

Abstract
Glomerulosclerosis is a common pathologic finding that often progresses to renal failure. The mechanisms of chronic kidney disease progression are not well-defined but may include activation of numerous vasoactive and inflammatory pathways. We hypothesized that podocytes are susceptible to filtered plasma components including hormones and growth factors that stimulate signaling pathways leading to glomerulosclerosis. Gα12 couples to numerous G-protein-coupled receptors (GPCR) and regulates multiple epithelial responses including proliferation, apoptosis, permeability and the actin cytoskeleton. Herein, we report that genetic activation of Gα12 in podocytes leads to time dependent increases in proteinuria and glomerulosclerosis. To mimic activation of Gα12-pathways, constitutively active Gα12(QL) was conditionally expressed in podocytes using Nphs2-Cre and LacZfloxed QLα12 transgenic mice. Some QLα12LacZ+/Cre+ mice developed proteinuria at 4-6m, and most were proteinuric by 12m. Proteinuria increased with age, and by 12-14m many demonstrated glomerulosclerosis with ultrastructural changes including foot process fusion and both mesangial and subendothelial deposits. QLα12LacZ+/Cre+ mice showed no changes in podocyte number, apoptosis, proliferation, or Rho/Src activation. Real-time PCR revealed no significant changes in Nphs1, Nphs2, Cd2ap, or Trpc6 expression, but Col4a2 message was increased in younger and older mice while Col4a5 was decreased in older mice. Confocal microscopy revealed disordered collagen IVα1/2 staining in older mice and loss of α5 without changes in other collagen IV subunits. Taken together, these studies suggest that Gα12 activation promotes glomerular injury without podocyte depletion through a novel mechanism regulating collagen (α)IV expression, and supports the notion that glomerular damage may accrue through persistent GPCR activation in podocytes.
Glomerulosclerosis (GS) is a common pathologic finding in patients with progressive chronic kidney disease (CKD) and often leads to end stage renal disease (ESRD). Numerous conditions predispose patients to GS including diabetes, hypertension, IgA nephropathy, FSGS (focal segmental GS) and immune-mediated injury. In adults over 60 years-old, the prevalence of CKD Stage III (glomerular filtration rate, 30-59mL/min) is estimated to be >25% (1). Although risk factors such as hypertension and diabetes are linked to CKD, little is known about the signaling mechanisms that lead to progression with ageing. Post mortem and nephrectomy samples in otherwise “healthy“ adults reveal variable amounts of glomerulosclerosis and interstitial fibrosis, suggestive of age associated damage (2, 3). Recent studies show that primary podocyte injury is sufficient to induce GS (4, 5). Podocytes are exposed to filtered reactive oxygen species (ROS), lipid mediators, cytokines and hormones that could contribute to injury. Many of these molecules activate G protein-coupled-receptors (GPCR), which couple to multiple Gα subunits. Each of the 16 Gα subunits (four main families; Gαs, Gαi/o, Gαq and Gα12/13) couples to many different GPCRs (6); thus, defining specific pathways in vivo has been difficult.
Gα12/13 are expressed in podocytes and couple to angiotensin II (AII), thrombin, endothelin, and LPA receptors, that are important in renal injury (7). Gα12/13 can activate Rho or Src to regulate the actin cytoskeleton (8), in addition to proliferation, transformation (9), tight junction (TJ) assembly (10-12), cell-cell adhesion (13, 14), directed cell migration (15), apoptosis (16), and cell attachment (17). RhoGDIα knockout mice develop proteinuria and renal failure (18), and many mutations in hereditary FSGS affect proteins linked to the actin cytoskeleton (reviewed in (19)). Gα12 also upregulates TGFβ (20, 21) and, several gene profiling studies found upregulated Gα12 in proteinuric kidneys and post transplant CKD (via Nephromine (22, 23)).
Targeting activated Gα subunits to specific cells in vivo permits identification of downstream effector pathways independent of receptor activation and thus, permits insight in to disease mechanisms otherwise impossible to study in vivo. Herein, we confirm expression of endogenous Gα12 in the major podocyte processes. Constitutively activated Gα12 (QLα12) was expressed in podocytes using a transgenic model that results in mosaic expression and mimics the focal nature of GS pathology. QLα12LacZ+/Cre+ mice develop proteinuria and focal GS without differences in podocyte number, apoptosis, proliferation, or Rho/Src signaling over time. Col4a was disregulated and correlated with altered localization and ultrastructural changes. These findings indicate that Gα12 activation in podocytes leads to disregulated collagen α(IV) expression, and supports a model of altered glomerular structure and function resulting from time dependent stimulation of GPCR-Gα12 signaling pathways.
Methods
Transgenic Mice Creation
All animal procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at Harvard University. Gα12 (Q229L) EE tagged was cloned in to the CMV floxed LacZ cassette kindly provided by Dr. Larry Holzman (24). C57/B6 mice were injected at the Brigham and Women’s Hospital Transgenic Mouse Facility and were then crossed with Nphs2/Cre mice on the same C57/BL6 background.
Urine Microalbumin, Serum Creatinine, LPS and Tissue Harvesting
Male and female mice were analyzed for urine microalbumin/creatinine ratio at specified ages using Bayer DCA 200+ Analyzer with software version E3.11/01.04. Mice were defined as proteinuric when microalbumin/creatinine ratio ≥34 due to the detection limits of the analyzer. LPS (Invivogen, San Diego, CA) (IP 10μg/g) was administered to 2-6m old mice, and urine was collected 18h post-injection and microalbumin/creatinine ratio. Serum was collected and BUN and creatinine analyzed using an iStat System with CHEM8+ catridges. Mice were anesthetized using inhaled isofluorane (Phoenix Pharmaceuticals, St. Louis, MO). For whole kidney collection, organs were perfused with cold PBS. Kidneys were removed and processed according to methods below.
GST-TPR Pull Down Assay
The GST-TPR construct was kindly provided by Dr. M. Negishi, Kyoto University, Kyoto Japan. GST-TPR was prepared as described previously (16). Harvested kidneys were homogenized in lysis buffer with eComplete (Roche) protease inhibitors, lysed through 22G needle and normalized for protein concentration. 1 μg of GST-TPR coupled to glutathione-agarose beads (Amersham Biosciences) was added to 800 μg of total protein and rocked overnight at 4 °C. Beads were pelleted with low speed centrifugation and washed three times with PBS, 0.1% Triton X-100, resuspended in Laemmli sample buffer, and analyzed by SDS-PAGE and Western blot using a Gα12 antibody (Santa Cruz, Santa Cruz, CA).
Immunogold EM
Immunogold labeling and EM was performed at the Membrane Biology Program, Massachusetts General Hospital. Normal mouse kidney was fixed in 2% fresh-made paraformaldehyde and 0.5% glutaraldehyde in 0.1M phosphate buffer, pH 7.4 and processed according to standard conditions. Grids were incubated with Rabbit anti-Gα12 (Santa Cruz) overnight at 4°C followed by a 25μl droplet of anti-goat IgG conjugated with gold (10nm). Grids were washed and imaged using Philips CM10 electron microscope (Philips Electronics, Mahwah, NJ).
Histology
Fixed kidney tissue was paraffin-processed and 4μm sections were stained with PAS. Light microscopic evaluation included quantification of the total number of glomeruli present in one microscopic section as well as quantification of glomeruli with lesions (global sclerosis, segmental sclerosis, glomerular collapse, and glomerular hypertrophy).
Electron microscopy
Fixed specimens were trimmed, post-fixed with osmium tetroxide, dehydrated in serial ethanols, and embedded in epoxy resin. Ultrathin sections were cut at 80nm, mounted on 200 mesh copper grids, treated with uranyl acetate and lead citrate, and examined in a JEOL 1010 transmission electron microscope (Tokyo, Japan). Electron micrographs showing glomerular ultrastructure were analyzed in a blinded manner. Podocytes, endothelium, GBM, and mesangium were examined and scored from 0 (no abnormality) to 4 (severe abnormality). Podocytes were scored for foot process effacement and irregularity, microvillous degeneration, vacuolization, and subepithelial deposits. Endothelium was scored for: double contours, subendothelial deposits, and swelling. GBM abnormalities were scored for thickness and irregularity, and the mesangium was scored for deposits, expansion by matrix, and cellular expansion.
LacZ, TUNEL, and Immunostaining
Fixed kidneys were washed in PBS and rehydrated overnight in 30% sucrose. Kidneys were embedded in O.C.T Compound (TissueTek, Sakura Finetek, Torrence, CA) and frozen in liquid nitrogen. For WT-1 and TUNEL staining, sections were labeled using the “In Situ Cell Death Detection Kit” (Roche) according to the manufacturers protocol. Kidneys were incubated in methanol, blocked, and incubated in anti-WT-1 antibody (sc-192/Santa Cruz) at 4°C overnight. Sections were washed, incubated with appropriate secondary antibody, and mounted using Prolong Gold (Invitrogen). Kidneys were photographed on a Nikon E-1000 equipped with a SPOT digital camera. For collagen staining, kidneys were blocked in 1% BSA, and incubated with primary antibodies (generous gift of D.B. Borga, Vanderbilt) or (Rockland Gilbertsville, PA). All sections were incubated with appropriate secondary antibody for 1hr at room temperature, and mounted using Prolong Gold (Invitrogen). Images were taken on a Nikon C1 D-Eclipse confocal microscope.
Glomerular isolation
Glomerular isolation was performed using magnetic beads(25). Two hundred μL Dynabeads (M450 Tosylactivated, Invitrogen, Carlsbad, CA) were washed with 0.1% PBS and incubated overnight in 0.2M Tris pH8.5 with 0.1%BSA. Beads were washed and resuspended in 30mL HBSS. Mice were anesthetized and organs perfused with HBSS. Kidneys were digested in 1mg/ml collagenase A (Roche, Indianapolis, IN) and 100u/ml DNaseI (New England Biolabs, Ipswich, MA) at 37°C for 30min. Digests were strained though a 100μm cell strainer, centrifuged and resuspended in HBSS. Glomeruli were obtained using magnetic separator (New England Bio Labs, Ipswich, MA).
RhoA Activation ELISA and Western Blot
Isolated glomeruli were resuspended in lysis buffer with eComplete (Roche) protease inhibitors and lysed through 22G needle. Rho activity was determined using G-LISA™ RhoA Activation Assay Kit (Cytoskeleton Denver CO) and absorbance at 490nm of an HRP-active RhoA antibody. Additionally, samples were analyzed by SDS-PAGE and Western blot analysis using an anti-RhoA antibody (Cytoskeleton) as previously described (17).
Real Time PCR
Purified glomeruli were lysed in TRIzol reagent (Invitrogen). RNA concentrations were determined, and RNA was incubated in DNAse I (New England Biolabs) with RNAse Inhibitor (Roche). DNAse treated RNA was reverse transcribed using Transcriptor Reverse Transcriptase (Roche). Negative control without enzyme was included in each analysis. The cDNA template was treated with RNaseH (Invitrogen). TaqMan Gene Expression assays (Applied Biosystems Foster City, CA) were performed using an ABI 7300 (Applied Biosystems) with the following conditions: 2min at 50°C and 10 min at 95°C, followed by 50 cycles of 95°C for 15s and 60°C for 1min. Melting curve analysis and gel electrophoresis of PCR products verified that a single product of the expected size was generated with each primer set. Data analysis used the ΔΔCt method. Ct was normalized to 18S ribosomal subunit.
Statistical Analyses
Data are expressed as medians or means ± SEM as indicated. Statistical analysis was performed using Prism 4 for Macintosh (GraphPad La Jolla, CA) using two-way ANOVA followed by Bonferroni’s post hoc test. Statistical significance was identified at P < 0.05.
Results
Gα12 is Expressed in Glomeruli
Although Gα12 is expressed in proximal and distal tubular epithelial cells (26) and glomerular proteomics identified Gα12 (27), its localization in glomeruli has not been described. Endogenous Gα12 was localized in normal mouse glomeruli by immunohistochemistry. At lower power, Gα12 was detected in glomeruli (Figure 1A; arrow heads) and throughout the nephron. At higher power (Figure 1A, black/white arrows), several cell types including podocytes, appear to express Gα12 with only background staining in the controls (antibody preincubated with peptide) (Figure 1A, b, d). To confirm podocyte Gα12 localization, immunogold electron microscopy (EM) was performed (Figure 1B). Gold particles were visible in several locations including foot processes (FP), major processes (MP), and at the branch points proximal to the foot processes (arrows). The localization of Gα12 in major processes suggests that Gα12 may have functions not directly related to slit diaphragm permeability.
Figure 1. Endogenous Gα12 is expressed in normal mouse kidney.
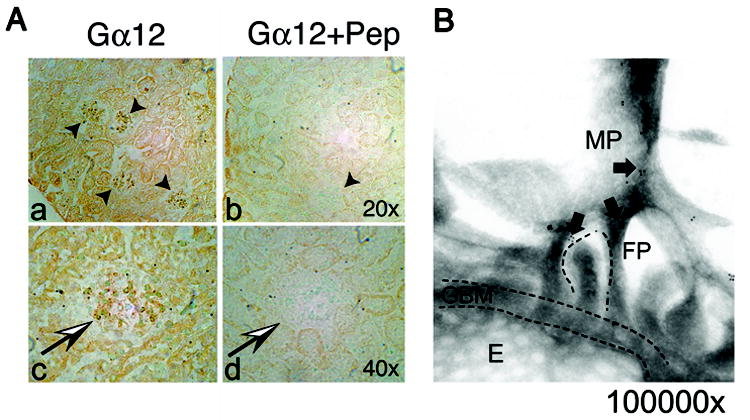
(A) Immunohistochemistry of normal mouse kidney demonstrates Gα12 expression in glomeruli. Sections were probed with rabbit anti-Gα12 and visualized with Vectastain. Two magnifications (20x and 40x) are shown (panel a, b). Negative controls were performed by pre-incubating the antibody with excess blocking peptide (Gα12 + Pep). Kidney sections using the blocked Gα12 antibody showed a significant reduction in staining (panel b, d). (B) Immunoelectron microscopy shows Gα12 localizes to interdigitating foot processes (FP) and major processes (MP). Immunogold labeling and electron microscopy were performed as described in materials and methods. Magnification ~100,000x. Arrows denote gold particles. Glomerular basement membrane (GBM), foot processes (FP), fenestrated endothelium (E), and larger major processes (MP) are labeled.
Establishing Transgenic Mice with Podocyte Expression of Activated Gα12 (QLα12)
Mice expressing EE tagged, human Gα12 (Q229L) were established using a LacZ/floxed transgenic approach ((24), Figure 2A). For conditional expression (28), QLα12LacZ+/Cre- mice were crossed with Nphs2-Cre mice, (Nphs2 is efficiently expressed in podocytes and no other glomerular cells, (29) Cre-mediated excision of the LacZ/stop in podocytes resulted in QLα12 expression (QLα12LacZ+/Cre+) (Figure 2). The CMV promoter is associated mosaic expression due to random inactivation (24) although Cre efficiently excises LacZ in these podocytes. QLα12LacZ+/Cre- (Control) and QLα12LacZ+/Cre+ mice at 2m of age (littermates) were stained for β-Gal using standard techniques. β-gal staining revealed that transgene expression was low in proximal tubules, but higher in the papilla [as reported (24), not shown]. QLα12LacZ+/Cre+ mice showed a major reduction in glomerular LacZ staining (Figure 2B) compared with Cre- controls. A semi-quantitative analysis of β-gal staining suggested that random inactivation of the CMV promoter in podocytes led to LacZ expression in approximately half of the targeted cells (Supplemental Figure 1). Note that all the Cre+ mice showed significantly less or no β-Gal staining while the controls were evenly distributed (Supplemental Figure 1). The small amount of residual LacZ staining in QLα12LacZ+/Cre+ mice likely reflects LacZ expression in mesangial or endothelial cells. To distinguish EE-QLα12 expression from endogenous Gα12, control and QLα12LacZ+/Cre+ kidneys were co-stained with anti-nephrin and anti-EE antibodies (Figure 2C). In control mice, nephrin was seen throughout the glomerulus, with no detectable EE staining (Figure. 2C, a). In QLα12LacZ+/Cre+ mice, both proteins were detected (Figure 2C, b), and as expected from immunoEM (Figure 1B), there was little overlap with nephrin in the slit diaphragm. ImmunoEM using the EE epitope antibodies confirmed transgenic QLα12 expression (not shown) in a similar distribution to the endogenous Gα12 (Figure 1B). To confirm that active Gα12 was expressed in these glomeruli, GST-TPR pulldowns were performed as previously described (16); the TPR domain of PP5 binds the active conformation of Gα12/13 (30). Figure 2D shows GST-TPR pulldowns of thrombin stimulated MDCK cells compared with cortical kidney lysates from QLα12LacZ+/Cre+ and QLα12LacZ+/Cre- mice.
Figure 2. Development of transgenic mice with podocyte specific expression of QLα12.
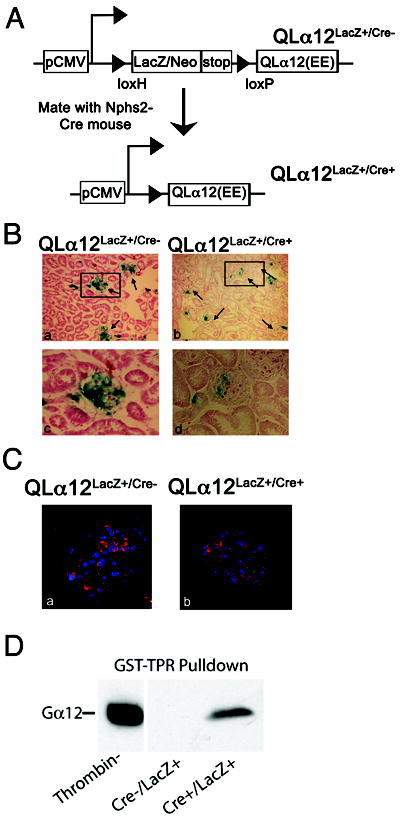
(A) Schematic of targeting epitope tagged (EE) human QLα12 to podocytes. The floxed LacZ/stop is driven by CMV promoter and Nphs-2 Podocin-Cre was used for podocyte expression. (B) Transgenic mice show mosaic expression of LacZ. Control and QLα12LacZ+/Cre+ mice at 2m of age (littermates) were stained for β-gal as describe in materials and methods. Insets (c, d) show an individual glomerulus. (C) QLα12LacZ+/Cre+ mice express EE-tagged QLα12 in podocytes. Immunofluorescent staining was performed on control (a) and QLα12LacZ+/Cre+ (b) mice using FITC conjugated goat anti-EE (shown in green) and guinea pig anti-nephrin (Progen) and Cy3 secondary antibody (shown in red.) (D) Activated Gα12 was pulled down from kidney lysates of QLα12LacZ+/Cre+ or thrombin stimulated MDCK cells using GST-TPR or GST alone.
QLα12 Expressed in Podocytes Leads to Age-Dependent Proteinuria
Development of microalbuminuria is a sensitive marker for podocyte injury (31). Urine was analyzed for albumin/creatinine ratio (Figure 3A) from QLα12LacZ+/Cre+ and control mice every two months. Microalbuminuria (albumin/creatinine ratios ≥34) appeared in a few control mice at 4-6m but was seen in ~40% of QLα12LacZ+/Cre+ mice (some with ratio >200, Figure 3, Table 1). Most the QLα12LacZ+/Cre+ mice developed proteinuria as they aged while only a few controls had mildly increased levels. Coomassie blue staining of urine confirmed the expression of albumin (Figure 3B). The magnitude of proteinuria continued to increase until sacrifice at 22-26m. Based on this phenotype, we divided the mice into young (<6m; occasional mild proteinuria) and older (>12m; frequent and often severe proteinuria) for further analysis.
Figure 3. QLα12LacZ+/Cre+ mice develop of proteinuria with age.
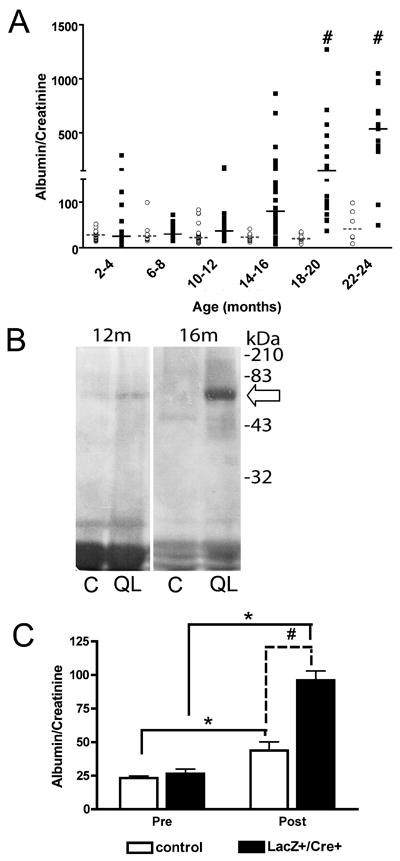
(A) Urine microalbumin/creatinine ratio in QLα12 mice is higher than in controls. Urine from control and QLα12 mice were monitored every two months using a BCA Analyzer. Albumin/Creatinine from individual control (○) and QLα12 (■) mice are shown. Lines indicate median value (dashed, control; solid, QLα12). (B) Urine from QLα12 mice contains high levels of albumin. Urine from 12 and 16 month QLα12 (QL) and littermate control mice (C) was collected and analyzed by SDS-PAGE and Coommassie Blue staining. The arrow denotes ~66kDa, the size of excreted albumin. Note that the 12m mice had more concentrated urine (based on the non-specific low molecular weight bands). (C) QLα12 mice are more susceptible to LPS induced injury. Control (n=17) and QLα12 (n=21) mice were injected with 10μg/g body weight LPS. Urine was collected 18h post injection and analyzed for urine microalbumin/creatinine ratio a BCA Analyzer. Statistical analysis was performed using two-way ANOVA followed by Bonferroni’s post hoc test (# p<0.001, * p<0.0001)
Table 1.
Transgenic mice develop microalbuminuria
| Age | 4-6 | 8-10 | 12-14 | 16-18 | 20-22 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Genotype | C | ++ | C | ++ | C | ++ | C | ++ | C | ++ | |
| A/C | Average±SEM | 28±1.7 | 65±20 | 32±5.5 | 42±4.0 | 26±2.4 | 80±10 | 20±1.3 | 105±17 (#) | 29±8.1 | 340±98 (#) |
| Range | 16-52 | 8-292 | 12-99 | 15-118 | 10-54 | 8-322 | 12-32 | 12-475 | 8-79 | 73-427 | |
| Mice with ≥34 | 3/24 | 6/15 | 5/22 | 14/30 | 2/17 | 28/40 | 0/18 | 27/28 | 3/9 | 8/9 | |
C=control Mice
=QLα12LacZ+/Nphs2-Cre+ mice
p<0.001, by two-way ANOVA followed by Bonferroni’s post hoc test
The lack of proteinuria in most younger mice suggests that podocytes compensate for the expression of activated Gα12. To test whether QLα12 expression predisposed younger mice (<6m) to injury, proteinuria was examined after LPS stimulation (a model of transient podocyte injury) (Figure 3C)(32). Baseline proteinuria was similar, and 18h after LPS stimulation, control mice increased proteinuria 1.9-fold, while QLα12LacZ+/Cre+ mice showed a significantly larger increase (3.6-fold). However, these short-term experiments did not detect any differences in ultrastructural or light microscopy findings (not shown). These findings are consistent with LPS-stimulated podocyte actin cytoskeleton changes leading to proteinuria and reveal that podocyte expression of QLα12 in young mice enhances the injury response.
Focal GS Develops with Age in QLα12LacZ+/Cre+ Mice
In mice aged <6m, no significant renal histopathologic changes were observed in control or QLα12LacZ+/Cre+ mice (Figure, 4 A, E). However, by 12-18m, QLα12LacZ+/Cre+ mice showed numerous globally and segmentally sclerosed glomeruli while controls exhibited only rare sclerosed glomeruli. Quantifying affected glomeruli showed a >6-fold increase in sclerosed glomeruli from kidneys of QLα12LacZ+/Cre+ mice (n=7) compared with controls (n=7) (B, F). Segmentally sclerosed glomeruli were characterized by hyalinosis, glomerular basement membrane (GBM) reduplication, isolated epithelial cells containing PAS-positive protein reabsorption granules, and adhesion of the tuft to Bowman’s capsule. Mesangial areas of QLα12LacZ+/Cre+ mice exhibited mild to focally moderate expansion by matrix and cells, whereas the mesangial areas of control mice exhibited minimal expansion (B, F, arrow). The kidneys of 18m mice exhibited changes similar to the 12-18m group, but more prominent. On average, 4.79% of glomeruli in QLα12LacZ+/Cre+ mice were globally or segmentally sclerosed, whereas only 0.16% of glomeruli in control mice were sclerosed (Figure 4 C, D, E, H,). Over 73% of these sclerosed glomeruli were juxtamedullary. Focal segmental double contours (areas of GBM redundancy) were more commonly seen in QLα12LacZ+/Cre+ mice than in controls. Occasional PAS-positive hyaline casts were observed in the medulla to varying degrees in older animals as well as focal interstitial mononuclear inflammation, usually in association with focal GS. (Figure 4 G, H arrows). Features of focal tubular injury, including tubular luminal distension, epithelial flattening, and microcyst formation, were seen in a subset of the oldest QLα12LacZ+/Cre+ mice and these findings were not seen in the age matched controls. The non-lesional glomeruli (H; arrowhead) exhibit moderate mesangial expansion by matrix and cells (*). Other features of active or chronic tubulointerstitial or vascular pathology were not observed in any animals. There were no detectable differences in serum creatinine between the 14-19 month old QLα12LacZ+/Cre+ mice and controls as all values were ≤0.2 mg/dL.
Figure 4. Light micrographs show focal and segmental glomerulosclerosis in the juxtamedullary region of older QLα12LacZ+/Cre+ mice.
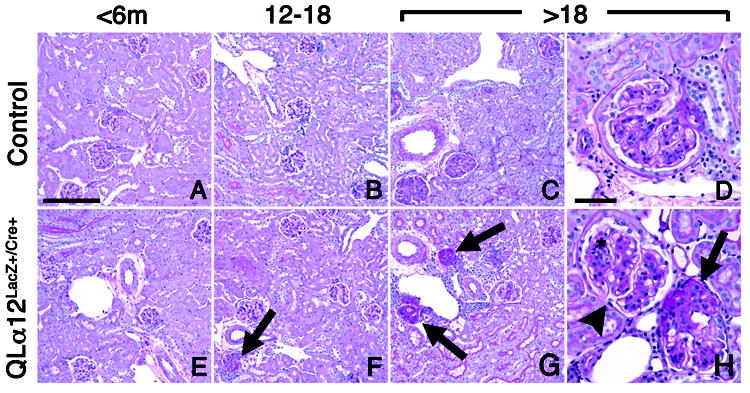
Representative light micrographs of murine juxtamedullary kidney cortex in control (top row) and QLα12LacZ+/Cre+ (bottom row) mice aged 4.5, 13, and 24 m are shown (A,E) Kidneys of mice aged <6 m, regardless of genotype, show no significant pathologic changes in glomeruli, tubulointerstitium, or vasculature. (B, F) Kidney of QLα12LacZ+/Cre+ mice (F) aged 12-18 months exhibit focal glomerulosclerosis involving juxtamedullary glomerulus (arrow). The parenchyma is otherwise well-preserved. Age-matched controls (B) show no significant pathologic changes. (C,D,G,H) Kidneys of QLα12LacZ+/Cre+ mice aged >18m show focal global (G) and segmental (H) glomerulosclerosis involving multiple juxtamedullary glomeruli (arrows), accompanied by focal interstitial mononuclear inflammation. The non-lesional glomeruli (H; arrowhead) exhibit moderate mesangial expansion by matrix and cells (*). Age-matched controls (C,D) show mild mesangial expansion but no other cortical parenchymal lesions are apparent. PAS; bar = 100 microns (left 3 columns), 50 microns (right column).
Older QLα12LacZ+/Cre+ Mice Develop GBM Irregularities, Mesangial Expansion and Foot Process Fusion
EM of QLα12LacZ+/Cre+ and control mice <6m, 12-18m and >18m were scored for ultrastructural abnormalities. The youngest QLα12LacZ+/Cre+ mice (Figure 5A) appeared normal. At 12-18m, there was little difference in the GBM or the podocytes, but most QLα12LacZ+/Cre+ mice had focal features of endothelial injury including double contours, subendothelial granular electron dense deposits, and cell swelling (Figure 5E arrowhead; not seen in the controls). Additionally, QLα12LacZ+/Cre+ scored higher for mesangial expansion by matrix and cells (Figure 5E,*; Table 2). Older QLα12LacZ+/Cre+ mice (>18m) (Figure 5F) mice exhibited a greater degree of foot process effacement and irregularity presumably associated with aging (Figure 5F, arrows) than was seen in the controls (although controls did reveal some age-related changes). There were few differences in the GBM, but there were an increased number of subepithelial GBM membrane projections in the oldest transgenics (Figure 5F, ◆). All mice exhibited mild mesangial expansion at >18m; however QLα12LacZ+/Cre+ mice showed more severe signs of mesangial abnormalities (Figure 5F, *), and overall scores (Table 2) for glomerular injury were twice as high in the QLα12LacZ+/Cre+ mice (Figure 5C).
Figure 5. QLα12LacZ+/Cre+ develop foot process fusion and ultrastructural changes that worsen with age.
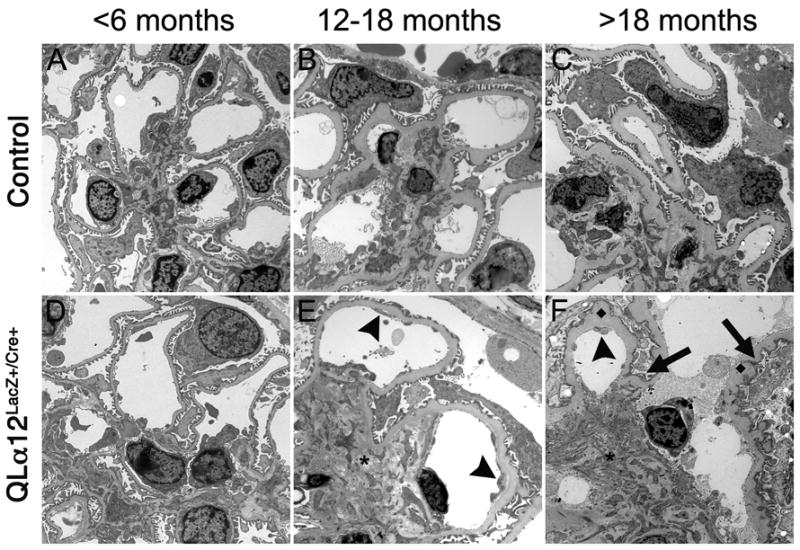
Transmission electron microscopy (EM) was performed on kidneys from control and QLα12LacZ+/Cre+ mice at <6m(A, D), 12-18m (B, E), and >18 m (C, F) were analyzed in a blindly and scored for severity of injury. Representative micrographs in control (top row) and QLα12LacZ+/Cre+ (bottom row) mice aged 4, 14, and 23m are shown. At <6m, both control (A) and QLα12LacZ+/Cre+ (D) show normal glomerular structure. By 12-18m, the QLα12LacZ+/Cre+ mice (E) show more signs of endothelial injury (arrowhead) and mesangial expansion (*) than controls (B). All of the oldest mice examined show significant GBM thickening but the QLα12LacZ+/Cre+ (F) show increased development of subepithelial basement membrane projections (◆) along the GBM. The podocytes have more foot process effacement and irregularity (arrow) in addition to the mesangial expansion (*) and endothelial injury arrowhead) seen the 12-18m mice compared with controls (C).
Table 2.
QLα12LacZ+/Cre+ mice score higher for glomerular injury
| Injury | PODOCYTES | GBM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FP EFF | FP IRREG | MICROVIL | KNOBS | DCs | |||||||
|
| |||||||||||
| Genotype | C | ++ | C | ++ | C | ++ | C | ++ | C | ++ | |
| Scores by Age (m) | <12 | 0 | 0.33 | 0.75 | 1 | 0.5 | 0.33 | 0.25 | 0.67 | 0 | 0 |
| 12-18 | 1 | 0.6 | 1.3 | 1 | 1 | 1 | 1.67 | 1.4 | 0.33 | 1 | |
| 18-24 | 0 | 1.5 | 0.33 | 1.8 | 0.66 | 1.25 | 1 | 2 | 0 | 1.2 | |
| Injury | ENDOTHELIUM | ||||||||
| SUBEND DEP | SWELLING | MES MATRIX | MES CELLS | ||||||
|
| |||||||||
| Genotype | C | ++ | C | ++ | C | ++ | C | ++ | |
|
| |||||||||
| Scores by Age (m) | <12 | 0 | 0 | 0 | 0.33 | 0.75 | 1 | 0.5 | 0.67 |
| 12-18 | 0 | 0.6 | 0 | 0.6 | 1 | 1.8 | 1 | 1.2 | |
| 18-24 | 0 | 0.88 | 0 | 1 | 1.3 | 2.6 | 1.7 | 2.4 | |
C=control Mice
++=QLα12LacZ+/Nphs2-Cre+ mice
FP=foot process
EFF=effacement
DC=double contours
MES=mesangial
QLα12LacZ+/Cre+ Mice Have Normal Numbers of Podocytes
Based on the morphologic changes, we examined podocyte number by Wilms’s Tumor-1 (WT-1) staining (33). Surprisingly, podocyte number/glomerulus showed no difference between QLα12LacZ+/Cre+ and controls >12m (Figure 6A, B). As Gα12 can stimulate both proliferation and apoptosis (16), WT-1 positive cells were quantified for apoptosis (TUNEL) and proliferation (Ki67) to exclude the possibility that podocyte number was preserved through QLα12 costimulation of proliferation and apoptosis. No apoptotic podocytes were seen in either QLα12LacZ+/Cre+ or controls (Figure 6A, middle panels), nor any difference observed in proliferation (not shown). Additionally, there were no differences in the number of glomeruli in kidneys obtained from QLα12 LacZ+/Cre+ mice as compared to controls (data not shown). This indicates that development of proteinuria and glomerulosclerosis does not result from developmental effects on glomeruli number or podocyte depletion, and other mechanisms must be responsible.
Figure 6. QLα12LacZ+/Cre+ mice have normal numbers of podocytes.
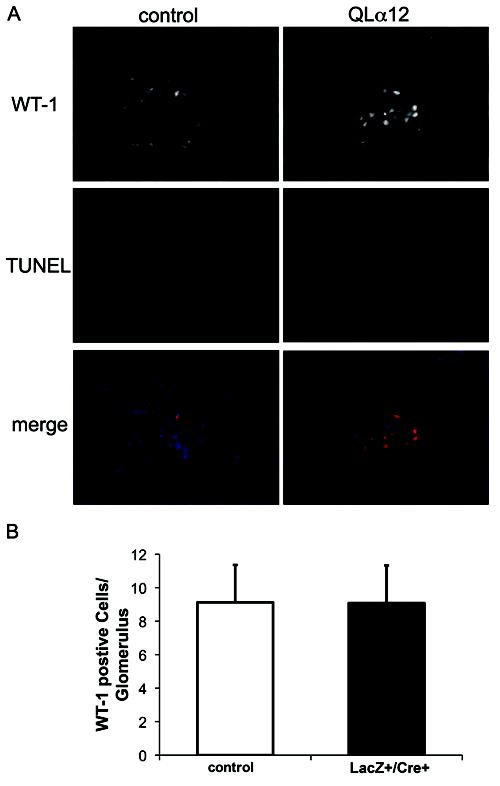
(A) WT-1 staining shows little podocyte apoptosis in both control and QLα12 mice. 24 fields and total >200 podocytes were counted for control and QLα12LacZ+/Cre+ mice. TUNEL assays were performed on kidney sections from QLα12 and control mice 2-6 and 12-16m of age. Additionally, sections were probed for WT-1 and stained for DAPI. (B) WT-1 quantification shows similar number of podocytes in both control and QLα12 mice. The number of cells per glomeruli stained for both WT-1 and DAPI quantified for 100 glomeruli and averaged.
QLα12 Expression in Podocytes Does Not Lead to Rho or Src Activation
Next, Rho activity was examined in QLα12Lac+/Cre+ and control mice. Rho activity was determined by ELISA on glomerular isolates from young and old mice. There were no significant differences in Rho activity from QLα12Lac+/Cre+ mice compared with controls, nor was there any difference in Rho protein expression (Figure 7A). Gα12 also activates Src (34), and Western blot using pY419 antibodies failed to demonstrate any differences in Src activation (not shown). This finding suggests that podocytes compensate for persistent QLα12 expression and employ other mechanisms to prevent sustained Rho or Src activation.
Figure 7. RhoA activity is not altered in QLα12LacZ+/Cre+ mice.
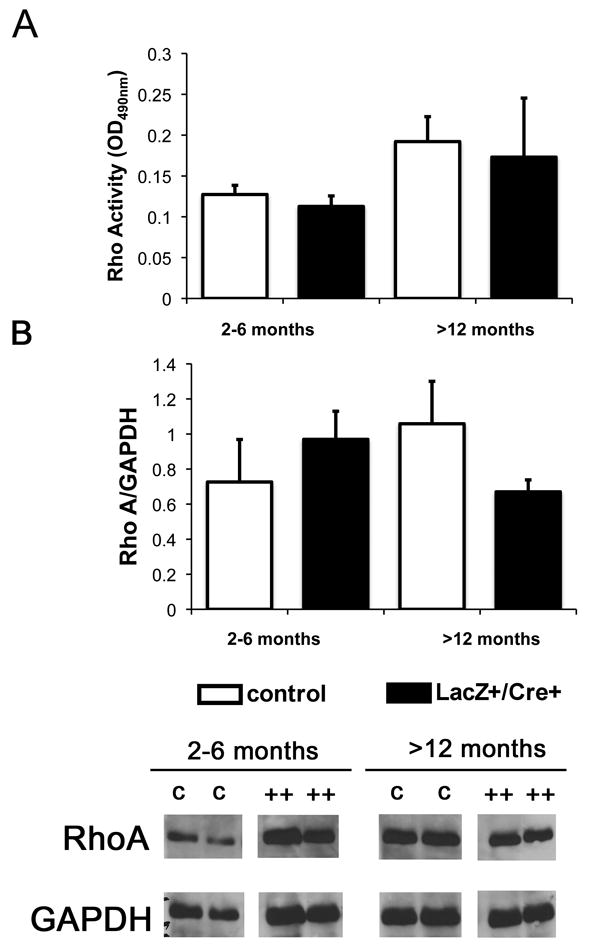
(A) ELISA for activated RhoA was performed on young (2-6mo) and old (>12mo) QLα12LacZ+/Cre+ and control mice. (B) Total RhoA did not change in QLα12LacZ+/Cre+ mice as they age. Western blot analysis was performed to examine total RhoA. Blots were stripped and re-probed for GAPDH and ImageJ was used to determine RhoA/GAPDH expression.
Real Time PCR Showed No Changes in Podocyte Specific Genes, but QLα12LacZ+/Cre+ Mice Exhibit Disregulated Glomerular Collagen Expression
Real time PCR of glomerular isolates failed to detect changes in expression levels of several major podocyte genes implicated in FSGS (Nphs1, Nphs2, CD2AP, and TRPC6) (Supplemental Figure 2). The normal adult GBM is composed predominantly of α3, α4 and α5(IV) collagen and laminin-11 (α5β2γ1)(35). During development, the normal GBM is composed of α1 and α2(IV) collagen that is followed by a switch to α3, α4 and α5(IV) expression in mature glomeruli. Real time PCR of Col4a showed increased Col4a2 transcript expression levels in younger mice (Figure 8A) although the variability in phenotype led to wide confidence intervals. These age-dependent changes are likely to contribute to the observed variability and since this analysis was performed on isolated glomeruli, it is possible that the most severely sclerotic glomeruli were not included and would thus tend to underestimate the differences. In addition, the real time results were reanalyzed in male and female mice, and no differences were observed to account for the differences in phenotype seen between transgenic and control mice in any age group (not shown). Nevertheless, when considered together, these results suggest a change in the relative balance of specific collagen α(IV) chains, and immunofluorescence microscopy confirmed mildly increased collagen α1/2(IV) expression (Figure 8C). In older mice, Col4a2 expression abnormalities persist and by 12-14m there is also a 2-fold increase in Col4a1 and decreased Col4a5 (Figure 8C). Consistent with the real-time results, collagen α1/2(IV) staining was increased in older mice, and the localization was disorganized without clear capillary loop staining (Figure 8D). Decreased expression of α5 was confirmed although the pattern of staining appeared similar to the control (Figure 8D). No other α(IV) chains showed differences in staining. The α3/4/5 antibody showed decreased intensity compared with the control and based on the α3 staining and real-time results, this difference is most likely explained by the decreased α5 collagen expression. To determine if QLα12 directly regulates Col4 gene expression, a previously characterized inducible (tet off) QLα12-MDCK cell line (10, 11, 17) was analyzed by microarray. Supplemental Figure 3 shows 7 and 10 fold increase in Col4a1 and Col4a2 respectively within 72h of QLα12 expression. Taken together, this analysis indicates that QLα12 expression leads to disregulated Col4a gene expression prior to the onset of proteinuria and suggests a mechanism where activated Gα12 alters Col4 gene expression.
Figure 8. Collagen (α)IV is misregulated QLα12LacZ+/Cre+ mice.
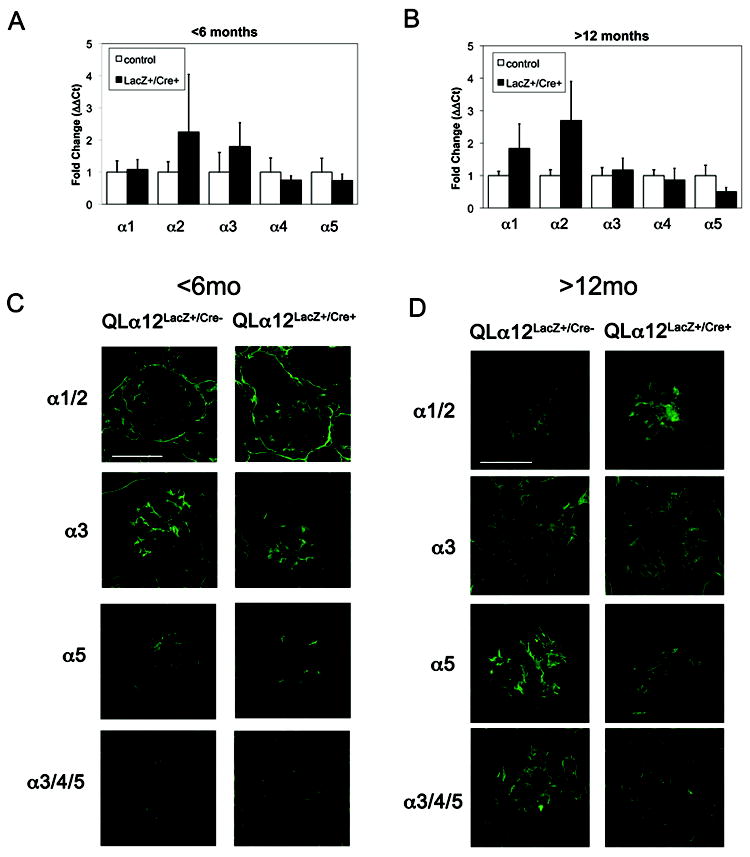
(A-B) Col4a1, Col4a2, and Col4a5 transcript expression are altered in QLα12LacZ+/Cre+ mice >12m of age. Various α chains of COL4 were examined in A (2-6 month) or B (>12 month) mice. Results were normalized to the 18S ribosomal subunit and graphed as relative expression compared to non-targeting control (normalized to 1). n≥ 6 mice for each experiment. (C) Immunofluorescent staining was performed on frozen kidney sections using collagen antibodies [α1/2, Rockland; α3NC1 (mAb 8D1), α3α4α5NC1 (mAb 26-20), α5 (polyclonal)] (52) and Alexa488 anti-mouse or anti-rabbit secondary antibody. Representative images are shown from QLα12LacZ+/Cre- and QLα12LacZ+/Cre+ mice 2-6 month or 12-16 month mice. Scale bar=100 microns
Discussion
Understanding the mechanisms of CKD progression is important for finding new therapeutic targets. Podocytes are exposed to approximately 180L of ultrafiltrate/day that contains biologically active molecules, including hormones, cytokines, and filtered proteins. Herein, we demonstrate that activation of Gα12 in podocytes leads to age-dependent proteinuria and focal GS through a distinct mechanism involving disregulated collagen α(IV) expression without podocyte depletion. This supports the hypothesis that filtered agonists activate podocyte signaling pathways and can contribute to progressive glomerular injury. Although Gα12 regulates numerous processes that could lead to podocyte damage including apoptosis, proliferation, cell attachment, actin cytoskeletal changes, and junctional regulation, these do not explain the time-dependent phenotype in these mice. Rather, a novel mechanism of collagen α(IV) abnormalities appears to be responsible.
Testing the notion that podocytes respond to filtered molecules is difficult to examine in vivo. Expressing a constitutively active G-protein allows for identification of downstream effector pathways, and this approach led to the identification of h-Ras as an oncogene (36). This strategy could identify novel therapeutic targets to prevent GS without the need to identify the specific GPCR or its ligand. Analogous to our studies, activated (QL) Gαq was targeted to podocytes (37) and these mice developed proteinuria through a different mechanism. QLαq mice had developmental defects including smaller kidneys and reduced nephron number. There was downregulation of nephrin and other podocyte genes although Col4a was not examined. QLαq mice were more susceptible to puromycin aminonucleoside injury, and a subset of mice developed GS at 6m of age, a finding that could occur from reduced nephron number. In contrast, QLα12 in podocytes did not affect renal development, nephron, or podocyte number and revealed altered regulation of collagen and GBM abnormalities.
Proteinuria developed in some QLα12LacZ+/Cre+ mice by 4-6m, prior to morphologic changes (Figures 3-5). By 12-14m, there was some foot process fusion that could account for the increased in proteinuria; however, the CMV promoter results in mosaic expression, and we estimate that only about half of podocytes express QLα12 (Figure 2). This focal expression is likely to account for the mild phenotype, but also recapitulates local differences in glomerular response to stress and the focal nature of GS. In fact, the majority of sclerotic glomeruli were located at the cortico-medullary junction, a region where glomeruli are more susceptible to hemodynamic stress (38, 39). Why some younger mice manifest proteinuria before morphologic changes are detected is not clear. They do not show enhanced baseline Rho or Src activity, but they are more susceptible to LPS induced proteinuria (without changes in Col4 gene expression (not shown), suggesting that QLα12 can promote functional effects on permeability. The absence of ultrastructural changes in young mice or after LPS suggests that perhaps through Gα12 localization in the major foot processes, it regulates cortical actin and permeability. Although only about 5% of the glomeruli develop GS, proteinuria is likely to arise from a more global effect on significantly more glomeruli. This is based on the observation that QLα12 is expressed in approximately 50% of glomeruli (Supplemental Fig. 1) and QLα12 expression increased proteinuria in LPS treated mice without morphologic or ultrastructural changes.
Mesangial expansion was prominent in the QLα12LacZ+/Cre+ mice, and mesangial expansion is particularly characteristic of FSGS, membranous and diabetic nephropathy, Alport’s syndrome, and Denys-Drash syndrome (40). Mesangial expansion can contribute to proteinuria through a mechanism that does not require podocyte depletion (as seen in this study) (41). Additionally, both nephropathy and Alport’s syndrome are both characterized by changes in collagen expression (42-44). Transcription of Col4a1 and A2 is coregulated by the same promoter due to head-head orientation in chromosome 13q34 (45). As a result, diseases affecting collagen IV expression will be associated with changes in both α1(IV) and α2(IV)) as seen in both the above disease and QLα12 mice (Figure 8). However, Alport’s syndrome is caused by mutations in α3, α4, or α5(IV) collagen leading to sustained α1 and α2(IV) expression whereas membranous nephropathy results from auto-antibodies (46) that promote podocyte injury and subepithelial deposits.
Based on the time course of proteinuria and morphologic changes, we speculate that Gα12 modifies COL4 gene expression and the resulting basement membrane and endothelial abnormalities accrue with time. However, whether these changes reflect age-dependent effects or are the result of cumulative Gα12 activation will require additional study. Since C57/B6 mice develop mild ultrastructural changes, cytokine activation and proteinuria with age (as seen in some control mice), the phenotypic differences seen in the older mice with Gα12 activation could represent indirect effects on cumulative age-related changes in this mouse strain, or a cumulative time effect of Gα12 activation. Although the control mice do develop mild changes with age, the dramatic differences seen in the age and sex matched QLα12 mice strongly implicates the persistent Gα12 activation as the etiology. Furthermore, the finding of up regulated COL4A1/2 seen with QLα12 expression in MDCK cells (Supplemental figure 3) supports an important link, between Gα12 activation and Col4A1/2 gene expression.
Although Gα12 stimulates TGFβ (20, 21) and could promote GS, we were unable to detect increased TGFβ in either young or old mice (not shown). However, an alternative mechanism is suggested by the increased number of subepithelial GBM membrane projections (Fig. 5) similar to findings seen in DDR1 and integrin α2 knockout mice (47, 48). DDR1 is a tyrosine kinase receptor for collagen IV, and we previously showed that Gα12 regulates α2β1 integrin signaling and attachment (17), thus suggesting that persistent Gα12 activation might lead to podocyte changes in integrin signaling. The onset of proteinuria is during adulthood and virtually all mice have proteinuria by mid-life (12-16m, normal C57/B6 mouse life span ~3y (49)). With ageing, podocytes may accumulate damage from a variety of sources including GPCR linked inflammatory and vasoactive mediators. ROS, inflammation, and vascular changes occur in hypertensive and diabetic kidney disease, and ROS directly activates Gα subunits (50). Based on these observations, we suggest that this model of slowly progressive kidney disease will be valuable for understanding CKD progression in humans. Consistent with GPCR pathways regulating age related kidney damage, AT1 receptor knockout mice exhibit decreased oxidative damage and outlive their wild-type littermates (51). The glomerular filtration rate declines with age, but it is unclear whether this is part of normal ageing or represents injury from cumulative exposures. If decreased renal function and GS occur through repetitive activation of podocyte signaling pathways, it is tempting to speculate that inhibiting Gα12 pathways in podocytes may provide new treatments to protect renal function and delay CKD progression.
Supplementary Material
Acknowledgments
We would like to thank Dr. Dorin-Bogdan Borza for his consultation and the gift of the collagen antibodies, Drs. Johannes Schlondorff and Michael Ross for their knowledge of podocytes and glomerular isolation, and Andrea Bernhardy for her technical assistance. Additionally, we thank Dr. Dennis Brown and Margaret McLaughlin for help with the immunoelectron microscopy, and Dr. Hui Chen and Colleen Ford for their technical assistance with EM of the transgenic mice.
This research was supported by NIH grants R21DK065932and R01GM055223 to B.M.D. I.B was supported by NIH training grant T32DK007527. IB conceived and carried out experiments, analyzed data, prepared figures, and contributed to writing and editing the paper; WY, SB, HN and MT conceived and carried out experiments and analyzed data; MP contributed to editing the paper; JH carried out experiments, analyzed data, prepared figures, and contributed to editing the paper; and BMD conceived of experiment and contributed to writing and editing the paper.
Footnotes
Conflict of Interest: None
References
- 1.U S Renal Data System. USRDS 2010 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2010. [Google Scholar]
- 2.Zhou XJ, Rakheja D, Yu X, et al. The aging kidney. Kidney Int. 2008;74(6):710–20. doi: 10.1038/ki.2008.319. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt R, Cantley LG. The impact of aging on kidney repair. Am J Physiol Renal Physiol. 2008 Jun 1;294(6):F1265–F72. doi: 10.1152/ajprenal.00543.2007. [DOI] [PubMed] [Google Scholar]
- 4.Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005 Oct 1;16(10):2941–52. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 5.Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005 Apr 1;16(4):1013–23. doi: 10.1681/ASN.2004080720. [DOI] [PubMed] [Google Scholar]
- 6.Worzfeld T, Wettschureck N, Offermanns S. G12/G13-mediated signalling in mammalian physiology and disease. Trends Pharmacological Sci. 2008;29(11):582. doi: 10.1016/j.tips.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Riobo NA, Manning DR. Receptors coupled to heterotrimeric G proteins of the G12 family. Trends Pharmacol Sci. 2005 Mar;26(3):146–54. doi: 10.1016/j.tips.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Buhl AM, Johnson NL, Dhanasekaran N et al. Gα12 and Gα13 Stimulate Rho-dependent Stress Fiber Formation and Focal Adhesion Assembly. J Biol Chem. 1995 Oct 20;270(42):24631–4. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H, Wu D, Simon MI. The transforming activity of activated Gα12. FEBS Lett. 1993;330(3):319–22. doi: 10.1016/0014-5793(93)80896-3. [DOI] [PubMed] [Google Scholar]
- 10.Meyer TN, Hunt J, Schwesinger C, et al. Gα12 regulates epithelial cell junctions through Src tyrosine kinases. Am J Physiol Cell Physiol. 2003 Nov 1;285(5):C1281–93. doi: 10.1152/ajpcell.00548.2002. [DOI] [PubMed] [Google Scholar]
- 11.Sabath E, Negoro H, Beaudry S, et al. Gα12 regulates protein interactions within the MDCK cell tight junction and inhibits tight-junction assembly. J Cell Sci. 2008 Mar 15;121(6):814–24. doi: 10.1242/jcs.014878. [DOI] [PubMed] [Google Scholar]
- 12.Meyer TN, Schwesinger C, Denker BM. Zonula Occludens-1 Is a Scaffolding Protein for Signaling Molecules: Gα12 directly binds to the Src homology 3 domain and regulates paracellular permeability in epithelial cells. J Biol Chem. 2002 Jul 12;277(28):24855–8. doi: 10.1074/jbc.C200240200. [DOI] [PubMed] [Google Scholar]
- 13.Meigs TE, Fields TA, McKee DD, et al. Interaction of Gα12 and Gα13 with the cytoplasmic domain of cadherin provides a mechanism for β-catenin release. Proc Natl Acad Sci U S A. 2001 Jan 16;98(2):519–24. doi: 10.1073/pnas.021350998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meigs TE, Fedor-Chaiken M, Kaplan DD, et al. Ga12 and Ga13 Negatively Regulate the Adhesive Functions of Cadherin. J Biol Chem. 2002 Jun 28;277(27):24594–600. doi: 10.1074/jbc.M201984200. [DOI] [PubMed] [Google Scholar]
- 15.Goulimari P, Kitzing TM, Knieling H, et al. Gα12/13 Is Essential for Directed Cell Migration and Localized Rho-Dia1 Function. J Biol Chem. 2005 Dec 23;280(51):42242–51. doi: 10.1074/jbc.M508690200. [DOI] [PubMed] [Google Scholar]
- 16.Yanamadala V, Negoro H, Gunaratnam L, et al. Gα12 Stimulates Apoptosis in Epithelial Cells through JNK1-mediated Bcl-2 Degradation and Up-regulation of IKBa. J Biol Chem. 2007 Aug 17;282(33):24352–63. doi: 10.1074/jbc.M702804200. [DOI] [PubMed] [Google Scholar]
- 17.Kong T, Xu D, Yu W, et al. Gα12 inhibits α2 β1 integrin-mediated Madin-Darby canine kidney cell attachment and migration on collagen-i and blocks tubulogenesis. Mol Biol Cell. 2009;20(21):4596–610. doi: 10.1091/mbc.E09-03-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Togawa A, Miyoshi J, Ishizaki H, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIα. Oncogene. 1999 Sep 23;18(39):5373–80. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 19.Pollak MR. Focal segmental glomerulosclerosis: recent advances. Curr Opin Nephrol Hypertens. 2008;17(2):138–42. doi: 10.1097/MNH.0b013e3282f5dbe4. [DOI] [PubMed] [Google Scholar]
- 20.Tsunoda S, Yamabe H, Osawa H, et al. Cultured rat glomerular epithelial cells show gene expression and production of transforming growth factor-β: expression is enhanced by thrombin. Nephrol Dial Transplant. 2001 Sep 1;16(9):1776–82. doi: 10.1093/ndt/16.9.1776. [DOI] [PubMed] [Google Scholar]
- 21.Lee SJ, Yang JW, Cho IJ et al. The gep oncogenes, Gα12 and Gα13, upregulate the transforming growth factor-β1 gene. Oncogene. 2009;28(9):1230. doi: 10.1038/onc.2008.488. [DOI] [PubMed] [Google Scholar]
- 22.Martini S, Eichinger F, Nair V, et al. Defining human diabetic nephropathy on the molecular level: Integration of transcriptomic profiles with biological knowledge. Rev Endocr Metab Disord. 2008;9(4):267–74. doi: 10.1007/s11154-008-9103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007 Feb;9(2):166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moeller MJ, Soofi A, Sanden S, et al. An efficient system for tissue-specific overexpression of transgenes in podocytes in vivo. Am J Physiol Renal Physiol. 2005 Aug 1;289(2):F481–8. doi: 10.1152/ajprenal.00332.2004. [DOI] [PubMed] [Google Scholar]
- 25.Takemoto M, Asker N, Gerhardt H, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161(3):799–805. doi: 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng S, Yu P, Zeng C, et al. Gα12- and Gα13-Protein Subunit Linkage of D5 Dopamine Receptors in the Nephron. Hypertension. 2003 Mar 1;41(3):604–10. doi: 10.1161/01.HYP.0000057422.75590.D7. [DOI] [PubMed] [Google Scholar]
- 27.Miyamoto M, Yoshida Y, Taguchi I, et al. In-depth proteomic profiling of the normal human kidney glomerulus using two-dimensional protein prefractionation in combination with liquid chromatography-tandem mass spectrometry. J Proteome Res. 2007 Sep;6(9):3680–90. doi: 10.1021/pr070203n. [DOI] [PubMed] [Google Scholar]
- 28.Moeller MJ, Sanden SK, Soofi A, et al. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis. 2003 Jan;35(1):39–42. doi: 10.1002/gene.10164. [DOI] [PubMed] [Google Scholar]
- 29.Moeller MJ, Sanden SK, Soofi A, et al. Two Gene Fragments that Direct Podocyte-Specific Expression in Transgenic Mice. J Am Soc Nephrol. 2002 Jun 1;13(6):1561–7. doi: 10.1097/01.asn.0000015614.68893.0b. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi Y, Katoh H, Mori K, et al. Gα12 and Gα13 Interact with Ser/Thr Protein Phosphatase Type 5 and Stimulate Its Phosphatase Activity. Current Biology. 2002;12(15):1353. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- 31.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13(12):3005–15. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 32.Reiser J, von Gersdorff G, Loos M, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004 May;113(10):1390–7. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mundlos S, Pelletier J, Darveau A, et al. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development. 1993 Dec;119(1)(4):1329–41. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- 34.Ma YC, Huang J, Ali S, et al. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102(5):635–46. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 35.Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83(1):253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 36.White MA, Nicolette C, Minden A, et al. Multiple ras functions can contribute to mammalian cell transformation. Cell. 1995;80(4):533–41. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Fields TA, Pazmino K, et al. Activation of Gαq-coupled signaling pathways in glomerular podocytes promotes renal injury. J Am Soc Nephrol. 2005;16(12):3611–22. doi: 10.1681/ASN.2005020167. [DOI] [PubMed] [Google Scholar]
- 38.Olson J. Renal disease caused by hypertension (Chapter 21) In: Jennette J, Olson J, Schwartz M, Silva F, editors. Heptinstall’s Pathology of the Kidney. 6. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 937–90. [Google Scholar]
- 39.Brezis M, Rosen S, Silva P, et al. Renal ischemia: A new perspective. Kidney Int. 1984;26(4):375–83. doi: 10.1038/ki.1984.185. [DOI] [PubMed] [Google Scholar]
- 40.Lee HS. Pathogenic role of TGF-beta in the progression of podocyte diseases. Histol Histopathol. Jan;26(1):107–16. doi: 10.14670/HH-26.107. [DOI] [PubMed] [Google Scholar]
- 41.Wiggins JE, Goyal M, Sanden SK, et al. Podocyte Hypertrophy, “Adaptation,” and “Decompensation” Associated with Glomerular Enlargement and Glomerulosclerosis in the Aging Rat: Prevention by Calorie Restriction. J Am Soc Nephrol. 2005 Oct;16(1)(10):2953–66. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- 42.Zhang YZ, Lee HS. Quantitive changes in the glomerular basement membrane components in human membranous nephropathy. J Pathol. 1997;183(1):8–15. doi: 10.1002/(SICI)1096-9896(199709)183:1<8::AID-PATH1079>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 43.Kalluri R, Shield CF, Todd P, et al. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997 May 15;99(10):2470–8. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim TS, Kim JY, Hong HK, et al. mRNA expression of glomerular basement membrane proteins and TGF-β1 in human membranous nephropathy. J Pathol. 1999;189(3):425–30. doi: 10.1002/(SICI)1096-9896(199911)189:3<425::AID-PATH454>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 45.Sado Y, Kagawa M, Naito I, et al. Organization and Expression of Basement Membrane Collagen IV Genes and Their Roles in Human Disorders. Journal of Biochemistry. 1998 May 1;123(5):767–76. doi: 10.1093/oxfordjournals.jbchem.a022003. [DOI] [PubMed] [Google Scholar]
- 46.Beck LH, Bonegio RGB, Lambeau G, et al. M-Type Phospholipase A2 Receptor as Target Antigen in Idiopathic Membranous Nephropathy. N Engl J Med. 2009;361(1):11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross O, Beirowski B, Harvey SJ, et al. DDR1-deficient mice show localized subepithelial GBM thickening with focal loss of slit diaphragms and proteinuria. Kidney Int. 2004;66(1):102–11. doi: 10.1111/j.1523-1755.2004.00712.x. [DOI] [PubMed] [Google Scholar]
- 48.Girgert R, Martin M, Kruegel J, et al. Integrin α2-deficient mice provide insights into specific functions of collagen receptors in the kidney. Fibrogenesis & Tissue Repair. 3(1):19. doi: 10.1186/1755-1536-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison D. [2011 March 30];Baseline life span data: commonly used JAX Mice and crosses. http://research.jax.org/faculty/harrison/ger1vi_LifeStudy1.html.
- 50.Nishida M, Maruyama Y, Tanaka R, et al. Gα(i) and Gα(o) are target proteins of reactive oxygen species. Nature. 2000;408(6811):492–5. doi: 10.1038/35044120. [DOI] [PubMed] [Google Scholar]
- 51.Benigni A, Corna D, Zoja C, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009 Mar;119(3):524–30. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang JS, Wang X-P, Miner JH, et al. Loss of α3/α4(IV) collagen from the glomerular basement membrane induces a strain-dependent isoform switch to α5α6(IV) collagen associated with longer renal survival in Col4a3-/- Alport mice. J Am Soc Nephrol. Jul;200617(1)(7):1962–9. doi: 10.1681/ASN.2006020165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.