

Abstract
We report on a genomic and functional analysis of a novel marine siphovirus, the Vibrio phage SIO-2. This phage is lytic for related Vibrio species of great ecological interest including the broadly antagonistic bacterium Vibrio sp. SWAT3 as well as notable members of the Harveyi clade (V. harveyi ATTC BAA-1116 and V. campbellii ATCC 25920). Vibrio phage SIO-2 has a circularly permuted genome of 80,598 bp, which displays unusual features. This genome is larger than that of most known siphoviruses and only 38 of the 116 predicted proteins had homologues in databases. Another divergence is manifest by the origin of core genes, most of which share robust similarities with unrelated viruses and bacteria spanning a wide range of phyla. These core genes are arranged in the same order as in most bacteriophages but they are unusually interspaced at two places with insertions of DNA comprising a high density of uncharacterized genes. The acquisition of these DNA inserts is associated with morphological variation of SIO-2 capsid, which assembles as a large (80 nm) shell with a novel T=12 symmetry. These atypical structural features confer on SIO-2 a remarkable stability to a variety of physical, chemical and environmental factors. Given this high level of functional and genomic novelty, SIO-2 emerges as a model of considerable interest in ecological and evolutionary studies.
INTRODUCTION
Viruses are found wherever life occurs and current estimates of 1031 viruses indicate that they are the most abundant biological entities on the planet (Suttle, 2005). The discovery that the world's ocean comprises half of the global virus abundance has strongly stimulated research on marine viruses (Fuhrman, 1999; Suttle, 2005). Over the two past decades, it has become evident that viruses, by killing a large proportion of marine plankton have a profound impact on the structure and the functioning of the microbial food web (as reviewed in Danovaro et al., 2011; Fuhrman, 1999; Suttle, 2007). Metagenome analyses also show that viral communities comprise the largest reservoir of genetic diversity in the ocean (Rohwer, 2003). Most of these environmental sequences have no obvious homologues in public databases, suggesting that known viruses might not be representative of the actual diversity of marine virioplankton (Breitbart and Rohwer, 2005; Angly et al., 2006). The genome analysis of representative members of the marine viral community is thus essential to improve understanding of the functional significance of this enormous genetic diversity.
Most marine viruses are bacteriophages that belong to the order Caudovirales (dsDNA tailed phages), which is divided into three families based on the morphologies of their tails: the Myo-, the Podo-, and the Siphoviridae. As of this writing, the complete genome sequences of ~ 35 marine phages, mostly Myo- and Podoviridae, have been published (e.g. Rohwer et al., 2000; Paul and Sullivan, 2005; Sullivan et al., 2005; Angly et al., 2009; Sullivan et al., 2010). These studies have revealed the presence of host-derived genes that are not widespread in the genomes of non-marine phages. For example, genes encoding functions in phosphate transport (PstS), or induced in response to phosphate starvation (PhoH), were identified in numbers of marine phage genomes (e.g. Rohwer et al., 2000; Miller et al., 2003; Sullivan et al., 2005; Angly et al., 2009). Phosphorus is an essential resource for phage replication, yet this nutrient is present in growth-limiting amounts for marine microbes in most parts of the ocean (Cotner et al., 1997; Karl and Tien, 1997; Thingstad et al., 2005). The presence of genes involved in phosphorus acquisition suggests that phages might have developed adaptations to life in the oligotrophic ocean. Similarly, the genomes of phages infecting the cyanobacteria Synechococcus and Prochlorococcus often contain host derived genes involved in the photosynthetic machinery (Mann, 2003; Lindell et al., 2004; Sullivan et al., 2005). Expression of these genes enables cyanophages to maintain host photosynthesis throughout the infection cycle (Clokie et al., 2006; Lindell et al., 2007), possibly to provide the host with sufficient energy until the release of phage progeny. The frequent occurrence of the photosynthetic gene in cyanophages' genome indicates the evolutionary importance of the acquisition of this characteristic.
In comparison to myo- and podoviruses, genomics of marine siphoviruses are poorly documented. Despite their prevalence in the ocean (Edwards and Rohwer, 2005), there are, to our knowledge, only 3 complete genome sequences of marine siphovirus reported in the literature. These include ΦJL001, a bacteriophage infecting a sponge associated α-Proteobacteria (Lohr et al., 2005), the Vibrio phage ΦHSIC (Paul et al., 2005), and the cyanophage P-SS2 (Sullivan et al., 2009). Based on comparative genomics, these phages appear to be considerably divergent from non-marine siphoviruses, with a high proportion of uncharacterized genes (72% on average) in their genome. Interestingly, marine siphoviruses studied to date lack identifiable homologues to genes involved in phosphate acquisition and/or photosynthesis machinery. These observations point to these marine phages as having distinct ecology and evolution compared to marine and non-marine counterparts.
Here, we report on the characterization of a novel marine siphovirus, the Vibrio phage SIO-2, which was isolated from the surface water of the Northern coastal Pacific Ocean. This Vibrio phage is lytic to the Vibrio sp. SWAT3 (hereafter SWAT3), a host of considerable ecological interest. The bacterium SWAT3 can actively produce inhibitory molecules (including the antibiotic andrimid), which impede the growth of a large number of marine bacterial isolates, including the human pathogen V. cholerae (Long and Azam, 2001; Long et al., 2005). This antagonistic bacterium may thus play an important role in structuring the bacterioplankton community (Long and Azam, 2001). Equally interesting, a parallel structural study by Lander et al. (submitted) showed that the capsid of Vibrio phage SIO-2 exhibits a novel architecture as revealed by cryo-electron microscopy. Given the novel structure of this phage and the ecological importance of its host, SIO-2 emerges as a marine virus model of considerable interest in ecological and evolutionary studies. This manuscript presents a functional and genomic analysis of SIO-2 and interprets these data in an ecological and evolutionary context.
RESULTS and DISCUSSION
A novel marine siphovirus
Morphology
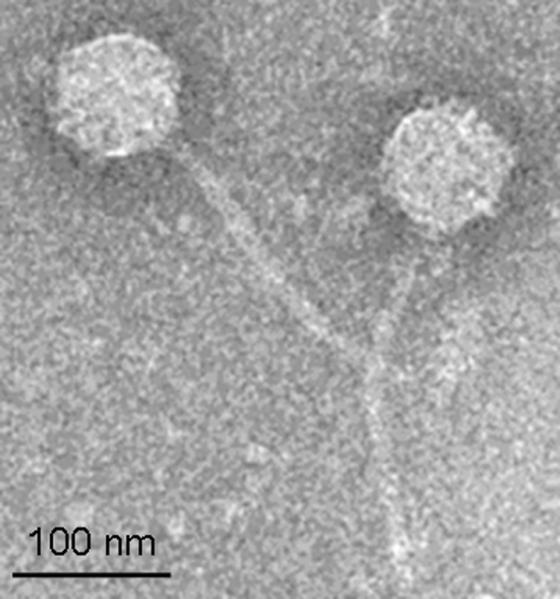
Vibrio - phage SIO-2 (hereafter, SIO-2) formed clear well resolved plaques that produced a high titer suspension. TEM examination indicated that SIO-2 has the morphology of a siphovirus with an isometric capsid, 81 ± 4 nm in diameter, connected to a long, flexible, and non-contractile tail of 209 ± 16 nm in length and 11 ± 2 nm in width (Fig 1). Chloroform treatment did not affect SIO-2 infectivity suggesting that this phage lacks a lipid membrane, as expected for a member of the Siphoviridae family.
Figure 1.
Host – virus interactions
This phage could readily propagate on Vibrio sp. SWAT3 host with a latent period of approx. 45 – 60 min and an average burst size of 60 viral particles per infected cell (Fig 2). The increase in SIO-2 abundance was accompanied by a rapid collapse of the infected host culture followed by the development of a population of resistant cells. Phages were never detected in cultures of resistant cells and their treatment with a prophage-inducing antibiotic (mitomycin C) did not lead to the production of SIO-2 particles. It is thus unlikely that the observed phage resistance is due to lysogeny. We further investigated the ability of SIO-2 to switch from a lytic to a lysogenic lifestyle using a host culture adapted in nutrient limited conditions (ZoBell/1000). For the case of phage λ, the lytic cycle was indeed shown to prevail in rapidly growing cells, whereas lysogeny was favored under low nutrient conditions (Gottesman and Oppenheim, 1994). Adaptation of SWAT3 host in ZoBell/1000 resulted in a significant reduction in cell growth rate (0.03 h−1 vs. 0.3 h−1 in nutrient replete conditions). Even under these conditions, SIO-2 remained lytic as it induced the lysis of the nutrient limited host culture with a relatively longer latent period (4 – 8 h) than in nutrient replete conditions.
Figure 2.

Plaque assays using a broad range of potential hosts indicated that SIO-2 could infect relatively closely related Vibrio species of considerable ecological interest (Table S1). Besides the antagonistic bacterium SWAT3, this bacteriophage caused the lysis of different
members of the Harveyi clade including Vibrio harveyi ATCC BAA-1116 and Vibrio campbellii ATCC 25920. The efficiency of plating (EOP) of SIO-2 on these alternate hosts was relatively unchanged with values of 0.7 and 0.8 for Vibrio harveyi ATCC BAA-1116 and Vibrio campbellii ATCC 25920, respectively (Table S1). Because cultured bacteria might not reflect the natural diversity of uncultured relatives, we also assessed SIO-2 hosts' abundance in natural microbial assemblages using fluorescently labeled particles (Table 1). Potential hosts, mostly vibrioid heterotrophic prokaryotes (Fig S2), were detected in all sampling sites with abundance ranging from 0.2 – 8.0×103 cells mL−1, which corresponds to an average fraction of 0.2% of the studied prokaryote communities (Table 1). We could not detect a clear spatial pattern in SIO-2 host dynamics due to the low number of samples and differing sampling periods. Nonetheless, these results indicated that SIO-2 not only infects hosts of ecological interest but also has the potential to impact bacterial community composition in different coastal systems.
Table 1.
Summary table of SIO-2 potential hosts as determined from FLVs assay applied in different marine ecosystems.
| Sampling site | Bacterial cells abundance (mL−1) | SIO2-tagged cells abundance (mL−1) |
|---|---|---|
| Coastal Pacific, SIO pier (n = 4) | 1.1 − 2.8×106 | 0.2 − 6.0×103 |
| Coastal Atlantic, SOMLIT-Brest (n = 1) | 1.0 ×106 | 8.0×103 |
| English Channel, SOMLIT-Roscoff (n = 2) | 0.8 − 1.7×106 | 2.7 − 4.7×103 |
| Coastal English Channel, Bay of Morlaix (n = 1) | 1.6×106 | 2.2×103 |
A divergent genome sequence
General properties
The sequence of SIO-2 genome assembles as a circle, indicating that the DNA in the virions is either circularly permuted or has a non-permuted terminal redundancy. Examination of the assembled sequence reads allowed us to conclude that the DNA is circularly permuted (see Experimental procedures section), implying a headful DNA packaging mechanism. The non-redundant length of the sequence is 80,598 bp and the G+C content is 45.0%, with no large-scale variations in G+C content across the sequence. This G+C value is similar to SIO-2 hosts SWAT-3 and V. harveyi ATCC BAA-1116 (45.4%). GeneMark predicted a total of 116 open reading frames (ORFs); all are oriented in the same direction (Fig 3). Of these 116 ORFs, only 38 (32%) had recognizable homologues in Genbank, and only half of these encoded protein with known function (Table 2). Thus, the SIO-2 gene inventory is largely uncharacterized, similar to the siphoviruses of marine origins that have previously been sequenced.
Figure 3.

Table 2.
Summary table of SIO-2 predicted proteins that contained relevant annotation information as determined from significant BLASTP hits (e-value < e-3) against the GenBank non-redundant database or experimental proteomics on the virus particle.
| SIO-2 ORF # | Size (aa) | Putative function | Species | e-value |
|---|---|---|---|---|
| 1 | 251 | unknown | ✦ | |
| 2 | 241 | unknown | ✦ | |
| 3 | 265 | unknown | ✦ | |
| 4 | 673 | terminase large subunit | ✦ Rhodococcus phage ReqiDocB7 | 8e−53 |
| 5 | 82 | unknown | ||
| 6 | 89 | unknown | ||
| 7 | 82 | unknown | ||
| 8 | 160 | unknown | ||
| 9 | 681 | pyruvate phosphate dikinase | Listeria monocytogenes | 1e−73 |
| 10 | 160 | unknown | ||
| 11 | 158 | protein phosphatase | Sulfolobus solfataricus | 3e−05 |
| 12 | 344 | unknown | ✦ | |
| 13 | 284 | unknown | ✦ | |
| 14 | 784 | DNA polymerase I | ✦ Gordonia phage GTE2 | 1e−53 |
| 15 | 621 | capsid portal protein | ✦ Haliangium ochraceum DSM 14365 | 1e−37 |
| 16 | 357 | head assembly protein* | Myxococcus xanthus | 1e−04 |
| 17 | 217 | methyltransferase | Hoeflea phototrophica | 6e−30 |
| 18 | 142 | unknown | ||
| 19 | 154 | unknown | ||
| 20 | 98 | unknown | ||
| 21 | 87 | unknown | ||
| 22 | 90 | unknown | ||
| 23 | 162 | hypothetical protein | Erwinia phage phiEa21-4 | 6e−5 |
| 24 | 98 | unknown | ||
| 25 | 61 | unknown | ||
| 26 | 152 | unknown | ||
| 27 | 104 | hypothetical protein | Citrobacter sp. 30_2 | 2e−09 |
| 28 | 165 | unknown | ||
| 29 | 225 | unknown | ||
| 30 | 85 | unknown | ||
| 31 | 94 | unknown | ||
| 32 | 135 | unknown | ||
| 33 | 285 | unknown | ||
| 34 | 145 | unknown | ||
| 35 | 161 | unknown | ||
| 36 | 152 | unknown | ||
| 37 | 564 | unknown | ||
| 38 | 198 | unknown | ||
| 39 | 197 | unknown | ||
| 40 | 84 | unknown | ||
| 41 | 79 | unknown | ||
| 42 | 118 | unknown | ||
| 43 | 162 | unknown | ||
| 44 | 118 | unknown | ||
| 45 | 330 | unknown | ||
| 46 | 67 | unknown | ||
| 47 | 84 | unknown | ||
| 48 | 150 | unknown | ||
| 49 | 256 | hypothetical protein | Vibrio cholerae NCTC 8457 | 2e−15 |
| 50 | 81 | unknown | ||
| 51 | 110 | unknown | ||
| 52 | 262 | unknown | ||
| 53 | 105 | unknown | ||
| 54 | 120 | unknown | ||
| 55 | 150 | unknown | ||
| 56 | 142 | unknown | ||
| 57 | 88 | unknown | ||
| 58 | 278 | unknown | ||
| 59 | 144 | unknown | ||
| 60 | 167 | hypothetical protein | Pseudomonas aeruginosa PA7 | 3e−06 |
| 61 | 114 | unknown | ||
| 62 | 111 | unknown | ||
| 63 | 159 | unknown | ||
| 64 | 106 | unknown | ||
| 65 | 342 | unknown | ||
| 66 | 117 | hypothetical protein | Pseudomonas phage M6 | 1e−06 |
| 67 | 112 | unknown | ✦ | |
| 68 | 158 | unknown | ||
| 69 | 89 | unknown | ||
| 70 | 196 | unknown | ||
| 71 | 275 | unknown | ||
| 72 | 112 | unknown | ||
| 73 | 135 | unknown | ||
| 74 | 77 | unknown | ||
| 75 | 95 | unknown | ||
| 76 | 100 | unknown | ||
| 77 | 176 | unknown | ||
| 78 | 90 | unknown | ||
| 79 | 270 | hypothetical protein | Yersinia enterocolitica | 9e−20 |
| 80 | 554 | unknown | ||
| 81 | 107 | unknown | ||
| 82 | 204 | Capsid protease* | Lactococcus phage u136.k1t1 | 9e8 |
| 83 | 391 | Scaffolding protein* | ||
| 84 | 314 | major capsid protein | Bacillus cereus 95/8201 | 4e−62 |
| 85 | 109 | unknown | ||
| 86 | 207 | unknown | ||
| 87 | 153 | Tail completion protein* | ||
| 88 | 152 | unknown | ||
| 89 | 265 | Major tail protein* | Ralstonia sp. 5_7_47FAA | 3e−27 |
| 90 | 144 | Tail assembly chaperone G* | ||
| 91 | 210 | Tail assembly chaperone T* | ||
| 92 | 1377 | tail tape measure protein | Clostridium botulinum Ba4 str. 657 | 3e−41 |
| 93 | 127 | unknown | ✦ Delftia acidovorans SPH1 | 8e−8 |
| 94 | 324 | unknown | ✦ Magnetococcus sp. MC-1 | 4e−5 |
| 95 | 299 | Tail baseplate protein* | ✦ | |
| 96 | 428 | unknown | Magnetococcus sp. MC-1 | 1e−18 |
| 97 | 169 | unknown | Magnetococcus sp. MC-1 | 2e−04 |
| 98 | 100 | unknown | Magnetococcus sp. MC-1 | 5e−05 |
| 99 | 75 | unknown | ||
| 100 | 145 | hypothetical protein | Desulfotalea psychrophila LSv54 | 7e−16 |
| 101 | 183 | hypothetical protein | Desulfotalea psychrophila LSv54 | 2e−08 |
| 102 | 413 | AAA+ ATPase | Acinetobacter junii SH205 | 1e−28 |
| 103 | 492 | unknown | ||
| 104 | 192 | unknown | ||
| 105 | 501 | DNA helicase | Bacteroides sp. 2_1_33B | 1e−17 |
| 106 | 207 | unknown | ||
| 107 | 328 | DNA primase | Geobacter lovleyi SZ | 5e−10 |
| 108 | 313 | unknown | ||
| 109 | 475 | helicase | Ectocarpus siliculosus virus 1 | 3e−38 |
| 110 | 136 | unknown | ||
| 111 | 138 | unknown | ||
| 112 | 273 | protein RecA | Slackia exigua ATCC 700122 | 2e−25 |
| 113 | 151 | unknown | ||
| 114 | 330 | unknown | ||
| 115 | 188 | protein RuvC | Bacillus pumilus ATCC 7061 | 3e−11 |
| 116 | 287 | unknown | ✦ |
annotation aided by the use of synteny
hit to VHS1 genome fragments
Among the 38 genes with detected homologues, 43% appeared to be related to viruses, 3% were related to archaea, and the remaining 54% had homologues in several groups of eubacteria (Table 2). Some or all of the cellular hits may be due to prophages in the cellular genomes. In contrast, there were no best hits to the SIO-2 host genomes nor to marine phage sequences excepting Vibrio harveyi siphovirus 1 (VHS1), which was isolated from shrimp culture ponds in Thailand (Pasharawipas et al., 2005). VHS1 sequences made up a third of the total matches in SIO-2, indicating a close genetic relatedness between both bacteriophages, although they were isolated from temporally and geographically separated locations. However, available sequences for VHS1 only account for 20% of this phage genome (Pasharawipas et al., 2005). This fragmentary representation of VHS1 genome may thus distort comparison between both phage genome and, thereby, the interpretation of SIO-2 genomics. We therefore chose to exclude VHS1 hits from the following analysis.
Transcription and translation machinery
We investigated putative promoter sequences using the E. coli σ70 consensus sequence as a probe. Twenty-one of the first 35 candidate promoter sequences were located in intergenic regions, which in aggregate accounted for 4.7% of the genome (Fig 3). This asymmetric distribution to intergenic regions strengthens the case that these are functional promoters rather than chance matches to the promoter consensus. We also examined the intergenic regions for possible factor-independent (stem-loop) transcription terminators and found 22 such sequences (Fig 3). A search for direct repeats in the sequence identified five sequences ranging in size from 22 bp to 45 bp that occurred identically two or more times on the genome. These repeated sequences all occurred in intergenic regions, overlapping with transcription promoters, terminators and Shine-Dalgarno sequences (Figs 3 and S4). The functional significance of the exact identity of these sequence repeats, if any, is unknown.
The translational start sites for almost all of the genes appeared unequivocal because of the presence of an appropriately located Shine-Dalgarno translation initiation sequence. The two exceptions are gene 91, which we discuss below, and gene 108, where an upstream Shine-Dalgarno sequence might conceivably be brought into position by formation of RNA secondary structure, as has been proposed for gene 38 of coliphage T4 (Gold, 1988) and gene 6 of coliphages HK97 and HK022 (Juhala et al., 2000). The start codon for 110 of the 116 genes was AUG; the remaining 6 used GUG. There was no detectable tRNA gene in the SIO-2 genome sequence. The function of these genes in bacteriophages is not fully understood. Bailly-Bechet et al. (2007) have recently proposed that tRNAs that are kept in phage genomes typically correspond to codons that are highly used by the phage genes while rare in the host genome. Codon usage pattern in the genome of SIO-2 was very similar to those of the SWAT3 and ATCC BAA-1116 hosts (Fig S5), suggesting that this phage has adapted well to the translation apparatus of its hosts.
DNA metabolism genes
Most of SIO-2 genes encoding protein with robust functional homologues in Genbank are involved in DNA metabolism (Table 2). We identified 6 core genes, found in most bacteriophages, responsible for DNA replication, repair, and recombination including two helicases (gene 105 and 109), a primase (gene 107), a DNA-dependent ATPase RecA (gene 112), a Holliday junction resolvase RuvC (gene 115) as well as a gene encoding a type A DNA polymerase with an associated 3′-5′ exonuclease domain (gene 14). The reconstruction of the molecular phylogeny of DNA pol A protein suggests that SIO-2 is distantly related to bacteriophage of differing morphology (myo-, podo-, and siphovirus), which infect unrelated hosts (Fig. 4). Interestingly, this phylogeny showed a consistent tendency for phage to cluster with counterparts sharing similar replication strategies (Fig. 4). Cluster I, which includes SIO-2, was composed of strictly virulent bacteriophages (e.g. Wang et al., 2005; Naryshkina et al., 2006; Stewart et al., 2009) with the exception of VHS1 which was shown to be non-obligately lytic (Khemayan et al., 2006; Pasharawipas et al., 2008). This observation supports the results from the laboratory experiments (see above “host - virus interactions”) indicating that SIO-2 has a virulent replication strategy. It is furthermore consistent with the lack of detectable elements associated with lysogeny control (repressors, anti-repressors, integrase) in this phage genome. As such, SIO-2 appears to be the first representative of marine siphovirus sequenced to date with a hypothesized strictly virulent lifestyle. Additional investigations will help to refine our understanding of the interactions between SIO-2 and its host.
Figure 4.

Structural and assembly genes
Sequences of the head and tail structure and assembly proteins are often the most strongly conserved among the predicted proteins of a newly sequenced phage, and their identification is usually made more secure by the fact that their genes are most often arranged in a stereotyped order on the genome (Casjens and Hendrix, 1974; Hatfull et al., 2008). For SIO-2, only three of these proteins, the terminase large subunit (gene 4), the portal protein (gene 15), and the tail length tape measure protein (gene 92) gave identifying hits better than an e=10−5 cutoff in a BlastP search. The identification of the tape measure protein is strengthened by the fact that structure predictions based on the amino acid sequence indicate that the protein is expected to be almost entirely α-helical, and arranged in a coiled coil; these are characteristic properties of tape measure proteins. In addition, the predicted length of the tail, based on the length of the tape measure protein arranged as an α-helix (Katsura, 1987; Pedulla et al., 2003), is 207 nm, which agrees well with the measured length of the tail at 209 ± 16 nm.
We were able to assign putative functions to six additional head and tail proteins for which there were good BlastP hits to proteins of unknown function that could be expanded to include known examples of head or tail proteins using PSI-Blast. These included the head accessory protein (gene 16), capsid maturation protease (gene 82), major capsid protein (gene 84), a tail completion protein (gene 87), major tail subunit (gene 89) and a tail baseplate protein (gene 96). The identifications for these six genes are rather weak in terms of sequence identity, but our confidence in their correctness is substantially strengthened by the fact that they are exactly in the “canonical” order on the genome map that has been documented for their homologues in many other tailed phages.
Finally, we were able to identify three additional head and tail proteins based on the positions of their genes and on special features of their sequences. The gene encoding the scaffolding protein (a capsid assembly chaperone) is typically located between the gene for the capsid protease and the gene for the major capsid subunit, which in SIO-2 describes gene 83. Although there is no useful sequence match to the protein encoded by gene 83, it is predicted to be almost entirely α-helix, which is characteristic of scaffolding proteins (Němeček et al., 2009). We have accordingly made this assignment. In the majority of long-tailed phages it has been possible to identify a pair of overlapping ORFs in the space between the genes encoding the major tail protein and the tape measure protein in which the downstream ORF is expressed as the result of a ribosomal frameshift in the region of overlap (Xu et al., 2004). SIO-2 genes 90 and 91 fit this description: a transfer of the ribosome from 90 to 91 would require a −1 frameshift, and we find a convincing −1 frameshift sequence (A.AAA.AAG) appropriately positioned in the overlap region. Taken together, we considered this strong evidence for correct identification of these genes, which in the prototypical example of coliphage λ have been shown to encode tail assembly chaperones (Xu et al., 2004). We have thus labeled genes 90 and 91 with the names of the λ analogs, G and T (Fig. 3). There were six additional genes interspersed in the main group of head and tail genes, namely genes 85, 86, 88, 93, 94 and 95. We have no direct information about their functions but we strongly suspect them to be also head and tail genes by analogy with the numbers and distributions of head and tail genes in more extensively characterized phages.
SDS-PAGE analysis of mature SIO-2 virion resolved 14 structural proteins including 6 major polypeptides of 12, 29, 31, 34, 55 and 137 kDa and 8 minor polypeptides with molecular masses ranging from 15 to 121 kDa (Fig. 5). Two of the bands on the gel can be confidently matched with functions, namely the 29 kDa band, which as the most abundant protein on the gel must be the major capsid protein, and the 137 kDa band, for which the tape measure protein is the only protein encoded in the genome that is big enough to account for it. The predicted size of the major capsid protein is 35.1 kDa, which suggests that it is proteolytically processed into the 29 kDa form during capsid maturation, in agreement with the presence of a gene for the capsid maturation protease. Similarly, the tape measure protein, at a predicted 149 kDa, is predicted to be somewhat larger than the 137 kDa form on the gel. There is evidence for assembly-dependent processing of the tape measure protein in some other phages such as coliphage λ (Hendrix and Casjens, 1974). Similar processing may be occurring in the SIO-2 tape measure protein.
Figure 5.

Distinctive features in SIO-2 genome
As discussed above, SIO-2 genome sequence per se shows substantial divergence to known bacteriophages. The 80 kb genome of SIO-2 is relatively larger than previously sequenced siphoviruses whose genome sizes range between 20 to 50 kb, with a few exceptions including the marine cyanophage P-SS2 (108 kb), the enterophage T5 (121.7 kb), or the mycobacteriophage Omega (111 kb). Furthermore, this large genome comprises a high proportion of genes, including core and non-core genes, lacking homologues in databases (68%). These uncharacterized genes are substantially more abundant in SIO-2 than in the more extensively characterized siphoviruses where often half the predicted proteins have detectable homologues (Brüssow and Desiere, 2001; Proux et al., 2002; Pedulla et al., 2003). A similar pattern was observed in the 3 marine siphovirus genomes presently available, pointing to the paucity of sequences of marine siphoviruses in the databases and/or probable adaptations of these phages to survive and propagate in the ocean (Lohr et al., 2005; Paul et al., 2005; Sullivan et al., 2009).
Another great divergence to known siphoviruses is also manifested by the origin of the core genes in SIO-2 genome. Several core genes encoding functions in DNA metabolism (genes 14, 105, 107, 109, 112, 115) and virion structure (genes 4, 15, 84, 89, 92) were indeed identified based on Blast matches. Unexpectedly, most of these genes shared robust similarities (evalue range: 10−10 to 10−62) with unrelated viruses (Rhodococcus phage, Ectocarpus siliculosus virus) as well as bacteria spanning a wide range of phyla (Proteobacteria, Bacteroidetes, Firmicutes, Actinobacteria). In contrast, there were no best hits to SIO-2's hosts' genome or to marine bacteriophage sequences (other than VHS1). Caudovirales are known for their ability to easily replace genes with completely unrelated sequences that are functionally equivalent (Lawrence et al., 2002). However, the gene repertoire of SIO-2 comprises a particularly wide variety of origins, presumably reflecting frequent subversion of bacterial genes and, possibly, a facilitated ability for gene recombination and expression.
In addition to the sequence content and origins, the genome architecture is also divergent in SIO-2. As described earlier, the order and location of genes involved in virion structure and assembly are typically conserved in siphovirus genomes (Casjens and Hendrix 1974, Hatfull et al., 2008). In SIO-2, these genes are in the same order as in most viruses, but they are interspaced at two places in an unusual way. First, the terminase gene (gene 4) and the portal gene (gene 15), adjacent in many phages, are separated by an apparent 9 kb insertion in the SIO-2 genome. Similar displacement has been observed in the myovirus SPO1 and some of its relatives (Stewart et al., 2009). In SIO-2, this apparent insert encodes 10 predicted proteins, including a pyruvate phosphate dikinase (gene 9), a tyrosine phosphatase (gene 11), 2 phage related proteins (genes 12 and 13) and the core gene DNA pol (gene 14). The 5 remaining genes are unique to SIO-2. Interestingly, genes 12 and 13 are also detected in VHS1 genome but in no other bacteriophage sequences, suggesting that these putatively inserted genes may encode important functions as they are maintained in this related phage. The second displacement of interest is located between the head accessory gene (gene 16) and the capsid protease gene (gene 82). These genes, typically adjacent in most tailed-phage genomes, are interspaced with a 34 kb insertion in SIO-2. This large insert has a distinctly different character from the rest of the genome. Most strikingly, the average gene size for this region (160 codons) is only 54% of the average size for the genes in the remainder of the genome (297 codons). Only one of the 65 predicted proteins encoded in this interval can be assigned a putative function (the methyl transferase in gene 17). The remaining 6 proteins encoded in this region that make significant BlastP hits match only hypothetical proteins of unknown function. Curiously, one of these (gene 60) matches over its N-terminal half to an abundantly synthesized early protein of phage λ of unknown function, and another (gene 23) matches in its C-terminal half to a hypothetical protein found in five different enterobacterial and Vibrio phages. These smaller genes of largely unknown function are reminiscent of the non-core genes reported in large bacteriophage genomes as in T4-like phages (Comeau et al., 2007), cyanophages (Millard et al., 2009), and also Bacillus phage SPO1 (Stewart et al., 2009). Inserts detected in these bacteriophages are highly variable in gene composition and origins within a phage superfamilly. These regions of hyperplasticity were therefore suggested to be a preferred target for gene shuffling (Comeau et al., 2007; Stewart et al., 2009). The functions encoded by these genes, the way they got inserted in phage genome, and their origins are still largely unknown, though a remarkable mechanism for short insertions has been proposed recently for the T4-like phages (Arbriol et al., 2010). The sequencing of novel SIO-2 like phage genomes should serve to better understand the evolution of these intriguing inserts and their possible implications for phage fitness.
The acquisition of these extra pieces of DNA during the course of its evolution can explain the relatively larger genome size in SIO-2 (80 kb) compared to most known siphoviruses (typically 20 to 50 kb). For these latter bacteriophages, the genome is usually densely packed in capsids typically in the order of 40 to 60 nm (as reviewed in Hatfull et al., 2008; Sullivan et al., 2009). This, of course, raises the question as to whether the acquisition of extra DNA in SIO-2 genome is associated with morphological variations of its capsid. And indeed, not only the size but also the overall architecture of SIO-2 appear to be divergent from known siphoviruses. TEM revealed that the SIO-2 capsid is bigger than previously reported for most known siphophages (80 nm vs. 40–60 nm). Importantly, a parallel structural study revealed that SIO-2 displays a novel capsid architecture (T=12) decorated with accessory proteins, presumably reinforcing this phage structure (Lander et al., submitted). We do not know whether the 65 genes of the 34 kb insert were acquired in a single event or incrementally over time, though we favor a single event because it is easier to reconcile with the fact that all 65 genes are inserted at the same place in the head gene order. If the 65 genes were added incrementally, the change to a T=12 capsid from a presumed ancestral T=7 or T=9 capsid could have been an adaptation to increasingly tight DNA packing. If the 65 genes were acquired in a single event, we suggest that the phage must have first acquired the ability to make larger capsids, which would have made it possible to accommodate a sudden large increase in genome size. We note that the constraints imposed by the geometry of triangulation numbers mean that, in contrast to genome size, capsid size cannot change in small increments (except in the case of changes in the length of prolate capsids (Lane and Eiserling, 1990), which does not apply here).
Insights into the functionality of SIO-2 structure
In the light of the high level of structural novelty in SIO-2 (see above discussion), we investigated the stability of these particles against a variety of chemical, physical, and environmental factors. These assays revealed a remarkable tolerance of SIO-2 to these treatments. This phage could survive temperatures from −196 to 65°C, it remained infectious after 24h exposure over a broad range of pH (3 to 10), salt concentrations (0 – 84 g L−1) and there was no evidence of inactivation caused by pressures ranging from 14.6 psi to 7251 psi corresponding to hydrostatic pressure from 1 to 5000 m depth (Table 3). A series of 24h incubations were also deployed to assess the effect of natural sunlight and UV-radiations, known to be the main cause of virus inactivation in the ocean, as well as viricidal agents in natural seawater (Suttle and Chen, 1992; Noble and Fuhrman, 1997). Under these conditions, SIO-2 had a global decay rate of 73% day−1. Not surprisingly, SIO-2 survival was mostly affected by natural sunlight, which accounted for most of the observed loss rate (72% d−1), with UV-radiations responsible for a decay of 12% d−1. In absence of light, no detectable loss in SIO-2 viability was observed suggesting that natural viricidal agents, such as hydrolytic enzymes from bacterial origins or grazing, had negligible effect on particle stability. The few marine bacteriophages investigated for their tolerance to solar radiations were all shown to be sensitive to this factor and decayed at slightly higher rates than those observed for SIO-2 (Suttle and Chen, 1992; Wommack et al., 1996; Noble and Fuhrman, 1997). However, the experimental set ups of these studies differed, which prevents further comparison or speculation. Collectively, these assays indicate that the structural information encoded in the SIO-2 genome confers on this phage a remarkable stability in nature. This structural stability and the observed abundance of potential host in different marine environments (see “host-virus interactions”) suggest that SIO-2 has a relatively large fundamental niche although best adapted to light attenuated ecosystems.
Table 3.
SIO-2 particle stability to physical, chemical treatments (+ no loss of infectivity, +− partial loss of infectivity, − complete inactivation)
| Treatment | Details | Stability |
|---|---|---|
| Temperature (°C, 15 min) | 4 – 60 | + |
| 65 | + − | |
| 70 – 90 | − | |
| Freezing (°C, 24 hours) | − 196 | + |
| − 80 | + − | |
| − 20 | + | |
| pH (24 h) | 2 | − |
| 3 | + − | |
| 4 – 10 | + | |
| NaCl concentration (g L−1, 24h) | 0 – 84 | + |
| Hydrostatic pressure (psi, 30 min) | 14.5 – 7251 | + |
Prevalence of SIO-2 in the ocean
The accumulation of marine virome datasets provides a unique opportunity to relate the fundamental niche of SIO-2 to their actual distribution (i.e. realized niche). In spite of the observed ability of this phage to withstand a wide variety of environmental factors, SIO-2 sequences appeared to be rare in oceanic viromes publicly available. There were very few reads (7 in total) with a best hit to SIO-2 genome (Table S3) and the alignment lengths of these hits were short (24 – 31 bp). Similarly, homolog sequences to SIO-2 hosts (SWAT-3, V. harveyi BAA-1116 and V. campbelli ATCC 25920) were not detected in oceanic metagenomes. Marine Vibrios, which are widely distributed in marine environments, can occur either free-living in the water column or associated to marine aggregates or organisms (e.g., Kushmaro et al., 2001; Thompson et al., 2004). Vibrio sp. SWAT3 was interestingly isolated from an organic aggregate (Long and Azam 2001), whereas strains of V. harveyi can be found on the surface or within marine animals such as shrimps, corals or fishes (Thompson et al., 2004; Austin and Zhang, 2006; Austin, 2010). Likewise, marine viruses can occur at high local concentrations on aggregates and in /on organisms (Comeau et al., 2006; Comeau and Suttle, 2007; Mari et al., 2007). It is thus possible that SIO-2 hosts, and presumably SIO-2 itself, have a particle-attached rather than a free-living lifestyle. Yet, marine metagenomes have essentially described the genetic diversity of the free-living communities, whereas particle-attached micro-organisms are overlooked. The observed absence of SIO-2-like phages and its hosts in metagenomic datasets may thus be explained by an inadequate sampling of particle attached viruses and their hosts. The presence of strikingly similar relatives, such as VHS1, in contrasting marine environments argues that SIO-2-like phages may, at least locally, be important members of the marine virioplankton.
Concluding remarks
Undoubtedly, viruses comprise the largest reservoir of genetic diversity in the ocean as revealed recently by metagenomic analysis. Most virus community sequences are, however, uncharacterized, suggesting that known viruses might not be representative of the actual diversity of marine virioplankton. In particular, genome sequences of marine siphoviruses are largely under-represented in current databases. This study reports a comprehensive characterization of a novel marine lytic siphovirus, Vibrio phage SIO-2, which infects hosts of great ecological interest including the antagonistic bacterium Vibrio sp. SWAT3 and notable members of the Harveyi clade. The complete genome analysis of this phage revealed distinctive features, which manifest at three levels. First, the genome sequence as such is rather unusual. It is relatively larger than most known siphoviruses genomes (80 kb vs. 20 – 50 kb) and comprises a high proportion of genes that are unique to SIO-2, presumably involved in this phage's adaptation to survive and propagate in the ocean. Second, the origins of the genes with robust homologues constitute another intriguing divergence. Although siphoviruses are known to easily shuffle genetic information through lateral gene transfers, the repertoire of genes in SIO-2 genome spans an astonishingly wide range of origins from archaea to eukaryotic viruses. We suspect that this phage-host system could occur on marine aggregates and/ or organisms, which are often densely colonized by taxonomically diverse and active microbial communities (Smith et al., 1992; Smith et al., 1995; Acinas et al., 1999; Simon et al., 2002). Whether such promiscuity was a driving force in SIO-2 genome evolution remains unclear, but it certainly points to the need of better characterizing the viral diversity associated with these quasi-unexplored environments. Third, the siphovirus SIO-2 harbors an unusual genome architecture where core genes are unexpectedly interspaced with insertions of DNA comprising a high density of unknown genes. These intriguing inserts are reminiscent of hyperplastic regions in T4-like myoviruses where at least some of these genes are thought to improve bacteriophage fitness. Although the function and origins of these inserts remain unknown, they are interestingly associated with morphological variation of the SIO-2 capsid, which was shown to assemble as a large (80 nm) shell with an unusual T=12 symmetry (Lander et al., submitted). Given the high level of novelty of this phage and the ecological implication of its host, Vibrio phage SIO-2 emerges as a model of considerable interest. The isolation and sequencing of additional SIO-2-like phage should serve to improve our understanding of the diversity, the evolution, and the ecology of this intriguing group of siphoviruses.
EXPERIMENTAL PROCEDURES
Phage isolation and purification
Vibrio phage SIO-2 was isolated from coastal Pacific surface waters off the pier of the Scripps Institution of Oceanography (SIO Pier), La Jolla, California on September 14, 2007. A seawater sample was filtered through 0.2 μm and concentrated (ca. 10-fold) using a 100 kDa hollow fiber ultrafiltration unit (Amersham). The concentrated sample was used directly in a plaque assay with the marine bacterium Vibrio sp. SWAT-3 (Long and Azam, 2001) grown in ZoBell medium (5g L−1 peptone, 1 g.L−1 yeast extract). After 48 h incubation, a large, well-resolved, plaque (Fig S1) was picked from the lawn of host cells, eluted in 0.02μm filtered autoclaved seawater (FASW), and combined with host culture in a plaque assay. This procedure was repeated two more times to ensure isolation of a clonal phage population. A clonal lysate of SIO-2 was stored at 4°C until use.
Transmission electron microscopy
An aliquot (4 μL) of clonal phage suspension was applied to a glow-discharged copper EM grid (400 mesh size) with a nitrocellulose backed carbon surface. Following a 10 sec incubation, the grid was blotted with filter paper and stained with 2% uranyl acetate and lead–citrate, blotted again and allowed to air dry. Specimens were imaged using a Tecnai F20 Twin transmission electron microscope operating at 120 keV at a magnification of 50,000×.
Phage – Host interactions
The life cycle of SIO-2 was studied on SWAT3 host cultures grown in ZoBell (not nutrient-limited) and also in 1,000-fold diluted ZoBell (ZoBell/1000, nutrient-limited). For the non-limited conditions, a host culture in exponential phase grown in ZoBell was inoculated with a phage suspension at multiplicity of infection (moi) of 50 and incubated for 10 min to let the phages adsorb on host membrane. A control culture received 0.02μm FASW and it was incubated under similar conditions. After incubation, both suspensions were pelleted (2,000×g, 5 min, 18°C) to remove unadsorbed viruses and resuspended in liquid ZoBell to a dilution of 100-fold from the initial infection conditions. Control and infected cultures were incubated in the dark at room temperature with gentle agitation. Samples for microscopy counts of viruses and bacteria were taken every 15 min to 6 h during a 24 h-incubation period. These samples were fixed with 0.02-μm filtered formaldehyde (2% v:v final concentration) for 10 min at 4°C followed by freezing in liquid nitrogen and storage at −80°C until analysis. Thawed samples were stained with Sybr Green according to Noble and Fuhrman (1998, see SI). Slides were examined at 100× magnification under blue-light excitation using an Olympus epifluorescence microscope. More than 200 bacteria and viruses were counted in 20 fields of view. The bacteriophage burst size was determined by calculating the ratio of the net increase in viral abundance to the maximum net decline in bacterial abundance. The latent period corresponded to the time period until extracellular release of SIO-2 progeny.
For the nutrient limited conditions, the bacterial host was acclimated in ZoBell/1000 for three cycles of 48 h at room temperature. After 48 h, 1 volume of the culture was added to 9 volumes of ZoBell/1000. This novel culture was grown for another 48 h. The process was repeated one more time. When acclimated, the host culture was inoculated with a suspension of SIO-2 (moi = 5) while a control culture received a similar amount of 0.02μm FASW. The effect of phage addition on host growth was verified using a SIO-2 resistant culture of SWAT3 previously acclimated on ZoBell/1000 as described above.
Host range
SIO-2 host specificity was first determined by plaque assay using a broad range of Vibrio species and related genus cultures (Table S1). Dilution series of SIO-2 suspension (102 to 108 PFU mL−1) were incubated with the potential host cultures for 15 min. After incubation, the sample was mixed with the top agar and plated onto a lawn of ZoBell agar. Efficiency of plating (EOP) was quantified by calculating the ratio of SIO-2 plaque titer obtained with the heterologous host to that obtained with Vibrio sp. SWAT3. To complement this culture based assay, fluorescently labeled virus (FLVs) were also used to enumerate SIO-2 hosts in natural seawater based on the protocol of Comeau and Noble (2010, see SI Appendix). An aliquot of FLVs suspension (1×108 FLVs mL−1, final concentration) was incubated in 10 mL natural seawater for 30 min, this mixture was filtered onto 0.2 μm filters (Anodisc or Nuclepore, Whatman) and examined by epifluorescence microscopy. FLVs-tagged cells were considered as potential SIO-2 hosts (Fig S2). This assay was applied in different marine systems including the coastal Pacific Ocean (SIO-pier, 32° 52'N, 117°15'W) from January to March 2009, and the English Channel (SOMLIT Roscoff, 48°46′N, 3°57′W), the Bay of Morlaix (48°40'N, 3°53'W) and coastal Atlantic Ocean (SOMLIT Brest, 48°21'N; 4°33'W) in October 2008.
Stability to chemical, physical, and environmental factors
For all the stability tests, we used a SIO-2 suspension of 106 PFU mL−1. The loss in infectivity after treatment was determined from PFU in plaque assay conducted under host culture conditions. The different treatments are detailed below.
Chloroform
SIO-2 suspension were treated with 10 and 50% (v:v) chloroform and incubated for 60 min at 15°C. The phages were recovered from the aqueous phase and loss in infectivity was determined.
Temperature
To test SIO-2 stability at low temperatures, 0.5 mL SIO-2 lysate were incubated in duplicate at −196°C (liquid nitrogen), −80°C and −20°C for 24 h. Subsequently, samples were thawed at 30°C and tested in plaque assay. Heat stability was tested on viral aliquots (1 mL) for temperatures ranging from 15 to 90°C for 15 min. Samples were then cooled on ice for 5 min and immediately used in plaque assay.
pH
SIO-2 suspension (1% v/v) was inoculated into 5 mL-FASW adjusted at pH 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 for 24h at 4°C. After incubation, SIO-2 infectivity was determined from plaque assays conducted in host culture conditions.
Salinity
SIO-2 suspension (1% v/v) was added to 5 mL-artificial seawater (Harrison et al., 1980) with different NaCl concentration (0×, 0.5×, 1× and 2× of original NaCl concentration). Samples were incubated for 24h at 4°C and tested in plaque assay.
Hydrostatic pressure
Fresh lysate was used to fill 4.5-mL polyethylene transfer pipettes (Samco). Transfer pipettes were heat sealed and incubated at 14.5, 725, 1450, 4351, and 7251 psi in stainless-steel pressure vessels for 30 min at room temperature. After incubation, the pressure was released and SIO-2 infectivity was immediately tested by plaque assay.
Environmental parameters
We deployed a series of incubations to determine the stability of SIO-2 to natural environmental factors, including natural sunlight, UV-radiations, and viricidal agents naturally present in seawater. All glassware was acid cleaned (1N HCl) and autoclaved (or otherwise rinsed 3 times with boiling MilliQ water). A freshly produced lysate was added to 1 L of seawater collected from the surface water at Scripps Pier at a final concentration of 1×106 PFU mL−1. This mixture was distributed, in duplicate, into 50 mL tubes made of quartz (UV-transparent), Pyrex (UV-opaque) and wrapped into 3 layers of aluminium foil (light-proof). Controls without phage addition were prepared in parallel. All samples were incubated at ambient seawater temperature on the roof of the Hubbs Hall building at Scripps Institution of Oceanography on a cloudless day (May 5, 2009). Decay in SIO-2 infectivity was monitored over 24 hours by plaque assay. During these incubations, the phage suspension was exposed to a temperature range of 14 – 19.9°C and average PAR of 1000 μmol sec−1 m−2 (as recorded at the SIO Pier weather station, 500 m from the site of the experiment, courtesy R. van Boxtel).
Genome sequencing
Culture of the bacterial host SWAT3 was combined with SIO-2 in 25 plaque assays giving confluent lysis. Once plaques appeared, viruses were eluted in 10 kDa FASW for 1 h. Eluates were 0.2 μm-filtered (Nuclepore), concentrated by ultracentrifugation (SW28, 141,000× g, 2 h at 8°C) and the subsequent phage pellet was purified by linear sucrose gradient (10 – 40% in 10 kDa FASW) centrifugation (SW41, 17,000 rpm, 1 h at 8°C). Purified SIO-2 particles were resuspended in 10 kDa FASW and stored at 4°C until use. Prior to sequencing, the nature and the size of SIO-2 genome were determined by pulsed field gel electrophoresis as described elsewhere (Baudoux and Brussaard, 2005). The dsDNA genome of approx. 80 kb (Fig S3) was extracted from sucrose-purified SIO-2 particles using standard procedure (Sambrook et al., 1989) and sent for sequencing to the Broad Institute in the context of Marine Phage, Virus, and Virome Sequencing Project launched by the Gordon and Betty Moore Foundation (GBMF). After passing the Broad Institute's quality control steps, 454 libraries were prepared and pyrosequenced as described in Henn et al. (2010). Briefly, SIO-2 genomic DNA was sheared using Covaris AFA technology and DNA fragments larger than 200 bp were concentrated by adding 0.8× the volume of AMPure PCR purification system (Agencourt Bioscience corporation). Construction of 454-clone libraries was carried out using the GS20 FLX Library preparation kit (454 Life Sciences, Brandford CT) according to Margulies et al. (2005). The DNA single strand template was recovered using the Dynal MPC-S magnet, denaturated with 250 mM sodium hydroxide, and neutralized with 10% acetic acid. After concentration with AMPure beads, the DNA template was quantified using the Pico 600 chip (Agilent, Santa Clara CA). PCR reactions and sequencing were performed according to the GS20 of FLX protocol. The sequence of SIO-2 genome has been submitted to the GenBank database under accession number PRJNA42177 ID: 42177.
Genome assembly and annotation
Raw sequence reads, generated at the Broad Institute, were assembled using Phrap and examined in Consed at the Pittsburgh Bacteriophage Institute. The final draft assembly was 81,184 bases, with an average coverage depth of 57 reads and a quality score greater than 60 for 99.86% of the bases. This draft sequence had a terminal duplication of 589 bases, indicating that the virion DNA either is circularly permuted or contains a terminal redundancy. Manual examination of the assembly showed no evidence of “pile-ups” of sequence reads with a common end, as is seen when the virion DNA contains non-permuted terminal redundancy. We therefore concluded that the virion DNA is circularly permuted. The resulting non-redundant sequence is 80,598 bp long. For a circularly permuted genome, the assignment of the start of base numbering is arbitrary. Since the cumulative GC Skew analysis (window size 100 bp, Grigoriev, 1998) failed in predicting a site of replication origin, we placed the origin in a large intergenic space three genes upstream from the terminase gene.
Initial gene calls were obtained from Glimmer (Delcher et al., 1999) and GeneMark (Besemer and Borodovsky, 2005) by DNA Master 5.22.1. In addition, gene coordinates were determined manually, guided by the GeneMark coding potential output and the presence of appropriately located Shine-Dalgarno translation start sequences. The three methods of assigning gene starts agreed for all but 6 genes, and the disagreements were resolved by manual inspection. Functional assignments of the predicted amino acid sequences were made by BlastP, automated by DNA Master, followed by manual PSI-Blast for all sequences making strong matches in BlastP. For some of the virion structure and assembly proteins, additional information, principally relative gene positions, was used to infer probable functions, as described in the text. Preliminary assignments of taxonomic origin for predicted proteins were made using the DarkHorse program, version 1.3 (Podell et al., 2008). Searches for potential tRNA and tmRNA genes were carried out with Aragorn (Laslett and Canback, 2004) and tRNAScanSE (Lowe and Eddy, 1997). Promoter candidates were found in DNA Master, using a search template for E. coli sigma70 promoters. Putative factor-independent transcription terminators were found initially by manual examination of intergenic regions. Subsequent automated search of the entire sequence for hairpins gave no additional convincing terminator candidates.
Structural proteins
Sucrose gradient purified suspension was diluted in Laemmli buffer (Laemmli, 1970) and heated at 95 °C for 5 min. A subsample was resolved on SDS-PAGE gel (Ready gel, 4 – 12% Tris-HCl, Bio-Rad, Hercules, CA, USA) using a Mini Protean 3 Cell (Bio-Rad). Protein molecular weight standards (Precision plus protein standard, Bio-Rad) were used for size calibration. The gel was stained with Coomassie Blue (Bio-Safe, Bio-Rad) for 30 min and destained for 30 min in MilliQ water prior to imaging using a white light table.
DNA polymerase A (DNA pol A) protein phylogeny
DNA pol A sequences of known bacteriophages (Table S2) were used to investigate SIO-2 phylogeny. Multiple alignment has been automatically performed using the specific family DNA pol A profile, the raw HMM guide PF00476 from the Pfam database (Finn et al., 2010) and run under HMMER v.3 software (http://hmmer.janelia.org). Alignments were manually edited as needed and used for conducting molecular phylogenies based on Maximum Likelihood and Neighbour-Joining, which lead to congruent topologies. An unrooted tree has been produced with FastTree program (Price et al., 2009) using a Maximum Likelihood method and exhibits three main clusters, arbitrarily named I, II, and III.
Marine microbial metagenome analysis
To determine SIO-2 biogeography, this phage genome was used to recruit homologous fragments from different microbial metagenomes publicly available from Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis (CAMERA) database (http://camera.calit2.net). SIO-2 genome was blasted using BLASTN (E <10−3) against several marine viromes available in this database. Additionally, we looked for homologs of the partial 16S rRNA gene sequence of SIO-2 hosts in CAMERA with a percentage of identity > 95% (GenBank accession nr: AF366022, X7469, and CP000789 for Vibrio sp. SWAT3, V. campbellii ATCC 25920, and V. harveyi ATCC BAA-1116, respectively).
Supplementary Material
Acknowledgments
The Captain and the crew of R.V. Mysis are acknowledged for sampling opportunity in the Bay of Morlaix and the English Channel. We thank E. Eloe and Å. Hagström for providing strains to determine SIO-2 host range, D. Russell for SIO-2 genome assembly, O. Holm and E. Kisfaludy for their help to set up the incubations on Hubbs Hall rooftop, Y. Bozec, M. Vernet and M. Porrachia for their technical assistance and E. Allen for stimulating discussion on bacteriophage genomics. The reviewers are acknowledged for their constructive comments on a previous version of this manuscript. This research was funded in part by the Gordon and Betty Moore Foundation Marine Microbiology Initiative to F.A. and to the Broad Institute for SIO-2 genome sequencing (sample G2314), by a Scripps post-doctoral fellowship to A-C. B. and by the NIH NCRR P41 program (RR017573) to the National Resource for Automated Molecular Microscopy.
References
- Acinas SG, Anton J, Rodriguez-Valera F. Diversity of free-living and attached bacteria in offshore western Mediterranean waters as depicted by analysis of genes encoding 16S rRNA. Appl Environ Microbiol. 1999;65:514–522. doi: 10.1128/aem.65.2.514-522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly F, Youle M, Nosrat B, Srinagesh S, Rodriguez-Brito B, McNairnie P, et al. Genomic analysis of multiple Roseophage SIO1 strains. Environ Microbiol. 2009;11:2863–2873. doi: 10.1111/j.1462-2920.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, et al. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbriol C, Comeau AM, Kutateladze M, Adamia R, Krisch HM. Mobile regulatory cassettes mediate modular shuffling in T4-type phage genomes. Genome Biol Evol. 2010;2:140–152. doi: 10.1093/gbe/evq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin B. Vibrios as causal agents of zoonoses. Vet Microbiol. 2010;140:310–317. doi: 10.1016/j.vetmic.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Austin B, Zhang XH. Vibrio harveyi: a significant pathogen of marine vertebrates and invertebrates. Letters Appl Microbiol. 2006;43:119–124. doi: 10.1111/j.1472-765X.2006.01989.x. [DOI] [PubMed] [Google Scholar]
- Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoux AC, Brussaard CPD. Characterization of different viruses infecting the marine harmful algal bloom species Phaeocystis globosa. Virol. 2005;341:80–90. doi: 10.1016/j.virol.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33:W451–W454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–284. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Brüssow H, Desiere F. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol Microbiol. 2001;39:213–223. doi: 10.1046/j.1365-2958.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- Casjens S, Hendrix R. Comments on Arrangement of Morphogenetic Genes of Bacteriophage-Lambda. J Mol Biol. 1974;90:20–23. doi: 10.1016/0022-2836(74)90253-8. [DOI] [PubMed] [Google Scholar]
- Clokie MRJ, Shan JY, Bailey S, Jia Y, Krisch HM, West S, Mann NH. Transcription of a 'photosynthetic' T4-type phage during infection of a marine cyanobacterium. Environ Microbiol. 2006;8:827–835. doi: 10.1111/j.1462-2920.2005.00969.x. [DOI] [PubMed] [Google Scholar]
- Comeau AM, Suttle CA. Distribution, genetic richness and phage sensitivity of Vibrio spp. from coastal British Columbia. Environ Microbiol. 2007;9:1790–1800. doi: 10.1111/j.1462-2920.2007.01299.x. [DOI] [PubMed] [Google Scholar]
- Comeau AM, Noble RT. Preparation and application of fluorescently labeled virus particles. In: Wilhelm SW, Weinbauer MG, Suttle CA, editors. Manual of Aquatic Viral Ecology. ASLO; 2010. pp. 19–29. [Google Scholar]
- Comeau AM, Chan AM, Suttle CA. Genetic richness of vibriophages isolated in a coastal environment. Environ Microbiol. 2006;8:1164–1176. doi: 10.1111/j.1462-2920.2006.01006.x. [DOI] [PubMed] [Google Scholar]
- Comeau AM, Bertrand C, Letarov A, Tetart F, Krisch HM. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virol. 2007;362:384–396. doi: 10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Cotner JB, Ammerman JW, Peele ER, Bentzen E. Phosphorus-limited bacterioplankton growth in the Sargasso Sea. Aquat Microb Ecol. 1997;13:141–149. [Google Scholar]
- Danovaro R, Corinaldesi C, Dell'Anno A, Fuhrman J, Middelburg J, Noble R, Suttle CA. Marine viruses and global climate change. FEMS Microbiol Rev. 2011;35:993–1034. doi: 10.1111/j.1574-6976.2010.00258.x. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards RA, Rohwer F. Viral metagenomics. Nature Rev Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunesekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res Database Issue. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–548. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- Gold L. Post-transcriptional regualtory mechanisms in E. coli. Annual Rev Biochem. 1988;57:199–233. doi: 10.1146/annurev.bi.57.070188.001215. [DOI] [PubMed] [Google Scholar]
- Gottesman M, Oppenheim A. Lysogeny and prophage. In: Webster RG, Granoff A, editors. Encyclopedia of Virology. Academic Press; London: 1994. pp. 814–823. [Google Scholar]
- Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26:2286–2290. doi: 10.1093/nar/26.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Waters RE, Taylor FJR. A broad spectrum artifical seawater medium for coastal and open ocean phytoplankton. J Phycol. 1980;16:28–35. [Google Scholar]
- Hatfull GF, Cresawn SG, Hendrix RW. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol. 2008;159:332–339. doi: 10.1016/j.resmic.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn MR, Sullivan MB, Stange-Thomann N, Osburne MS, Berlin AM, Kelly L, et al. Analysis of High-Throughput Sequencing and Annotation Strategies for Phage Genomes. PLos One. 5 doi: 10.1371/journal.pone.0009083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW, Casjens SR. Protein cleavage in bacteriophage λ tail assembly. Virol. 1974;61:156–159. doi: 10.1016/0042-6822(74)90250-5. [DOI] [PubMed] [Google Scholar]
- Juhala RJ, Ford ME, Duda RL, Yulton A, Hatfull GF, Hendrix RW. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol. 2000;299:27–51. doi: 10.1006/jmbi.2000.3729. [DOI] [PubMed] [Google Scholar]
- Karl DM, Tien G. Temporal variability in dissolved phosphorus concentrations in the subtropical North Pacific Ocean. Mar Chem. 1997;56:77–96. [Google Scholar]
- Katsura I. Determination of bacteriophage lambda tail length by a protein ruler. Nature. 1987;327:73–75. doi: 10.1038/327073a0. [DOI] [PubMed] [Google Scholar]
- Khemayan K, Pasharawipas T, Puiprom O, Sriurairatana S, Suthienkul O, Flegel TW. Unstable lysogeny and pseudolysogeny in Vibrio harveyi siphovirus-like phage 1. Appl Environ Microbiol. 2006;72:1355–1363. doi: 10.1128/AEM.72.2.1355-1363.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmaro A, Banin E, Loya Y, Stackebrandt E, Rosenberg E. Vibrio shiloi sp nov., the causative agent of bleaching of the coral Oculina patagonica. Int J Sys Evol Microbiol. 2001;51:1383–1388. doi: 10.1099/00207713-51-4-1383. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of Structural Proteins During Assembly of Head of Bacteriophage-T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lane T, Eiserling F. Genetic Control of Capsid Length in Bacteriophage T4. VII. A model of length regulation based on DNA size. J. Struct. Biol. 1990;104:9–23. doi: 10.1016/1047-8477(90)90052-e. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Hatfull GF, Hendrix RW. Imbroglios of viral taxonomy: Genetic exchange and failings of phenetic approaches. J Bacteriol. 2002;184:4891–4905. doi: 10.1128/JB.184.17.4891-4905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F, Chisholm SW. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc Natl Acad Sci USA. 2004;101:11013–11018. doi: 10.1073/pnas.0401526101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell D, Jaffe JD, Coleman ML, Futschik ME, Axmann IM, Rector T, et al. Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature. 2007;449:83–86. doi: 10.1038/nature06130. [DOI] [PubMed] [Google Scholar]
- Lohr JE, Chen F, Hill RT. Genomic analysis of bacteriophage Phi JL001: Insights into its interaction with a sponge-associated alpha-proteobacterium. Appl Environ Microbiol. 2005;71:1598–1609. doi: 10.1128/AEM.71.3.1598-1609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long RA, Azam F. Antagonistic interactions among marine pelagic bacteria. Appl Environ Microbiol. 2001;67:4975–4983. doi: 10.1128/AEM.67.11.4975-4983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long RA, Rowley DC, Zamora E, Liu J, Bartlett DH, Azam F. Antagonistic interactions among marine bacteria impede the proliferation of Vibrio cholera. Appl Environ Microbiol. 2005;71:8531–8536. doi: 10.1128/AEM.71.12.8531-8536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann NH. Phages of the marine cyanobacterial picophytoplankton. FEMS Microbiol Reviews. 2003;27:17–34. doi: 10.1016/S0168-6445(03)00016-0. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari X, Kerros ME, Weinbauer MG. Virus attachment to transparent exopolymeric particles along trophic gradients in the southwestern lagoon of New Caledonia. Appl Environ Microbiol. 2007;73:5245–5252. doi: 10.1128/AEM.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard AD, Zwirglmaier K, Downey MJ, Mann NH, Scanlan DJ. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: implications for mechanisms of cyanophage evolution. Environl Microbiol. 2009;11:2370–2387. doi: 10.1111/j.1462-2920.2009.01966.x. [DOI] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naryshkina T, Liu J, Florens L, Swanson SK, Pavlov AR, Pavlova NV, et al. Thermus thermophilus bacteriophage phi YS40 genome and proteomic characterization of virions. J Mol Biol. 2006;364:667–677. doi: 10.1016/j.jmb.2006.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Němeček D, Overman SA, Hendrix RW, Thomas GJ., Jr Unfolding Thermodynamics of the Δ-Domain in theProhead I Subunit of Phage HK97: Determination byFactor Analysis of Raman Spectra. J Mol Biol. 2009;385:628–641. doi: 10.1016/j.jmb.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble RT, Fuhrman JA. Virus decay and its causes in coastal waters. Appl Environ Microbiol. 1997;63:77–83. doi: 10.1128/aem.63.1.77-83.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble RT, Fuhrman JA. Use of SYBR Green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat Microb Ecol. 1998;14:113–118. [Google Scholar]
- Pasharawipas T, Wetchakit N, Sriurairatana S. The cycle for a Siphoviridae-like phage (VHS1) of Vibrio harveyi is dependent on the physiological state of the host. Virus Res. 2008;135:332. doi: 10.1016/j.virusres.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Pasharawipas T, Thaikua S, Sriurairatana S, Ruangpan L, Direkbusarakum S, Manopvisetcharean J, Flegel TW. Partial characterization of a novel bacteriophage of Vibrio harveyi isolated from shrimp culture ponds in Thailand. Virus Res. 2005;114:63–69. doi: 10.1016/j.virusres.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Paul JH, Sullivan MB. Marine phage genomics: what have we learned? Curr Opin Biotech. 2005;16:299. doi: 10.1016/j.copbio.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Paul JH, Williamson SJ, Long A, Authement RN, John D, Segall AM, et al. Complete genome sequence of phi HSIC, a pseudotemperate marine phage of Listonella pelagia. Appl Environ Microbiol. 2005;71:3311–3320. doi: 10.1128/AEM.71.6.3311-3320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, et al. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–182. doi: 10.1016/s0092-8674(03)00233-2. [DOI] [PubMed] [Google Scholar]
- Podell S, Gaasterland T, Allen EE. A database of phylogentically atypical genes in archaeal and bacterial genomes, identified using the DarkHorse algorithm. BMC Bioinformatics. 2008;9:419. doi: 10.1186/1471-2105-9-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M, Dehal P, Arkin A. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol Biol Evol. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux C, van Sinderen D, Suarez J, Garcia P, Ladero V, Fitzgerald GF, et al. The dilemma of phage taxonomy illustrated by comparative genomics of Sfi21-like Siphoviridae in lactic acid bacteria. J Bacteriol. 2002;184:6026–6036. doi: 10.1128/JB.184.21.6026-6036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F. Global phage diversity. Cell. 2003;113:141–141. doi: 10.1016/s0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Segall A, Steward G, Seguritan V, Breitbart M, Wolven F, Azam F. The complete genomic sequence of the marine phage Roseophage SIO1 shares homology with nonmarine phages. Limnol Oceanogr. 2000;45:408–418. [Google Scholar]
- Sambrook JE, Fritsch EF, Maniatis T. Molecular cloning, a laboratory manual. Cold spring Harbor Press; 1989. [Google Scholar]
- Simon M, Grossart HP, Schweitzer B, Ploug H. Microbial ecology of organic aggregates in aquatic ecosystems. Aquat Microb Ecol. 2002;28:175–211. [Google Scholar]
- Smith DC, Simon M, Alldredge AL, Azam F. Intense Hydrolytic Enzyme-Activity on Marine Aggregates and Implications for Rapid Particle Dissolution. Nature. 1992;359:139–142. [Google Scholar]
- Smith DC, Steward GF, Long RA, Azam F. Bacterial Mediation of Carbon Fluxes During a Diatom Bloom in a Mesocosm. Deep-Sea Res II. 1995;42:75–97. [Google Scholar]
- Stewart CR, Casjens SR, Cresawn SG, Houtz JM, Smith AL, Ford ME, et al. The Genome of Bacillus subtilis Bacteriophage SPO1. J Mol Biol. 2009;388:48–70. doi: 10.1016/j.jmb.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLos Biol. 2005;3:790–806. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Krastins B, Hughes JL, Kelly L, Chase M, Sarracino D, Chisholm SW. The genome and structural proteome of an ocean siphovirus: a new window into the cyanobacterial `mobilome'. Environ Microbiol. 2009;11:2935–2951. doi: 10.1111/j.1462-2920.2009.02081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Suttle CA. Marine viruses - major players in the global ecosystem. Nature Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- Suttle CA, Chen F. Mechanisms and Rates of Decay of Marine Viruses in Seawater. Appl Environ Microbiol. 1992;58:3721–3729. doi: 10.1128/aem.58.11.3721-3729.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thingstad TF, Krom MD, Mantoura RFC, Flaten GAF, Groom S, Herut B, et al. Nature of phosphorus limitation in the ultraoligotrophic eastern Mediterranean. Science. 2005;309:1068–1071. doi: 10.1126/science.1112632. [DOI] [PubMed] [Google Scholar]
- Thompson FL, Iida T, Swings J. Biodiversity of vibrios. Microbiol Mol Biol Rev. 2004;68:403–431. doi: 10.1128/MMBR.68.3.403-431.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JB, Jiang Y, Vincent M, Sun YQ, Yu H, Wang J, et al. Complete genome sequence of bacteriophage T5. Virol. 2005;332:45–65. doi: 10.1016/j.virol.2004.10.049. [DOI] [PubMed] [Google Scholar]
- Wommack KE, Hill RT, Muller TA, Colwell RR. Effects of sunlight on bacteriophage viability and structure. Appl Environ Microbiol. 1996;62:1336–1341. doi: 10.1128/aem.62.4.1336-1341.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Hendrix RW, Duda RL. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004;16:11–21. doi: 10.1016/j.molcel.2004.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.