a Phenomenex Luna C18 HPLC column (250 × 10 mm, 5 μm particle size). Optical rotations were obtained using a Jasco Polarimeter.
Culturing
The isolated strain CR1642D is deposited at INBio, Costa Rica. Agar plugs of CR1642D were initially grown at 25 °C on yeast malt agar plates supplemented with 30 μg/mL streptomycin and 12 μg/mL chlortetracycline. After one week, 3 macerated agar plugs from these plates were placed in 75 mL of rich seed medium [tryptone peptone (5 g/L), dextrose (10 g/L), yeast extract (3 g/L), and malt extract (10 g/L)] in a 125 mL erlenmeyer (×2) with a pH value of 6.2. It was grown at 25 °C and 150 rpm for 6 days. 150 mL of 0.66% (w/v) malt extract and 5 g HP-20 resin were then added to each Erlenmeyer (250 mL each, ×8), which was inoculated with 15 mL of the rich seed media, and the fungi were cultured under the same conditions for 16 days. The fungal cultures were then held at 25 °C without shaking for 5 days.
Sequencing and species identification
For identification by internal transcribed spacer (ITS) sequencing, CR1642D was cultured in the above-mentioned rich seed medium (see the “Culturing” section) for 6 days. Mycelia were then retrieved by filtration and ground to a fine powder in liquid N2. Genomic DNA was extracted using the SurePrep™ RNA/DNA/Protein Purification Kit (Fisher Bioreagents), and large subunit rDNA was amplified by PCR using primers LR5 (5′-TCCTGAGGGAAACTTCG-3′) and LROR (5′-ACCCGCTGAACTTAAGC-3′). PCR products were sequenced at Genewiz (http://www.genewiz.com/). The DNA sequence data obtained from the fungal strain CR1642D have been deposited at GenBank with accession number JQ778844.
Extraction and separation
The cultures were filtered, the mycelial mat and HP-20 were extracted with 90% EtOH three times, and the extract was concentrated under vacuum. The crude extract (1.2 g) of CR1642D in 90% H2O-MeOH was passed through a C18 SPE and then washed with MeOH. The MeOH wash was evaporated to dryness on a rotary evaporator, redissolved and fractionated using a phenyl-hexyl column (2 mL/min; 70% CH3CN for 20 min then to 100% CH3CN in 10 min). Fractions 2, 4, 5, and 7 yielded compounds 4 (SC2-30-2, tR 13.5 min, 1.0 mg/L), 3 (SC2-30-4, tR 19 min, 19 mg/L), 1 (SC2-30-5, tR 23.5 min, 1.3 mg/L), and 5 (SC2-30-7, tR 27 min, 0.8 mg/L), respectively. Further purification of the most polar fraction, fraction 1, using a Phenomenex Luna C18 HPLC column (2 mL/min; 60% CH3CN for 30 min then to 100% CH3CN in 10 min) yielded compound 2 (SC2-32-3, tR 22.5 min, 0.4 mg/L).
Penexanthone A (1)
yellow powder; [α]23D −36 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 212 (4.23), 242 (sh), 337 (4.30) nm; IR (film) νmax 3419, 2966, 2934, 1744, 1605, 1562, 1435, 1410, 1373, 1324, 1229, 1048 cm−1; 1H NMR (600 MHz, CD3CN): δ 7.30 (1H, d, J = 7.8 Hz, H-3′), 7.18 (1H, d, J = 8.4 Hz), H-3, 6.50 (1H, d, J = 7.8 Hz, H-2′), 6.40 (1H, d, J = 8.4 Hz, H-4), 5.65 (1H, s, H-5), 5.35 (1H, s, H-5′), 4.53 (1H, d, J = 13.2 Hz, H-12α’), 3.99 (1H, d, J = 12.6 Hz, H-12α), 3.90 (1H, d, J = 12.6 Hz, H-12β’), 3.60 (1H, d, J = 13.2 Hz, H-12β), 2.30-2.50 (6H, m, H-6, H-6′, H2-7, H2-7′), 2.06 (3H, s, 5-OCOCH3), 2.03 (3H, s, 5′-OCOCH3), 1.76 (3H, s, 12′-OCOCH3), 1.03 (3H, d, J = 6.6 Hz, CH3-11), 0.95 (3H, d, J = 6.6 Hz, CH3-11′); 1H NMR (600 MHz, CDCl ) and 13C NMR (150 MHz, CDCl3) see Table 1; HRESIMS m/z 709.2126 ([M+H]+ calcd for C36H37O15, 709.2132).
Table 1.
| 1 | 2 | |||||
|---|---|---|---|---|---|---|
| # | δC, type (I) | δC, type (II) | δH (J in Hz) (I) |
δH (J in Hz) (II) |
δC, type | δH (J in Hz) |
| 1, 1’ | 160.2, C | 161.9, C | 162.0, C | |||
| 2, 2’ | 118.6, C | 110.3, CH | 6.58, d (8.4) | 108.1, CH | 6.35, d (8.4) | |
| 3, 3’ | 139.4, CH | 139.4, CH | 7.16, d (8.4) | 7.32, d (8.4) | 139.4, CH | 7.39, t (8.4) |
| 4, 4’ | 107.6, CH | 117.4, C | 6.43, d (8.4) | 109.7, CH | 6.47, d (8.4) | |
| 4a, 4a’ | 157.6, C | 155.0, C | 158.2, C | |||
| 5, 5’ | 72.1, CH | 69.8, CH | 5.55, br s | 5.30, br s | 70.6, CH | 5.90, br s |
| 6, 6’ | 28.0, CH | 27.9, CH | 2.47, m | 2.28, m | 27.5, CH | 2.57, m |
| 7, 7’ | 33.7, CH2 | 33.3, CH2 | 2.30~2.50, m | 2.30~2.50, m | 33.4, CH2 | 2.57, m; 2.37, m |
| 8, 8’ | 177.9, C | 177.9, C | 179.5, C | |||
| 8a, 8a’ | 101.5, C | 100.8, C | 100.3, C | |||
| 9, 9’ | −, C | −, C | −, C | |||
| 9a, 9a’ | 106.8, C | 106.7, C | 106.5, C | |||
| 10a, 10a’ | 80.7, C | 80.3, C | 80.8, C | |||
| 5,5’- OCOCH3 |
170.9, C 3 | 170.8, C | 169.8, C | |||
| 5,5’- OCOCH3 |
21.3, CH3 | 21.3, CH3 | 2.11, s | 2.05, s | 20.3, CH3 | 2.03, s |
| 11, 11’ | 17.9, CH3 | 17.6, CH3 | 1.07, d (6.0) | 0.97, d (6.0) | 17.0, CH3 | 1.03, d (6.0) |
| 12, 12’ | 65.6, CH2 | 64.2, CH2 | 3.94, m | 4.52, d (12.0) | 65.0, CH2 | 4.51, d (12.0) |
| 3.75, m | 3.86, d (12.0) | 4.27, d (12.0) | ||||
| 12,12’- OCOCH3 |
170.1, C | 169.5, C | ||||
| 12,12’- OCOCH3 |
21.1, CH3 | 1.83, s | 20.1, CH3 | 1.99, s | ||
| 1,1’-OH | 11.62, s | 11.20, s | ||||
† (ppm) 600 MHz; multiplicities; J values (Hz) in parentheses.
† (ppm) 150 MHz; chemical shifts from gHSQC and gHMBC.
Penexanthone B (2)
yellow powder; [α]23D +2.5 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 208 (3.89), 222 (sh), 267 (sh), 276 (3.28), 332 (3.90) nm; IR (film) νmax 3420, 1741, 1602, 1441, 1230, 1044 cm−1; 1H NMR (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) see Table 1; HRESIMS m/z 377.1243 ([M+H]+ calcd for C19H21O8, 377.1236).
Biological Assay
As previously described,1 stromal cells were plated and allowed to seed overnight for non-adherent tumor experiments. The following day, tumor cells were overlaid and treated with natural product extracts. Following 48 hrs of incubation, luciferin substrate was added, cultures incubated for 30 min and bioluminescence signal read on an Envision or Luminoskan luminometer. For adherent tumors, stromal cells were counted and plated with tumor cells, both allowed to seed overnight and treated with extracts the following day. Cultures were again incubated for 48 h in the presence of extracts and bioluminescence signal read on a luminometer following addition of luciferin substrate. Conditions were normalized to each respective non-treated control and each condition was performed in quadruplicate. High-throughput screening of natural product extract libraries was performed by screening MM.1S multiple myeloma (MM) tumor cells in the presence and absence of HS-5 bone marrow stromal cells (BMSCs). Natural product hits were identified as those extracts which resulted in enhanced anti-tumor killing in the presence of BMSCs compared to their absence, with minimal cytotoxic activity against the HS-5 BMSCs alone. Tumor cells were co-cultured with stromal cells for 48 h unless otherwise stated.
Supplementary Material
Figure 1.

Key HMBC correlations in monomer I of 1
Chart 1.

Structures of compounds 1-5
ACKNOWLEDGMENT
This work was generously supported by NIH U01 TW007404 (J.C.) and NIH R01 CA050947 (C.S.M). We thank Joshua A.V.D. Blodgett for taking the photo and helping deposit the DNA sequence into GenBank.
Footnotes
ASSOCIATED CONTENT
NMR spectra of penexanthone A (1) and penexanthone B (2). These materials are available free of charge via the internet at http://pubs.acs.org.
REFERENCES AND NOTES
- (1).a McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC, Mitsiades CS. Nat. Med. 2010;16:483–489. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McMillin DW, Delmore J, Negri JM, Ooi M, Klippel S, Miduturu CV, Gray NS, Richardson PG, Anderson KC, Kung AL, Mitsiades CS. PLoS ONE. 2011;6:e20226. doi: 10.1371/journal.pone.0020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).a Cao S, Ross L, Tamayo G, Clardy J. Org. Lett. 2010;12:4661–4663. doi: 10.1021/ol101972g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cao S, Clardy J. Tetrahedron Lett. 2011;52:2206–2208. doi: 10.1016/j.tetlet.2010.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Isaka M, Jaturapat A, Rukseree K, Danwisetkanjana K, Tanticharoen M, Thebtaranonth Y. J. Nat. Prod. 2001;64:1015–1018. doi: 10.1021/np010006h. [DOI] [PubMed] [Google Scholar]
- (4).a Aberhart DJ, Chen YS, de Mayo P, Stothers JB. Tetrahedron. 1965;21:1417–1432. [Google Scholar]; b Aberhart DJ, De Mayo P. Tetrahedron. 1966;22:2359–2366. [Google Scholar]; c Hopper JW, Marlow W, Whalley WB, Borthwick AD, Bowden R. J. Chem. Soc. Chem. Commun. 1971:111–112. doi: 10.1039/j39710003580. [DOI] [PubMed] [Google Scholar]; d Hopper JW, Marlow W, Whalley WB, Borthwick AD, Bowden R. J. Chem. Soc. C. 1971:3580–3590. doi: 10.1039/j39710003580. [DOI] [PubMed] [Google Scholar]; e Yang D-M, Takeda N, Iitaka Y, Sankawa U, Shibata S. Tetrahedron. 1973;29:519–528. [Google Scholar]; f Andersen R, Buechi G, Kobbe B, Demain AL. J. Org. Chem. 1977;42:352–353. doi: 10.1021/jo00422a042. [DOI] [PubMed] [Google Scholar]; g Kurobane I, Vining LC, McInnes AG. Tetrahedron Lett. 1978;19:4633–4636. [Google Scholar]; h Proksa B, Uhrin D, Liptaj T, Turdikova M. Phytochemistry. 1998;48:1161–1164. [Google Scholar]; i Wagenaar MM, Clardy J. J. Nat. Prod. 2001;64:1006–1009. doi: 10.1021/np010020u. [DOI] [PubMed] [Google Scholar]; j Pontius A, Krick A, Mesry R, Kehraus S, Foegen SE, Michael Müller M, Klimo K, Gerhäuser C, König GM. J. Nat. Prod. 2008;71:1793–1799. doi: 10.1021/np800392w. [DOI] [PubMed] [Google Scholar]; k Zhang W, Krohn K, Zia-Ullah, Flörke U, Pescitelli G, Di Bari L, Antus S, Kurtán T, Rheinheimer J, Draeger JS, Schulz B. Eur. J. Org. Chem. 2008;14:4913–4923. doi: 10.1002/chem.200800035. [DOI] [PubMed] [Google Scholar]
- (5).Lee CH, Kim JG, Lim CS, Kim JY. Repub. Korean Kongkae Taeho Kongbo. KR 2011021258 A 20110304. 2011
- (6).Elsässer B, Krohn K, Flörke U, Root N, Aust H-J, Draeger S, Schulz B, Antus S, Kurtán T. Eur. J. Org. Chem. 2005;11:4563–4570. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.