

Abstract
Background: Ornithine transcarbamylase deficiency (OTCD) is an X‐linked urea cycle disorder characterized by hyperammonemia resulting in white matter injury and impairments in working memory and executive cognition. Objective: To test for differences in BOLD signal activation between subjects with OTCD and healthy controls during a working memory task. Design, setting and patients: Nineteen subjects with OTCD and 21 healthy controls participated in a case‐control, IRB‐approved study at Georgetown University Medical Center. Intervention: An N‐back working memory task was performed in a block design using 3T functional magnetic resonance imaging. Results: In subjects with OTCD we observed increased BOLD signal in the right dorsolateral prefrontal cortex (DLPFC) and anterior cingulate cortex (ACC) relative to healthy age matched controls. Conclusions: Increased neuronal activation in OTCD subjects despite equivalent task performance points to sub‐optimal activation of the working memory network in these subjects, most likely reflecting damage caused by hyperammonemic events. These increases directly relate to our previous finding of reduced frontal white matter integrity in the superior extents of the corpus callosum; key hemispheric connections for these areas. Future studies using higher cognitive load are required to further characterize these effects. Hum Brain Mapp, 2013. © 2011 Wiley Periodicals, Inc.
Keywords: ammonia, dorsolateral prefrontal cortex (DLPFC), fMRI, ornithine transcarbamylase deficiency (OTCD), hyperammonemia (HA)
INTRODUCTION
Ornithine transcarbamylase deficiency (OTCD; OMIM #311250) is an X‐linked inborn error of metabolism and the most common of the urea cycle disorders, with an incidence of 1:14,000 [Kang et al.,1973]. Approximately 60% of hemizygous males present with newborn coma, while the remainder typically exhibit a less severe phenotype [Msall et al.,1984]. Female heterozygotes display a broad phenotype, owing to allelic heterogeneity and differential patterns of X‐inactivation. Roughly 85% are considered clinically asymptomatic despite subtle differences in metabolic and neurocognitive parameters of brain function, while the remainder exhibit neurocognitive abnormalities, stroke‐like episodes and protein intolerance [Gropman and Batshaw,2004; Gyato et al.,2004]. Deficient protein metabolism in OTCD results in episodes of hyperammonemia (HA) with acute elevations of ammonia that cause substantial injury to the brain's white matter [Gropman et al.,2010]. Additionally, “asymptomatic” OTCD is associated with altered neurochemical profile [Gropman, et al.,2008] in an array of cognitive subdomains based in the prefrontal cortex (PFC), such as working memory, executive cognition and attention [Gropman and Batshaw2004; Gropman et al.,2008]. These deficits contribute significantly to disability in OTCD [Gyato et al.,2004] despite normal global IQ [Gropman and Batshaw,2004].
Working memory, defined as the storage, manipulation, and retrieval of information in conscious awareness over brief intervals, is a critical component of executive cognition and is known to be impaired in OTCD [Baddeley,2003; Gyato et al.,2004]. A network of brain structures subserves working memory, with the dorsolateral prefrontal cortex (DLPFC) functioning as its central executive, regulating the encoding and manipulation of memory items [Fuster,1997; Postle,2006]. DLPFC activation follows an inverted U‐shaped curve in response to increases in cognitive load, with neural activation peaking when task demands maximally tap an individual's cognitive capacity, and decreasing after capacity is breached [Callicott et al.,1999]. DLPFC activation is classified as “inefficient” when an individual reaches peak activation at low levels of cognitive demand, relative to the normal population [Callicott et al.,2003; Rypma et al.,2006]. In such cases, increased neurophysiological input is required for a given level of neurocognitive output. A multitude of pathologic processes may result in this phenotypic endpoint, and prefrontal inefficiency has been documented in a variety of populations, including schizophrenia [Callicott et al.,2003] and healthy ageing [Mattay et al.,2006].
Over the past several decades, functional magnetic resonance imaging (fMRI) has emerged as a powerful tool for investigating human brain function in vivo. A noninvasive imaging modality, fMRI exploits the magnetic contrast between oxygenated and deoxygenated hemoglobin, using the blood oxygenation level‐dependent (BOLD) signal to index metabolically active brain regions as a function of regional cerebral blood flow. To date, no study has reported fMRI findings in patients with OTCD.
While current understanding of the pathophysiologic mechanisms of cognitive dysfunction in OTCD demands further study, evidence from clinical neuroimaging, rodent modeling, and in vitro cell culture studies converge on a plausible neurologic model, in which hyperammonemic cytotoxicity induces significant neurochemical stress on the central nervous system [for review, see Braissant,2010].
Glutamine synthesis is one of the brain's primary mechanisms for buffering against increases in ammonia, a process mediated by the astrocytic enzyme gluatmine synthetase. It has been shown in a cell cultures model that the osmotic activity of glutamine induces astrocytic swelling and cytotoxic edema [Bachmann et al.,2004], conferring considerable strain throughout the central nervous system. Clinically, MRI‐based investigations have revealed a structural phenotype characterized by basal ganglia atrophy, hypomyelination, cortical inshemia, cysts, and ventricular dilation [Braissant,2010].
This metabolic stress has consequences across an array of neural structures and chemical systems. In a mouse model that mutation of the OTCD gene results in significant loss of cholinergic neurons in the forebrain [Ratnakumari et al.,1994]. Loss of cholinergic neurons has also been demonstrated in cell cultures exposed to NH4Cl [Braissant et al.,2002]. Damage to the cholinergic system significantly disrupts the synthesis, neurotransmission and metabolism of dopamine, well known to subserve higher order cognitive processing in the human prefrontal cortex. Even subtle dopaminergic imbalances have been shown to affect cognitive performance in healthy subjects [Mattay et al.,2003], and are suspected as a key mechanism in the cognitive deficits associated with schizophrenia [Callicott et al.,2003].
Robust correlations have been identified between cognitive decline in OTCD and degree of exposure to hyperammonemic conditions [Braissant,2010]. While extensive work remains to characterize the specific nature of neurotransmitter imbalance and neurocognitive pathways affected in OTCD, it is plausible that structural damage to cholinergic neurons depletes these patients' stores of prefrontal dopamine, and that increased neural activation during prefrontally mediated cognitive operations is employed as a compensatory response.
In this study, we sought to determine whether neurologic insult conferred by HA is associated with occult manifestations of neurocognitive impairment in OTCD subjects. To this end, we used cognitive assessment to compare indices of executive function and fMRI to compare task‐dependent neural activation between subjects with OTCD and age‐matched controls. During fMRI testing, subjects completed an N‐back working memory paradigm, known to tap cognitive resources subserved by the PFC.
MATERIALS AND METHODS
Participants
OTCD subjects were recruited through the Online Rare Diseases Clinical Research Network (RDCRN) registry, the National Urea Cycle Disorders Foundation (NUCDF), the Society for Inherited Metabolic Disease (SIMD), and colleagues of the principal investigator known to serve OTCD patients in metabolic clinics throughout the country. Control subjects were recruited via IRB‐approved advertisements posted throughout the Georgetown University Hospital, medical school and graduate school. Control participants were matched to within five years of age of recruited OTCD cases. Medical histories for cases and controls were screened to exclude history of epilepsy, stroke, cognitive dysfunction, liver disease, and psychiatric illness. Subjects meeting these criteria who scored above 80 on the Wechsler Abbreviated Scale of Intelligence (WASI) were invited to participate in the study.
Nineteen subjects with a diagnosis of OTCD (16 female, 3 male, 33.2 ± 2.7 y) and 21 healthy controls (14 female, 7 male, 31.8 ± 2.7 y) gave informed consent approved by the Georgetown University Medical Center Biomedical Institutional Review Board before completing the experiment. An attempt was made to match subjects for age, handedness, and gender (Table I). However, OTCD subjects had significantly lower full‐scale IQ scores than controls (P = 0.001).
Table I.
Demographic information and task performance across OTCD and control groups
| OTCD group | Control group | |
|---|---|---|
| Age | 33.2 ± 2.7 y | 31.8 ± 2.7 y |
| Gender | 3 M, 16 F | 7 M, 14 F |
| Handedness | 16 R, 3 L | 17 R, 4 L |
| IQ | 111 ± 2.1** | 122 ± 2.7 |
| Digit Span Score | 10.4 ± 0.6* | 13.2 ± 0.6 |
| Stroop Test Reaction Time (Incongruent Trials) | 1089 ± 34** | 874 ± 42 |
| CTMT Composite Score | 40.68 ± 1.67** | 50.25 ± 1.77 |
| 1‐back Acc. | 0.992 ± 0.0050 | 0.991 ± 0.0027 |
| 1‐back RT | 500 ± 24 ms | 501 ± 23 ms |
| 2‐back Acc. | 0.972 ± 0.0074 | 0.974 ± 0.0063 |
| 2‐back RT | 528 ± 21 ms | 540 ± 24 ms |
The groups are well matched and only differ on full‐scale IQ (P = 0.001). Variance reflects ± 1 standard deviation. Digit span data were only available for six OTCD subjects.
P < 0.05,
P < 0.01.
Cognitive Assessment
We investigated neurocognitive performance differences between OTCD subjects and controls using three assessments of executive cognition: the digit span portion of the Wechsler Abbreviated Scale of Intelligence (WASI), an index of working memory capacity; the Stroop task, an index of cognitive conflict monitoring in which words for colors (e.g., “green”) are printed in the corresponding color (congruent trials) or a different color (incongruent trials); and the Comprehensive Trail‐Making Test (CTMT), an index of attention and cognitive flexibility. Digit span data were available for 22 healthy controls and 6 OTCD subjects, and STROOP and CTMT data were available for all subjects in the fMRI analysis. Independent samples t‐tests were performed to compare scaled scores between groups for each of these tests, using a significance threshold of P < 0.05.
Task Design
Participants completed an N‐back continuous performance working memory task with blocks of 1‐back and 2‐back loads. The task presented a sequence of individual uppercase letters, and the subject indicated when the current stimulus matched the one presented N steps earlier in the sequence. Each letter was presented for 1,000 ms and was separated from the following stimulus by 1,500 ms of blank screen. Six blocks each of alternating 1‐back and 2‐back conditions consisted of nine stimuli and were separated by 1,000 ms of block instruction label (i.e., “1‐back” or “2‐back”) creating 25 s blocks and 300 s task duration. Targets comprised 15 of 54 stimuli (27.8%) in each condition. Accuracy was measured by subtracting the sum of misses and false positives from the number of condition items then dividing by the number of condition items.
Stimuli were presented using EPrime software (Psychology Software Tools, Pittsburgh, PA) and projected onto a screen at the head of the scanner, visible to subjects through a mirror attached to the head coil. Stimuli were presented in bolded black 40‐point Arial text against a white background in the center of the visual field. Responses were collected from a thumb‐button box in the subject's right hand.
Scanning Parameters
All scanning was performed on a Siemens 3T Trio scanner (Erlengen, Germany) using a circularly polarized, transmit/receive head coil. Head movement was minimized by padding that was fitted to hold the subject's head in the coil firmly and comfortably. Before functional scanning, an anatomical image was collected using a magnetized prepared rapid acquisition gradient‐echo echo planar imaging (MPRAGE) sequence, TR/TE 1900/2.52 ms, 9° flip angle, 256 mm FOV, 256 × 256 matrix, 144 sagittal slices for an effective resolution of 1.0 mm3. Blood oxygen level‐dependent (BOLD) images were collected using gradient echo planar imaging (EPI), TR/TE 2,500/30 ms, 90° flip angle, 256 mm FOV, 64 × 64 matrix, 3.7 mm slice thickness with 0.3‐mm gap for an effective resolution of 4.0 mm3.
Image Analysis
Anatomical scans were segmented and normalized to MNI space using the segmentation routine of SPM5 software (http://www.fil.ion.ucl.ac.uk/spm/software/spm5/). EPI scans for each subject, also processed using SPM5, were realigned to correct for interscan head movement, coregistered to the grey matter segmentation output of the subject's anatomical scan, normalized using the anatomical normalization parameters, and smoothed with an 8 mm Gaussian kernel to prepare the data for analysis in a general linear model (GLM). We applied a high pass filter to correct for slow drifts in signal intensity. Regressors for the GLM design matrix were created by convolving block onset times and durations with a canonical hemodynamic response function. A linear regressor for subjects' full scale IQ scores was also included to account for between‐group IQ differences. Linear contrasts subtracting the 1‐back beta image from the 2‐back beta image for each subject were prepared and represent the individual increase in BOLD signal correlating to the increase in working memory load between the two conditions. These contrast images were compared in a random‐effects group analysis to create statistical parametric maps. Images were analyzed within groups using one‐way t tests, and between groups using two‐way t tests. To emphasize patterns of the most robust neural activity, presentation of the one‐way t test data is limited to a conservative threshold of P < 0.01, FWE corrected. To restrict the two‐way between groups t test to a cluster level correction of P < 0.05, a cluster size threshold of 160 mm3 was used. On the basis of extensive prior evidence of PFC involvement in executive cognition and N‐back task performance, between‐group second level analyses were restricted to the PFC (mask constructed in WFU Pickatlas using BA 9, 10, 32, 44, 45, 46, 47, dilated to 1) [Meyer‐Lindenberg et al.,2008].
RESULTS
Cognitive Assessment
Comparison of digit span scores demonstrated a significant decrease in performance among OTCD subjects relative to controls (P = 0.0407; Table I). OTCD subjects also showed significantly longer reaction times for incongruent trials on the STROOP test (P = 0.0124; Table I) and lower composite scores on the CTMT (P = 0.000435; Table I).
fMRI Findings
Performance on the N‐back task was not significantly different between groups for 1‐back or 2‐back accuracy (1‐back P = 0.859, 2‐back P = 0.854) or reaction time (1‐back P = 0.947, 2‐back P = 0.684, Table I). As expected, the average accuracy decreased and average reaction time increased for the 2‐back condition compared with the 1‐back condition for each group, with both groups exhibiting a significant decrease in accuracy (control group P = 0.0083, OTCD group P = 0.0144) and a nonsignificant increase in reaction time (control group P = 0.124, OTCD group P = 0.193). The decrease in accuracy and increase in reaction time were each nonsignificant between groups (accuracy P = 0.398, reaction time P = 0.355).
Statistical parametric maps of within‐group one‐way t‐tests revealed that increased working memory load correlated with increased activation across groups in bilateral parietal and frontal areas, as well as bilateral cerebellum (Fig. 1, Table II); the control group also increased activation bilaterally in the inferior temporal gyrus, insula, and basal ganglia. Two‐way between‐group t‐tests revealed that OTCD patients displayed significantly greater task‐dependent BOLD signal than controls in right Broadman's area (BA) 46 (DLPFC; 48 45 14, Tal; Z = 4.21), BA 10 (DLPFC; 24 61 17, Tal; Z = 3.77) and BA 32 (ACC; 8 19 30, Tal; Z = 3.11; Figure 2, Table 3). All clusters were significant at the whole‐brain cluster level, and the peak voxel value in BA 46 survived FWE‐correction when the analysis was constrained to the PFC. However, between‐group analysis did not reveal any areas with a significantly higher degree of activation in controls relative to OTCD patients.
Figure 1.
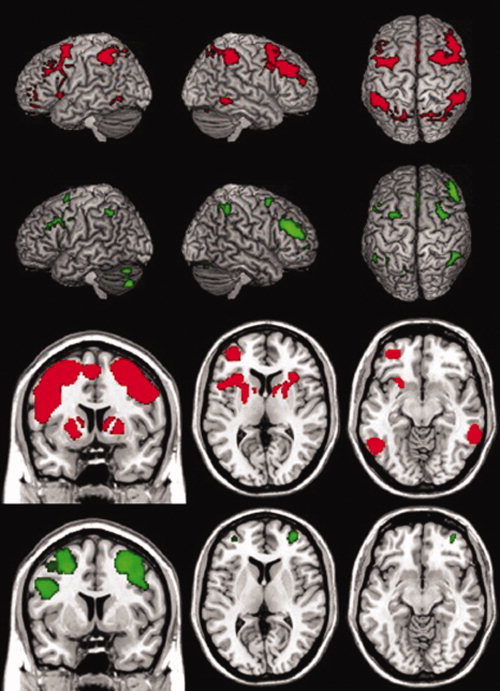
Task‐dependent BOLD activation in OTCD patients and healthy controls. Statistical parametric maps of one‐way within‐group t‐tests for the Control group (red) and OTCD group (green) for 2‐back greater than 1‐back task‐related BOLD signal change (FWE P < 0.01). Bottom two rows, left to right, show coronal slice at y = 7, axial slices at z = 7, and z = −10. The position of right and left hemispheres are as they appear.
Table II.
Within Peak voxel values and coordinates for whole‐brain voxelwise statistical parametric maps of one‐way t‐tests within the Control Group (N = 21) and OTCD Group (N = 19)
| Area | Talairach Coordinates | T‐statistic | Uncorrected P‐value | FWE corr. P‐value | |||
|---|---|---|---|---|---|---|---|
| X | Y | Z | |||||
| Control group | |||||||
| Cluster 1 | 51744 mm3 | ||||||
| R. Inf. Parietal Lob. | 37 | −47 | 42 | 10.4 | 5.06E‐13 | 6.42E‐08 | |
| L. Precuneus | −15 | −66 | 45 | 9.54 | 6.20E‐12 | 6.13E‐07 | |
| L. Inf. Parietal Lob. | −35 | −48 | 37 | 9.48 | 7.37E‐12 | 7.15E‐07 | |
| R. Sup. Parietal Lob. | 11 | −64 | 49 | 9.17 | 1.77E‐11 | 1.57E‐06 | |
| L. Inf. Parietal Lob. | −46 | −45 | 41 | 8.99 | 3.01E‐11 | 2.52E‐06 | |
| L. Mid. Occipital G. | −27 | −66 | 26 | 8.3 | 2.30E‐10 | 1.55E‐05 | |
| Cluster 2 | 42208 mm3 | ||||||
| L. Claustrum | −29 | 15 | 5 | 8.94 | 3.49E‐11 | 2.88E‐06 | |
| L. SMA | 0 | 13 | 45 | 8.39 | 1.75E‐10 | 1.21E‐05 | |
| L. Mid. Frontal G. | −22 | −1 | 54 | 8.06 | 4.82E‐10 | 2.98E‐05 | |
| L. Mid. Frontal G. | −44 | 18 | 36 | 8.05 | 4.83E‐10 | 2.98E‐05 | |
| L. Mid. Frontal G. | −28 | −4 | 52 | 8 | 5.68E‐10 | 3.45E‐05 | |
| L. Pallidum | −18 | 2 | 8 | 7.93 | 7.02E‐10 | 4.16E‐05 | |
| L. Precentral G. | −38 | −3 | 36 | 7.6 | 1.91E‐09 | 1.01E‐04 | |
| L. Precentral G. | −38 | −1 | 32 | 7.6 | 1.94E‐09 | 1.02E‐04 | |
| L. Inf. Frontal G. (p. Opercularis) | −48 | 6 | 22 | 7.36 | 4.04E‐09 | 1.95E‐04 | |
| L. SMA | −5 | −1 | 56 | 6.87 | 1.82E‐08 | 7.28E‐04 | |
| Cluster 3 | 25400 mm3 | ||||||
| R. Sup. Frontal G. | 22 | 2 | 57 | 8.48 | 1.36E‐10 | 9.67E‐06 | |
| R. Inf. Frontal G. (p. Triangularis) | 43 | 24 | 32 | 8.29 | 2.35E‐10 | 1.57E‐05 | |
| R. Mid. Frontal G. | 34 | 29 | 36 | 8.09 | 4.41E‐10 | 2.75E‐05 | |
| R. Mid. Frontal G. | 41 | 19 | 39 | 7.96 | 6.38E‐10 | 3.82E‐05 | |
| R.Precentral G. | 37 | 0 | 45 | 7.21 | 6.38E‐09 | 2.91E‐04 | |
| R.Precentral G. | 47 | 2 | 36 | 6.91 | 1.61E‐08 | 6.53E‐04 | |
| R. Mid. Frontal G. | 38 | 41 | 25 | 6.83 | 2.07E‐08 | 8.13E‐04 | |
| Cluster 4 | 5952 mm3 | ||||||
| L. Mid. Orbital G. | −27 | 42 | −5 | 8.17 | 3.45E‐10 | 2.22E‐05 | |
| L. Mid. Orbital G. | −34 | 47 | 6 | 7.54 | 2.32E‐09 | 1.20E‐04 | |
| Cluster 5 | 4064 mm3 | ||||||
| R. Claustrum | 27 | 19 | 6 | 8.09 | 4.35E‐10 | 2.72E‐05 | |
| R. Putamen | 17 | 3 | 10 | 7.14 | 7.86E‐09 | 3.49E‐04 | |
| Cluster 6 | 2728 mm3 | ||||||
| L. Inf. Temporal G. | −47 | −55 | −9 | 7.73 | 1.28E‐09 | 7.09E‐05 | |
| Cluster 7 | 4048 mm3 | ||||||
| R. Cerebelum (VIII) | 32 | −58 | −47 | 7.37 | 3.89E‐09 | 1.88E‐04 | |
| R. Cerebelum (Crus 1) | 30 | −63 | −21 | 7.36 | 4.07E‐09 | 1.96E‐04 | |
| R. Cerebelum (Crus 1) | 38 | −57 | −27 | 6.72 | 2.94E‐08 | 1.11E‐03 | |
| Cluster 8 | 2464 mm3 | ||||||
| L. Cerebelum (Crus 1) | −27 | −58 | −31 | 7.21 | 6.38E‐09 | 2.91E‐04 | |
| L. Cerebelum (VII) | −28 | −59 | −40 | 6.67 | 3.43E‐08 | 1.26E‐03 | |
| Cluster 9 | 1744 mm3 | ||||||
| R. Inf. Temporal G. | 56 | −46 | −8 | 6.99 | 1.27E‐08 | 5.32E‐04 | |
| Cluster 10 | 440 mm3 | ||||||
| R. Caudate Nucleus | 14 | 3 | 10 | 6.7 | 3.15E‐08 | 1.17E‐03 | |
| R. Caudate Nucleus | 8 | 3 | 4 | 6.65 | 3.62E‐08 | 1.33E‐03 | |
| R. Caudate Nucleus | 14 | 10 | 7 | 6.47 | 6.50E‐08 | 2.20E‐03 | |
| Cluster 11 | 256 mm3 | ||||||
| L. Caudate Nucleus | −16 | −1 | 17 | 6.53 | 5.31E‐08 | 1.85E‐03 | |
| L. Caudate Nucleus | −16 | −5 | 18 | 6.5 | 5.93E‐08 | 2.03E‐03 | |
| L. Caudate Nucleus | −12 | 5 | 8 | 6.23 | 1.36E‐07 | 4.17E‐03 | |
| L. Caudate Nucleus | −16 | 3 | 15 | 6.1 | 2.08E‐07 | 5.99E‐03 | |
| L. Caudate Nucleus | −12 | 8 | 5 | 6.04 | 2.49E‐07 | 7.00E‐03 | |
| L. Caudate Nucleus | −12 | −3 | 13 | 5.96 | 3.26E‐07 | 8.80E‐03 | |
| OTCD group | |||||||
| Cluster 1 | 12832 mm3 | ||||||
| R. Inf. Frontal G. (p. Triangularis) | 41 | 30 | 23 | 8.85 | 4.49E‐11 | 3.60E‐06 | |
| R. Mid. Frontal G. | 36 | 34 | 15 | 8.08 | 4.44E‐10 | 2.77E‐05 | |
| R. Mid. Frontal G. | 37 | 57 | 6 | 7.36 | 4.02E‐09 | 1.94E‐04 | |
| Cluster 2 | 9296 mm3 | ||||||
| R. Sup. Medial G. | 2 | 15 | 44 | 8.62 | 8.93E‐11 | 6.65E‐06 | |
| Cluster 3 | 5984 mm3 | ||||||
| R. Inf. Parietal Lob. | 35 | −51 | 42 | 8.08 | 4.41E‐10 | 2.75E‐05 | |
| Cluster 4 | 7176 mm3 | ||||||
| L. Cerebelum (Crus 1) | −29 | −58 | −32 | 7.88 | 8.08E‐10 | 4.71E‐05 | |
| L. Cerebelum (Crus 2) | −36 | −64 | −47 | 7.73 | 1.28E‐09 | 7.07E‐05 | |
| Cluster 5 | 6512 mm3 | ||||||
| R. Mid. Frontal G. | 24 | −1 | 51 | 7.5 | 2.59E‐09 | 1.32E‐04 | |
| R.Precentral G. | 36 | 0 | 43 | 6.85 | 1.99E‐08 | 7.86E‐04 | |
| Cluster 6 | 3736 mm3 | ||||||
| L. Mid. Frontal G. | −26 | 0 | 45 | 7.44 | 3.11E‐09 | 1.55E‐04 | |
| L. Sup. Frontal G. | −24 | −2 | 65 | 6.12 | 1.94E‐07 | 5.64E‐03 | |
| Cluster 7 | 5664 mm3 | ||||||
| L. Inf. Parietal Lob. | −35 | −56 | 42 | 7.38 | 3.82E‐09 | 1.85E‐04 | |
| L. Inf. Parietal Lob. | −52 | −52 | 40 | 6.45 | 6.79E‐08 | 2.29E‐03 | |
| L. Inf. Parietal Lob. | −48 | −47 | 42 | 6.33 | 1.02E‐07 | 3.25E‐03 | |
| L. Sup. Parietal Lob. | −24 | −70 | 44 | 5.91 | 3.72E‐07 | 9.87E‐03 | |
| Cluster 8 | 1648 mm3 | ||||||
| R. Cerebelum (Crus 1) | 34 | −67 | −27 | 7.25 | 5.64E‐09 | 2.61E‐04 | |
| Cluster 9 | 5736 mm3 | ||||||
| L. Inf. Frontal G. (p. Opercularis) | −49 | 12 | 35 | 7.08 | 9.49E‐09 | 4.12E‐04 | |
| L. Inf. Frontal G. (p. Triangularis) | −42 | 22 | 31 | 7.05 | 1.05E‐08 | 4.49E‐04 | |
| L. Precentral G. | −48 | 1 | 31 | 6.73 | 2.82E‐08 | 1.07E‐03 | |
| L. Precentral G. | −42 | −2 | 47 | 6.11 | 2.01E‐07 | 5.84E‐03 | |
| Cluster 10 | 368 mm3 | ||||||
| L. Sup. Parietal Lob. | −15 | −66 | 46 | 6.61 | 4.23E‐08 | 1.52E‐03 | |
These testing for activation correlated with an increase in working memory load. Clusters reported are for voxels surviving FWE corrected P‐value < 0.01, minimum of 240 mm3 per cluster and peaks 4‐mm apart.
Figure 2.
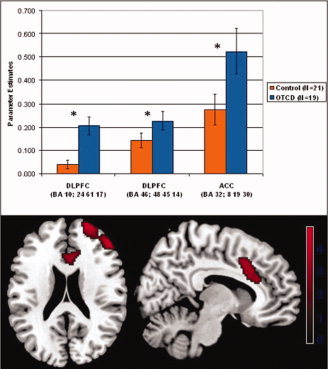
BOLD signal differences between OTCD patients and healthy controls. Statistical parametric maps of between‐group t‐tests, showing activation from the OTCD > control subject contrast. For each subject, the 2‐back > 1‐back contrast was used as the input image for group analysis (displayed at P < 0.005, uncorrected). Bars represent mean BOLD signal change, and error bars represent ± 1 standard error of the mean. Scale bar represents the BOLD signal t‐value.
Table III.
Peak voxel values and coordinates for whole brain, voxelwise, statistical parametric maps of two‐way t‐tests between the Control Group (N = 21) and OTCD Group (N = 19), testing for activation correlated with an increase in working memory load
| Area | Talairach coordinates | T‐statistic | Uncorrected P‐value | |||
|---|---|---|---|---|---|---|
| X | Y | Z | ||||
| Control Group—OTCD Group | ||||||
| No suprathreshold voxels | ||||||
| OTCD Group—Control Group | ||||||
| Cluster 1 | 4464 mm3 | |||||
| R. Mid. Frontal G. | 47 | 46 | 18 | 4.21 | 0.0000129 | |
| R. Sup. Frontal G. | 23 | 60 | 19 | 3.77 | 0.0000832 | |
| R. Sup. Frontal G. | 31 | 48 | 26 | 2.98 | 0.00145 | |
| Cluster 2 | 3856 mm3 | |||||
| R. Cingulate G. | 7 | 18 | 32 | 3.11 | 0.00095 | |
| R. Cingulate G. | 3 | 23 | 21 | 2.95 | 0.00159 | |
| L. Cingulate G. | −9 | 25 | 28 | 2.79 | 0.00264 | |
| Cluster 3 | 1352 mm3 | |||||
| R. Mid. Frontal G. | 26 | 30 | 41 | 3.08 | 0.00102 | |
| Cluster 4 | 544 mm3 | |||||
| R. Mid. Frontal G. | 31 | 51 | 0 | 3.01 | 0.00132 | |
| Cluster 5 | 760 mm3 | |||||
| R. Mid. Temporal G. | 60 | −24 | −11 | 2.97 | 0.0015 | |
| Cluster 6 | 248 mm3 | |||||
| L. Sup. Frontal G. | −30 | 53 | 23 | 2.94 | 0.00164 | |
| Cluster 7 | 664 mm3 | |||||
| L. Cerebellum | −41 | −68 | −21 | 2.86 | 0.00211 | |
| Cluster 8 | 280 mm3 | |||||
| R. Lingual G. | 2 | −78 | −5 | 2.82 | 0.00238 | |
| Cluster 9 | 240 mm3 | |||||
| L. Cerebellum | −11 | −84 | −16 | 2.79 | 0.00263 | |
Clusters reported are for voxels surviving uncorrected P‐value < 0.005, minimum of 160 mm3 per cluster and peaks 4‐mm apart.
DISCUSSION
We offer evidence of neurocognitive variance between OTCD patients and healthy controls during working memory. When load increased from 1‐ to 2‐back, OTCD patients evinced greater increased BOLD signal in right DLPFC and ACC relative to healthy controls. Importantly, this increase occurred despite a lack of between‐group differences in task accuracy or reaction time, suggesting that divergence between the groups does not reflect differences in behavioral output or task difficulty, but may instead be related to prefrontal inefficiency in the OTCD population. The additional finding of performance deficits among OTCD patients on three tasks of executive cognition further support an abnormal neurocognitive phenotype is this disorder.
It has been demonstrated that PFC activation during working memory follows an inverted U‐shaped curve, with the BOLD signal increasing with cognitive load until task demands overwhelm the individual's capacity, after which BOLD signal decreases [Callicott et al.,1999; Callicott et al.,2003]. It has also been shown that the apex of this curve may be shifted in disorders affecting neurocognition, such that peak activation is reached at relatively lower load in patients compared with control subjects [Callicott et al.,2000; Callicott et al.,2003; Mattay et al.,2006]. Our finding of increased DLPFC activity for a given level of task performance in OTCD patients may reflect such a pattern of prefrontal inefficiency, with OTCD patients experiencing peak DLPFC activation at lower cognitive load than controls. Additionally, OTCD patients exhibited higher ACC activation than controls. The ACC is a key regulator of cognitive control and error monitoring [Kerns et al.,2004] and its activation has been shown to increase along with that of the DLPFC when cognitive load is increased on an N‐back paradigm [Jansma et al.,2000]. It is likely that increased ACC activation signals a relative increase in cognitive demand in OTCD patients, and is consistent with a model of cognitive inefficiency.
Because both groups performed near 100% accuracy, it is possible that important neurocognitive features of OTCD were obscured by a ceiling effect on task performance. Our N‐back task did not incorporate a 3‐back condition, and further study is required to characterize group differences in performance and activation at high load. Our findings of possible prefrontal inefficiency in OTCD suggest that the demands of a 3‐back task may exceed the load level associated with peak prefrontal activation in OTCD patients, and that between‐group performance differences may occur at higher load.
Taken together, the increased demand on neuronal activity for equivalent performance and the decreased performance on three neuropsychological measures of PFC‐based executive cognition implicate a true neurocognitive deficit in OTCD, likely arising from damage to prefrontal neurons and white matter tracts caused by hyperammonemic events [Braissant,2010]. Not surprisingly, the areas with increased activation in the OTCD patients have hemispheric connections that overlap with our prior findings of reduced white matter integrity in the superior projections of the genu and rostrum of the corpus callosum [Gropman et al.,2010].
Previous work on the lateralization of working memory function in the prefrontal cortex posits the left PFC as primarily involved in verbal/non‐spatial working memory, and the right PFC as primarily involved in spatial working memory [D'Esposito et al.,1998; Smith et al.,1996]. We employed a verbal/nonspatial N‐back task and observed increased BOLD signal in the right DLPFC among OTCD subjects relative to controls. It is possible that OTCD subjects recruited additional working memory resources in the right DLPFC as a compensatory mechanism to supplement nonspatial working memory processes subserved by the left DLPFC.
Finally, while we observed altered BOLD signal in OTCD patients during N‐back performance, it is plausible that increased neural activation may be observed in OTCD patients across a variety of neurocognitive domains. Performance differences in nonverbal memory and attention have also been reported in OTCD patients,3 and it is possible that these deficits reflect neurophysiological inefficiency that increases neural activation within these domains, relative to the healthy population. As such, we propose that working memory is one in what may be an array of cognitive functions that are affected in OTCD.
Overall, these findings offer preliminary evidence that brain injury caused by biochemical dysregulation in OTCD may impact the functional neuroanatomy serving working memory processes. OTCD patients show relatively higher DLPFC activity, suggesting a pattern of prefrontal inefficiency possibly related to reduced white matter integrity between hemispheres.
It is possible that the results could be affected by unknown effects of the OTCD disease process on the variability of hemodynamic response function (HRF) coupling as has been demonstrated in Alzheimer Disease populations [D'Esposito,2003].
While this may be the case in subjects who have significant symptoms, the effects in our asymptomatic and mildly symptomatic OTCD subjects remains unknown. Further studies in this population will address this.
Further investigation at higher working memory load and across the other cognitive domains believed to be affected by this disorder are required to further characterize the neurocognitive differences between OTCD patients and healthy subjects.
Acknowledgements
The authors thank the patients and the National Urea Cycle Disorders Foundation for their participation.
References
- Bachmann C, Braissant O, Villard AM, Boulat O, Henry H ( 2004): Ammonia toxicity to the brain and creatine. Mol Genet Metab 81 Suppl 1: S52–S57. [DOI] [PubMed] [Google Scholar]
- Baddeley A ( 2003): Working memory: Looking back and looking forward. Nat Rev Neurosci 4: 829–839. [DOI] [PubMed] [Google Scholar]
- Braissant O ( 2010): Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab 100 ( Suppl 1): S3–S12. [DOI] [PubMed] [Google Scholar]
- Braissant O, Henry H, Villard AM, Zurich MG, Loup M, Eilers B, Parlascino G, Matter E, Boulat O, Honegger P, et al. ( 2002): Ammonium‐induced impairment of axonal growth is prevented through glial creatine. J Neurosci 22: 9810–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, Goldberg TE, Weinberger DR ( 2000): Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex 10: 1078–1092. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Bertolino A, Finn K, Coppola R, Frank JA, Goldberg TE, Weinberger DR ( 1999): Physiological characteristics of capacity constraints in working memory as revealed by functional MRI. Cereb Cortex 9: 20–26. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR ( 2003): Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry 160: 2209–2215. [DOI] [PubMed] [Google Scholar]
- D'Esposito M, Aguirre GK, Zarahn E, Ballard D, Shin RK, Lease J ( 1998): Functional MRI studies of spatial and nonspatial working memory. Brain Res Cogn Brain Res 7: 1–13. [DOI] [PubMed] [Google Scholar]
- D'Esposito M, Deouell L, Gazzaley A ( 2003): Alterations in the BOLD fMRI signal with ageing and disease: A challenge for neuroimaging. Nat Rev Neurosci 4: 863–m72. [DOI] [PubMed] [Google Scholar]
- Fuster JM ( 1997): The Prefrontal Cortex: Anatomy, Physiology, and Neuropsychology of the Frontal Lobe. Philadelphia: Lippincott‐Raven; pp 178–208; 234–270; 285–283. [Google Scholar]
- Gropman AL, Batshaw ML ( 2004): Cognitive outcome in urea cycle disorders. Mol Genet Metab 81 ( Suppl 1): S58–S62. [DOI] [PubMed] [Google Scholar]
- Gropman AL, Fricke ST, Seltzer RR, Hailu A, Adeyemo A, Sawyer A, van Meter J, Gaillard WD, McCarter R, Tuchman M, et al. ( 2008): 1H MRS identifies symptomatic and asymptomatic subjects with partial ornithine transcarbamylase deficiency. Mol Genet Metab 95 ( 1–2): 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropman AL, Gertz B, Shattuck K, Kahn IL, Seltzer R, Krivitsky L, Van Meter J ( 2010): Diffusion tensor imaging detects areas of abnormal white matter microstructure in patients with partial ornithine transcarbamylase deficiency. AJNR Am J Neuroradiol 31: 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyato K, Wray J, Huang ZJ, Yudkoff M, Batshaw ML ( 2004): Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol 55: 80–86. [DOI] [PubMed] [Google Scholar]
- Jansma JM, Ramsey NF, Coppola R, Kahn RS ( 2000): Specific versus nonspecific brain activity in a parametric N‐back task. Neuroimage 12: 688–697. [DOI] [PubMed] [Google Scholar]
- Kang ES, Snodgrass PJ, Gerald PS ( 1973): Ornithine transcarbamylase deficiency in the newborn infant. J Pediatr 82: 642–649. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Cohen JD, MacDonald AW III, Cho RY, Stenger VA, Carter CS ( 2004): Anterior cingulate conflict monitoring and adjustments in control. Science 303: 1023–1026. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Fera F, Tessitore A, Hariri AR, Berman KF, Das S, Meyer‐Lindenberg A, Goldberg TE, Callicott JH, Weinberger DR ( 2006): Neurophysiological correlates of age‐related changes in working memory capacity. Neurosci Lett 392 ( 1–2): 32–37. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF, Kolachana B, Callicott JH, Weinberger DR ( 2003): Catechol O‐methyltransferase val158‐met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci USA 100: 6186–6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Lindenberg A, Nicodemus KK, Egan MF, Callicott JH, Mattay V, Weinberger DR ( 2008): False positives in imaging genetics. Neuroimage 40: 655–661. [DOI] [PubMed] [Google Scholar]
- Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED ( 1984): Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea‐cycle enzymopathies. N Engl J Med 310: 1500–1505. [DOI] [PubMed] [Google Scholar]
- Postle BR ( 2006): Working memory as an emergent property of the mind and brain. Neuroscience 139: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumari L, Qureshi IA, Butterworth RF ( 1994): Evidence for cholinergic neuronal loss in brain in congenital ornithine transcarbamylase deficiency. Neurosci Lett 178: 63–65. [DOI] [PubMed] [Google Scholar]
- Rypma B, Berger JS, Prabhakaran V, Bly BM, Kimberg DY, Biswal BB, D'Esposito M ( 2006): Neural correlates of cognitive efficiency. Neuroimage 33: 969–979. [DOI] [PubMed] [Google Scholar]
- Smith EE, Jonides J, Koeppe RA ( 1996): Dissociating verbal and spatial working memory using PET. Cereb Cortex 6: 11–20. [DOI] [PubMed] [Google Scholar]