

Abstract
Spinocerebellar ataxia 36 has been recently described in Japanese families as a new type of spinocerebellar ataxia with motor neuron signs. It is caused by a GGCCTG repeat expansion in intron 1 of NOP56. Family interview and document research allowed us to reconstruct two extensive, multigenerational kindreds stemming from the same village (Costa da Morte in Galicia, Spain), in the 17th century. We found the presence of the spinocerebellar ataxia 36 mutation co-segregating with disease in these families in whom we had previously identified an ∼0.8 Mb linkage region to chromosome 20 p. Subsequent screening revealed the NOP56 expansion in eight additional Galician ataxia kindreds. While normal alleles contain 5–14 hexanucleotide repeats, expanded alleles range from ∼650 to 2500 repeats, within a shared haplotype. Further expansion of repeat size was frequent, especially upon paternal transmission, while instances of allele contraction were observed in maternal transmissions. We found a total of 63 individuals carrying the mutation, 44 of whom were confirmed to be clinically affected; over 400 people are at risk. We describe here the detailed clinical picture, consisting of a late-onset, slowly progressive cerebellar syndrome with variable eye movement abnormalities and sensorineural hearing loss. There were signs of denervation in the tongue, as well as mild pyramidal signs, but otherwise no signs of classical amyotrophic lateral sclerosis. Magnetic resonance imaging findings were consistent with the clinical course, showing atrophy of the cerebellar vermis in initial stages, later evolving to a pattern of olivo-ponto-cerebellar atrophy. We estimated the origin of the founder mutation in Galicia to have occurred ∼1275 years ago. Out of 160 Galician families with spinocerebellar ataxia, 10 (6.3%) were found to have spinocerebellar ataxia 36, while 15 (9.4%) showed other of the routinely tested dominant spinocerebellar ataxia types. Spinocerebellar ataxia 36 is thus, so far, the most frequent dominant spinocerebellar ataxia in this region, which may have implications for American countries associated with traditional Spanish emigration.
Keywords: spinocerebellar ataxia, NOP56, expansion mutation, founder effect, Galicia
Introduction
The spinocerebellar ataxias (SCAs) are a heterogeneous group of dominantly inherited neurodegenerative disorders, caused by diverse mutation types and complex pathogenesis (Schöls et al., 2004; Sequeiros et al., 2011). Large expansions of a GGCCTG hexanucleotide repeat in the first intron of NOP56 were recently reported as the cause of a SCA and motor neuron disease, SCA36, in 17 patients from 9 (3.6%) SCA families, originating from the Japanese district of Chugoku (Kobayashi et al., 2011).
NOP56 encodes a component of the ribonucleoprotein complex and plays a role in transcription and splicing processes. The recognition of large, non-coding expansions in an increasing number of neuromuscular disorders (Matsuura et al., 2000; Sato et al., 2009; Dejesus-Hernandez et al., 2011; Renton et al., 2011) has raised interest in the pathogenic routes mediated by repetitive RNA elements (Krzyzosiak et al., 2011). Although the pathogenic mechanism is not well understood, the presence of RNA foci has been related to sequestration of RNA-binding proteins (Daughters et al., 2009; Sato et al., 2009; Kobayashi et al., 2011). Furthermore, non-expansion mutations affecting RNA biogenesis and regulation have also been associated with disorders of neurodevelopment and neurodegenerative diseases affecting cerebellar and motor pathways (Edvardson et al., 2007; Sreedharan et al., 2008; Vance et al., 2009).
The Japanese patients with SCA36 showed a characteristic phenotype, with spinocerebellar symptoms starting in the fourth or fifth decade, associated with signs of upper as well as lower motor neuron involvement, manifested by skeletal muscle and tongue atrophy and fasciculations. Although they were apparently independent families, all mutation carriers showed the same haplotype, suggesting a common founder and leading the authors to hypothesize that SCA36 might be geographically limited to that western region of Japan (Kobayashi et al., 2011).
At the time of the Kobayashi et al. (2011) publication, we found the same mutation in two very large kindreds from a small Atlantic coastal region in Northwestern Spain. These patients had an autosomal-dominant, adult-onset and slowly progressive spinocerebellar disease, which had been named ‘Costa da Morte’ (‘Coast of Death’ in Spanish) ataxia (Arias et al., 2008), after the name of that coastal area of Galicia. We now report a thorough clinical and genetic characterization of the two original families, as well as eight additional SCA36 kindreds from this region. This new mutation represents the most frequent cause of SCA so far in Galicia.
Patients and methods
Patient recruitment and clinical characterization
Apparently unrelated families with an autosomal dominant SCA had been followed, since the early 1990s, at the Clinical University Hospital of Santiago de Compostela. Clinical similarities and close regional origin of the families led us to start an investigation to further delineate the clinical and genealogical characteristics. These initial families turned out to be branches of an extensive pedigree (Fig. 1, Family 1). Subsequently, another large kindred, from the same area, segregating a similar disease, was identified (Fig. 1, Family 2). Reconstruction of multigenerational genealogies was achieved through recall of elder family members, as well as from extensive documentation from birth and death certificates and property transmission certificates. The oldest family members from whom documented references were available lived in the 17th century.
Figure 1.

Schematic representation of the first six generations of the two largest SCA36 kindreds, Family 1 (left) and Family 2 (right); black diamonds = definitely affected by examination and genetic analysis; white diamonds = definitely unaffected by examination and genetic analysis or children of confirmed unaffected individuals; question mark = individuals at risk, status unknown; inner square = obligate carrier, not evaluated. Below each symbol the minimum number of subjects is indicated (i.e. for whom direct information was obtained by family history and/or examination).
The recruitment and study protocol was approved by the medical research ethics committee from Galicia. Individuals who accepted to participate in this study underwent a thorough neurological examination. The following workup was carried out: brain and spinal MRI, complete blood count and metabolic screen, CSF analysis, EMG and nerve conduction studies, audiometry, brainstem auditory evoked potentials and somatosensory evoked potentials. Peripheral blood was drawn and used for DNA purification by standard methods. In addition to Families 1 and 2, DNA samples from 158 index cases with SCA, all from Galicia, were available for this study. From the total of 160 ataxia kindreds, 47 families segregated the disease in an autosomal dominant manner, 38 had a family history of ataxia but no clear inheritance pattern, 34 were reported as sporadic cases, and no family information was available for 41. All these patients had previously been screened for Friedreich ataxia, SCA1, SCA2, Machado–Joseph disease/SCA3, SCA6, SCA7, SCA12, SCA17, dentatorubropallidoluysian atrophy and fragile-X-associated tremor ataxia syndrome (M. García-Murias et al., unpublished data).
Genotyping and linkage analysis
In a first phase, DNA samples from 68 individuals belonging to Families 1 and 2 were genotyped for a panel of 1000 microsatellite markers with average spacing of 4 cM along the genome (DeCode Genetics). Out of 31 repeat motives identified in the region of interest, 12 were found to be polymorphic and used for refinement of linkage analysis and haplotype reconstruction (Supplementary Tables 1 and 2). Subsequently, we selected 241 single nucleotide polymorphisms (SNPs) spanning the linkage region using information in dbSNP version 130 (http://www.ncbi.nlm.nih.gov/projects/SNP/) and HapMap (http://hapmap.ncbi.nlm.nih.gov/). In this tier, 87 individuals were genotyped together with those from Phase 1. Obligate carriers and healthy spouses were included in the haplotype analysis to establish phase. The iPLEX® assay for MassARRAY® platform (Sequenom® Inc.) was used for genotyping in multiplex format, and the analysis software (TYPER Analyzer®) was used to translate the observed mass into a genotype for each reaction. Parametric two-point and multipoint linkage analyses, and haplotype reconstruction, were performed using the software FastLink v4.1 and GeneHunter v2.1r5, implemented by the easyLINKAGE V5.05 platform (Lindner et al., 2005), under the assumptions of autosomal dominant inheritance, complete penetrance, a disease allele frequency of 0.00001 and equal allele frequencies for marker loci.
Haplotype reconstruction and estimation of mutation age
Merlin 1.1.2 was used for the inference of combined—microsatellite and SNP—haplotypes (Abecasis et al., 2002). Where family data were not informative, we used PHASE 2.1.1 (Stephens et al., 2001; Stephens and Donnelly, 2003) specifying the known haplotypes. The age of the most recent common ancestor of mutation carriers was estimated using a likelihood-based method with the ESTIAGE program, based on the length of the haplotype shared on each side of the disease locus (Genin et al., 2004). For that purpose, 14 microsatellites and one SNP spanning 4.98 Mb around the mutation were analysed in 10 mutation carriers representing each family. An attempt to date the mutation was also performed with DMLE+2.3 (http://dmle.org). The software implements a Bayesian method based on the observed linkage disequilibrium between a disease mutation and linked markers to infer the age of the mutation (Reeve and Rannala, 2002; Rannala and Reeve, 2003). Haplotypes in control individuals were estimated using PHASE, and the relative position of the mutation was set within the haplotype. Population growth rates of 0.05 and 0.07 were used for the calculations, based on previous data for European populations and on Galician historic census (http://www.ine.es/). Two alternative proportions of carrier chromosomes were estimated, considering a disease prevalence of 1:10 000 or 1:20 000.
Mutation screening and structure of the NOP56 intronic repeat
Mutation screening was carried out in patients and controls. Primer pairs were designed to polymerase chain reaction (PCR)-amplify and sequence all coding exons in 21 known and predicted genes, as well as nine small non-coding RNAs in the region, using an ABI3730 automatic analyser. The STADEN software (Staden, 1996) was used to analyse electrophoretic traces. To investigate potential deletions or multiplications, we carried out a high-throughput genotyping of affected and non-affected individuals, with the Affymetrix® 6.0 genome-wide array, which included 277 SNPs between the limits of recombination. Copy number files (CNCHP) were generated with the Genotyping Console® and visualized on the Affymetrix® Chromosome Analysis Suite. For a finer level of resolution, a 60 000-oligonucleotide array was custom-designed (qGenomics) covering 1.9 Mb and affected/non-affected sample pairs were hybridized. The Agilent Genomic Workbench® software (Agilent Technologies) was used to analyse the data.
In addition, 31 sequence elements of 3–7 bp and at least four repeat units were identified in the region and amplified with flanking primers to screen for potential expansions. Two primer pairs were used to amplify the NOP56 intron 1 region, one of them flanking the GGCCTG repeat only, while the other pair included a 6-bp CGGGCG insertion/deletion (rs28970277, NM_006392.2:c.3+22_3+27delCGGGCG) 44 bp upstream from the hexanucleotide repeat. Fluorescent triplet repeat primed polymerase chain reaction (TP-PCR) (Warner et al., 1996) was carried out on 25 µl reactions, as follows: 60 ng of DNA, 200 µM of each dNTP, 1.5 mM MgCl2, 5 µl of 360 GC Enhancer (Applied Biosystems), 1.25 U AmpliTaq Gold® 360 DNA Polymerase and 1× buffer (Applied Biosystems), 0.8 µM reverse universal primer, 0.8 µM forward primer and 0.08 µM repeat-specific primer (Kobayashi et al., 2011). The reaction mixture was denatured at 95°C for 10 min and subjected to 40 cycles (95°C × 1 min, 63.5°C × 1 min, 72°C × 4 min, adding 10 s each cycle). The mixture was maintained at 72°C, for 10 min, for final extension. Amplified fragments were separated in an ABI3730 analyser and alleles were determined with Fragment Analysis software. Both standard and TP-PCR were carried out in 136 individuals from Families 1 and 2. These analyses were then performed in another 158 index cases with SCA from Galicia. Subsequently, 23 additional individuals from Families 3–10 were also analysed, as well as 214 Spanish patients with spastic paraplegia of unknown aetiology and 234 Galician healthy individuals aged >65 years. To establish the variability in the structure of non-expanded SCA36 alleles, homozygous samples were sequenced by standard Sanger sequencing. We also sequenced the PCR products obtained through standard amplification from expansion carriers, thus characterizing the smaller, non-expanded allele in each patient. All primer sequences are available in Supplementary Table 1.
Southern blot
Detection and approximate assessment of the number of expanded GGCCTG repeats in affected individuals was carried out by Southern blot. Genomic DNA (10 µg) was digested with BsmI (New England Biolabs) according to the manufacturer’s instructions. Digestion products were separated by electrophoresis on a 0.8% agarose gel, depurinated with 0.25 M HCl, denaturized with 0.4 M NaOH and transferred with 10× SSC (saline-sodium citrate) buffer onto a Hybond-N+ nylon membrane (GE Healthcare). DNA was immobilized on the membrane by UV cross-linking. A 715-bp probe for hybridization was synthesized from genomic DNA, by PCR, using the Expand High Fidelity PCR System (Roche Applied Science) and primers flanking exons 2 and 3 of NOP56 (Supplementary Table 1). The probe was labelled with [α-32P]-dCTP with a specific activity of 3000 Ci/mmol (Perkin Elmer), using the Prime It® II Random Primer Labelling Kit (Agilent Technologies). Hybridization with the labelled probe was carried out overnight, at 65°C, and signal detection was performed by X-ray film exposure.
Statistical analysis
The size of the expanded repeat was compared between subsequent generations for 27 mutated alleles, in Families 1 and 2, both globally and according to the gender of the transmitting parent. Kaplan–Meier analyses were used to estimate the influence of allele size on disease-onset age. Onset age was compared between groups of patients with allele size of ∼600–850, 850–1200 and >1200 repeats. Log-rank test statistics were used to determine whether the Kaplan–Meier curves differed between subgroups. Mean onset age per generation was also analysed. Statistical significance was defined as P < 0.05, and calculations were performed with the statistical software package SPSS version 15.0 for Windows (SPSS Inc.).
Results
Linkage to chromosome 20 p and mutation screening
Two large kindreds with an autosomal dominant form of SCA originated from the most Northwestern part of Galicia (Spain), an area with, for many years, difficult access both by sea and by road. Extensive family interviews and document studies allowed us to reconstruct two large pedigrees with 673 individuals (Family 1) and 676 individuals (Family 2), spanning seven generations, and represented in Fig. 1. After genome-wide linkage analysis, a maximum two-point LOD (log10 of odds) score of 9.8 was obtained for marker D20S842. Subsequent microsatellite and SNP genotyping placed the critical interval between the telomeric marker rs2422752 and the centromeric marker D20S181, spanning ∼2 cM and ∼0.8 Mb. No point mutations co-segregating with the disease were found in exonic regions of 30 genes and microRNAs within the candidate region by direct sequencing of affected and unaffected subjects. The variants identified are available as Supplementary Table 2. Single nucleotide changes without an ‘rs’ number were submitted to dbSNP. Specifically, we did not find mutations in PDYN and TGM6, which lie within the linkage region and have been reported as mutated in SCA23 and SCA35, respectively. Dosage analysis by comparative genomic hybridization array and Affymetrix® 6.0 genome-wide SNP array disclosed no aberrations.
Unstable NOP56 hexanucleotide expansion in families with ataxia
Mendelian inconsistencies in allele transmission within nuclear families were observed upon fragment length analysis of a GGCCTG repeat in intron 1 of NOP56, while no patterns suggestive of repeat expansion were detected in additional repetitive elements in the region. A TP-PCR pattern suggesting the presence of an expansion was present in all affected members of Families 1 and 2; a similar pattern was then observed in eight additional index cases from a cohort of patients with SCA from our region (Fig. 2A). Ten out of 160 (6.3%) index cases from our cohort of families with SCA showed a SCA36 expansion. In all, 63 individuals from these 10 families tested positive by TP-PCR. Southern blot analysis was possible in 34 samples and confirmed the presence of very large abnormal alleles in all cases. While the normal restriction fragment is of 2.9 kb with our protocol, expanded alleles ranged from 6.8 to ∼18 kb, corresponding to ∼650–2500 additional hexanucleotide repeat units (Fig. 2B).
Figure 2.

Mutation analysis in patients with SCA36. (A) PCR amplification of the (GGCCTG)n in a nuclear family showing the lack of transmission of a normal size allele from the affected parent to the affected child, as well as a repeat expansion pattern in both by TP-PCR. AC = affected child; AP = affected parent; UC = unaffected child; UP = unaffected parent. Genotypes are indicated on the right end of each panel. (B) Southern blot analysis of four unaffected (left) and four affected subjects (right).
(GGCCTG)n size distribution in ataxia, spastic paraplegia and healthy individuals
No instances of the NOP56 mutation were detected among 234 healthy Galician controls. Likewise, no SCA36 expansions were present in 214 Spanish patients with spastic paraplegia, a group of genetically heterogeneous degenerative motor neuron disorders. To characterize normal variation of the NOP56 intron 1 repeat, allele size distribution was analysed in the three samples studied (Fig. 3). The number of hexanucleotide repeats in normal alleles varied from 3 to 14, with the nine-repeat allele being the most frequent. The repeat size distribution and allele frequencies were similar in normal alleles of SCA, patients with spastic paraplegia and in healthy controls.
Figure 3.

(A) Allelic distribution of NOP56 intron 1 GGCCTG hexanucleotide repeat in patients with SCA, patients with spastic paraplegia (SPG) and control individuals. Sequence structures identified for normal size alleles are represented inside the boxes, the total number of alleles sequenced with each structure indicated in brackets. Insertions (INS) and deletions (DEL) before the repeat sequence indicate alleles with the upstream CGGGCG indel polymorphism insertion or deletion, respectively. (B) Schematic representation of NOP56 exons 1 and 2, and intron 1, indicating the relative positions of the GGCCTG repeat expansion mutation and the CGGGCG indel polymorphism (rs28970277).
Clinical characteristics of SCA36 in Costa da Morte families
A total of 144 subjects belonging to Families 1 and 2 were clinically examined. Once the SCA36 mutation was identified, eight additional patients with SCA carrying the same mutation were detected in our cohort of undiagnosed ataxia index cases, and 23 other members of these families were recruited for further clinical and genetic characterization in the context of the present research. The main clinical features of 44 affected people (20 males, 24 females, mean age at examination: 63.8 years) are summarized in Table 1. In brief, the typical picture is one of gait imbalance, starting from the late 40 s to the mid-50 s. A few patients reported initial symptoms as episodes of vertigo, dizziness or blurred vision. Almost all noticed progressive hearing loss at onset, some even before the balance problems. Difficulties with speech were noticed generally a few years after disease onset, although some patients reported simultaneous onset of both ataxia and dysarthria. Midline-gait ataxia was the most prominent and invariable feature, with limb ataxia generally of lesser degree and more pronounced in lower than upper limbs. Abnormal eye movements were common, mostly consisting of slow saccades, slow pursuit and fixation instability. Some patients also had vertical or horizontal gaze limitation, with certain quality of ocular motor apraxia and a mild eyelid ptosis in a few cases. The dysarthria had mostly cerebellar, but also a somewhat pseudobulbar quality. Horizontal and vertical nystagmus was only occasionally seen. Almost all patients showed involuntary worm-like tongue movements suggestive of fasciculations or myokymia, and some had mild tongue atrophy. Although with disease progression some patients had mild dysphagia, especially for liquids, swallowing was not affected in a degree comparable with other diseases affecting motor neurons such as amyotrophic lateral sclerosis. Furthermore, no fasciculations or significant muscle atrophy, indicative of otherwise typical lower motor neuron disease, was detected in other facial muscles or in other body segments. Mild pyramidal signs were common, generally limited to brisk tendon reflexes and occasional Babinski sign, although an increased muscle tone in the lower limbs was also present in some patients (Table 1). Most patients were cognitively intact, and in only two cases could we observe mild cognitive decline of frontal-subcortical characteristics.
Table 1.
Summary of clinical and genetic data of 44 affected patients with SCA36
| Family | Expansion (fragment in kb) | Expansion (repeat number) | Gender | Onset age (years) | Age at exam | Onset symptom | Midline ataxia | Apendicular ataxia | Eye movements | Hypoacusis | Dysarthria | Pyramidal signs | Tongue fasciculations | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | 8 | ∼830 | F | 65 | 74 | WI | ++ | +++ | S↓ | ++ | ++ | B | + | – |
| F1 | 9 | ∼1000 | F | 64 | 68 | WI | + | + | − | + | + | B, HR, mild HTl | + | – |
| F1 | NA | M | 51 | 65 | H | +++ | ++ | − | +++ | ++ | B | + | – | |
| F1 | 8 | ∼830 | F | 60 | 66 | WI | + | + | LVG | + | + | B, HR | + | Ptosis |
| F1 | 15 | ∼2000 | F | 55 | 64 | WI | ++ | ++ | − | + | + | HR | ++ | – |
| F1 | 10 | ∼1170 | F | 53 | 58 | WI | + | + | − | + | + | – | − | – |
| F1 | NA | M | 52 | 57 | WI | + | + | − | +++ | + | – | + | – | |
| F1 | 7 | ∼670 | F | 37 | 37 | WI | + | − | − | − | − | – | − | – |
| F1 | 10 | ∼1170 | F | 29 | 29 | WI/Di | + | + | S↓ | − | + | – | + | – |
| F1 | NA | F | 50 | 72 | WI | +++ | +++ | S↓ | − | ++ | B | − | – | |
| F1 | 7.8 | ∼800 | M | 60 | 83 | WI | +++ | ++ | S↓ | +++ | ++ | B, HR, HTl | + | – |
| F1 | 9 | ∼1000 | M | 48 | 58 | WI | ++++ | ++ | S↓↓, hN | ++ | +++ | B, HR, HTl | + | – |
| F1 | 8.5 | ∼920 | M | 48 | 52 | WI | + | + | S↓ | + | + | B | + | – |
| F1 | NA | F | 50 | 39 | WI | +++ | +++ | S↓ | +++ | +++ | B | + | – | |
| F1 | NA | M | 55 | 79 | WI | +++ | ++ | S↓ | +++ | +++ | B | + | – | |
| F1 | NA | M | NN | 58 | - | + | − | − | ++ | ++ | – | − | – | |
| F1 | NA | F | 44 | 50 | WI | + | + | S↓↓, LHG | + | + | HR | ++ | Ptosis | |
| F1 | 8.5 | ∼920 | F | 50 | 53 | WI | + | + | S↓, mild LVG | + | + | HR | ++ | Ptosis, diplopia |
| F1 | NA | M | 44 | 45 | WI | + | − | mild S↓ | − | −/+ | – | − | Mild head tremor | |
| F1 | 7.9 | ∼820 | F | 45 | 65 | WI | +++ | +++ | S↓, hN | + | +++ | B | + | – |
| F1 | 7 | ∼670 | M | 50 | 63 | WI | +++ | ++ | S↓, hN | + | +++ | – | − | – |
| F1 | 6.9 | ∼650 | F | 49 | 62 | WI | +++ | ++ | S↓↓, LVG, LHG | +++ | ++ | B, HR, HTl | ++ | Ptosis |
| F1 | NA | M | 55 | 59 | WI | + | + | − | − | + | B | − | – | |
| F1 | 7 | ∼670 | M | 60 | 84 | WI | ++ | ++ | S↓ | ++ | ++ | – | + | – |
| F1 | 9.2 | ∼1033 | M | NN | 58 | - | +/− | + | − | − | − | HR | − | – |
| F2 | 8.8 | ∼970 | F | 60 | 74 | WI | +++ | ++ | S↓ | − | ++ | – | − | – |
| F2 | NA | M | 60 | 76 | WI | +++ | +++ | S↓↓ | − | +++ | – | + | – | |
| F2 | 8 | ∼830 | F | 60 | 63 | WI | ++ | ++ | − | + | + | – | − | – |
| F2 | 8.5 | ∼920 | F | 55 | 86 | WI | ++++ | ++ | S↓, hN | + | ++ | – | + | – |
| F2 | 10 | ∼1170 | F | 53 | 68 | Da | +++ | ++ | S↓, dysmetric | ++ | +++ | HR,HTl, B | + | – |
| F2 | 8 | ∼830 | F | 50 | 80 | WI | +++ | + | S↓ | ++ | ++ | – | +/− | – |
| F2 | NA | M | 56 | 75 | WI, tinnitus | +++ | ++ | S↓↓,LVG, OMA | +++ | +++ | HTl, HTu, HR, B | ++ | Urinary and bowel incontinence | |
| F3 | NA | F | 62 | 65 | WI/Di | +++ | + | S↓ | ++ | ++ | – | + | – | |
| F4 | 18 | ∼2500 | M | 46 | 62 | WI | +++ | ++ | S↓, LVG, OMA | +++ | ++ | HTl, HR, B | + | Mild cognitive dysfunction |
| F5 | 15 | ∼2000 | M | 50 | 60 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| F6 | 15 | ∼2000 | M | 63 | 69 | WI | ++ | ++ | S↓, LVG | ++ | ++ | HR, mild HTl | Tongue tremor | Mild attentional deficit |
| F7 | 8 | ∼833 | M | 54 | 72 | WI | ++ | + | NA | + | ++ | NA | NA | – |
| F7 | NA | F | 50 | 75a | H | +++ | NA | NA | ++ | +++ | NA | NA | – | |
| F8 | NA | F | 50 | 61 | Dp | + | − | − | −/+ | −/+ | mild HR | − | – | |
| F8 | NA | F | 61 | 65 | WI | + | + | − | + | + | – | − | Hand tremor, migraine | |
| F9 | 15 | ∼2000 | F | 50 | 72 | WI + H | +++ | ++ | NA | ++ | + | HR, B | NA | Dysphagia for solids |
| F9 | NA | M | 62 | 78 | Di | ++ | + | NA | NA | + | NA | NA | Hip fracture | |
| F9 | NA | F | 45 | 48 | WI | + | −/+ | − | −/+ | − | NA | NA | Vertigo | |
| F10 | 10.5 | ∼1250 | M | 55 | 60 | WI | +++ | +++ | S↓, LVG | − | ++ | HR | − | Dysphagia, intention tremor, myoclonus of limbs and neck |
a Deceased.
Signs or symptoms: − = absent; +/− = minimal; + = mild; ++ = moderate; +++ = severe; B = Babinski sign; Da = dysarthria; Di = dizziness; Dp = dysphagia; H = hearing loss; hN = horizontal nystagmus; HR = hyperreflexia; HTl = hypertonia lower limbs; HTu = hypertonia upper limbs; LHG = limitation of horizontal gaze; LVG = limitation of vertical gaze; NA = data not available; NN = symptoms not noticed by patient; OMA = oculomotor apraxia; S↓ = slow saccades; WI = walk imbalance.
Brain MRI, available for nine patients of different ages, showed superior cerebellar vermis atrophy in the younger cases, diffuse cerebellar atrophy in patients some years into the disease course and a pattern of olivo-ponto-cerebellar atrophy without abnormal T2-weighted image MRI signal in the pons or medulla in older patients (Fig. 4). Cortical brain atrophy was present only in some patients of more advanced age. No white matter or other lesions were generally observed, with the exception of one patient who also had high blood pressure. Cell count, proteins and glucose in the CSF were normal in four patients in whom it was examined. Audiometry studies, carried out in six patients, showed moderate to severe bilateral sensorineural hearing loss, with over 40 dB drop beyond 2500 Hz. Brainstem evoked potentials in the same cases disclosed absent or severe decrease of waves I and II. Somatosensory evoked potentials performed in five patients (three males, two females, ages 60–80 years) invariably showed prolonged latency and low amplitudes upon stimulus of the lower limbs, while normal in the upper limbs. Electroneuromyography, performed in nine cases at ages 57–90 years, showed normal sensory and motor nerve parameters. Invariably, signs of denervation (fibrillations, positive waves) were limited to the tongue, without evidence of spontaneous muscle activity in other territories, thus suggesting affectation limited to the second motor neurons in the lowest portion of the brainstem.
Figure 4.

Brain T1-weighted MRI images from three patients with SCA36 of different ages. Saggital and axial images are shown from patients aged 52 (A and B), 65 (C and D) and 85 (E and F) years. The disease affects manly the superior vermis in younger patients, later also affecting other cerebellar areas and the brainstem.
Repeat instability and genotype–phenotype correlation
The size of the mutant allele differs often within nuclear families. Analysis of the available Southern blot results for the two largest kindreds showed that the expansion tends to increase upon transmission in successive generations. Mean allele size increased from 7.4 to 9.2 kb when comparing three successive generations. While the average allele size was of 7.9 kb in patients who received it from their mother, it was 9.4 kb when received from an affected father (Table 2). In three mother–child pairs where the mutant allele could be measured, there was an increase of 0.4 kb in one case, while a contraction of 5–8 kb was observed in the other two. On the other hand, the increase of allele size in seven available father–child transmissions was 1.6 kb on average, with one case of invariable allele size, but no case of allele contraction among these transmissions. In Family 1, the largest pedigree we studied, mean age of onset was 56.3 years in generation IV and 52.4 years in generation V. While observation of single instances suggested an earlier onset in carriers of larger alleles, this association was not statistically significant by Kaplan–Meier analysis (data not shown). Carriers of larger alleles also had a tendency to show more severe symptoms, especially in gait ataxia, and a mild impairment of executive function.
Table 2.
Comparison of expanded allele size by generation and gender of the transmitting parent in 27 expanded alleles from Families 1 and 2
| Generation | Allele size total | Paternally transmitted alleles | Maternally transmitted alleles |
|---|---|---|---|
| IV | 7.4 ± 0.4 (2) | 7.8 (1) | 7 (1) |
| V | 8.8 ± 1.1 (16) | 9.3 ± 1.3 (10) | 7.8 ± 0.6 (6) |
| VI | 9.2 ± 0.8 (9) | 9.5 ± 0.6 (6) | 8.4 ± 1.0 (3) |
| Global | 8.8 ± 1.1 (27) | 9.4 ± 1.1 (17) | 7.9 ± 0.8 (10) |
Number of alleles in each category are indicated in brackets. Allele sizes are expressed in kilobase as mean ± SD.
Estimation of mutation age of the founder repeat
All 63 subjects from the 10 kindreds with the NOP56 hexanucleotide expansion carried the same core haplotype, suggesting that a founder chromosome was responsible for the Galician cases. Table 3 shows the overlap between 10 distinct extended haplotypes identified in the study families. The age of the most recent common ancestor was estimated by ESTIAGE in 25 generations (95% confidence interval, 16–41). Given that from the deduced haplotypes some families seemed to be more closely related, we sequentially removed each of the haplotypes from the analysis, with estimates varying from 24 to 29 generations. Considering an intergenerational time interval of 25 years, this ancestor would have lived 625 years ago (95% confidence interval, 400–1025). Estimates of the mutation age using the DMLE+2.3 software using different demographic and disease prevalence parameters are shown in Fig. 5. The estimated mutation age ranged from 37 to 74 generations, the lower and upper limits for these estimates being 23 and 122 generations, respectively. Given a disease prevalence of 1:10 000 and a population growth rate of r = 0.07, the origin was calculated to have occurred 51 generations ago (95% credible set ranging from 31 to 89 generations). According to this, the founder effect would have been more likely ∼1275 (775–2225) years ago.
Table 3.
Representative haplotypes in patients from the ten SCA36 families of the present study
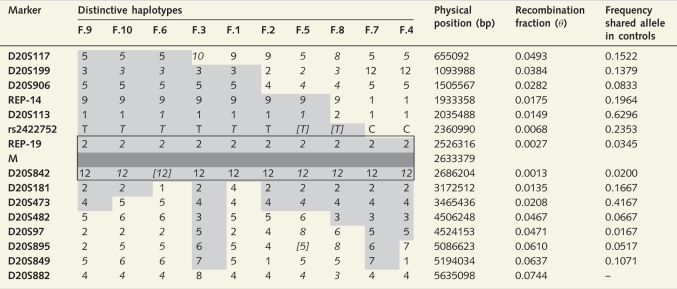 |
Framed haplotype indicates the common haplotype shared by all patients. Shaded haplotypes correspond to haplotypes shared by subsets of patients and part of a presumed ancestral haplotype. Allelic phase inferred by PHASE is shown in italics. In brackets are those alleles not genotyped, but inferred by PHASE. M = mutation. Physical position in base pairs according to GRCh37/hg19. Recombination fraction between each marker and the mutation was computed using Kosambi mapping function, considering a correspondence of 2.5 cM per Mb for the region studied. Frequency of the shared allele for each marker was estimated from 30 population-matched controls.
Figure 5.

Result from the DMLE analysis showing posterior probability density of the mutation age for different population growth rates (r = 0.05, r = 0.07) and different proportions of mutation bearing chromosomes sampled (f = 0.037, f = 0.074). The dashed lines correspond to the 95% credible set of values.
Discussion
We report here the presence of the NOP56 intronic GGCCTG hexanucleotide expansion in two large families from Costa da Morte (Arias et al., 2008) presenting with an SCA phenotype that we had previously linked to chromosome 20 p, and eight additional kindreds also from the Spanish region of Galicia. In a retrospective analysis of our total cohort of 160 SCA index cases, we observed that 10 (6.3%) had SCA36, 7 (4.4%) had an SCA2 mutation, 3 (1.9%) had SCA1, 3 (1.9%) SCA3 and 2 (1.3%) had SCA7. While SCA3 is accounted for as the most frequent SCA type worldwide (Silveira et al., 1996; Tang et al., 2000; Sequeiros et al., 2011), the frequency of SCA36 in Galicia exceeds that of any of the other SCA types. Further, > 80% of the Galician patients with SCA remain without a genetic diagnosis. Previous studies in Spanish patients showed a similar frequency of SCA2 and SCA3, each representing between 15% and 30% of all autosomal dominant ataxia (Pujana et al., 1999; Infante et al., 2005). If only families with definite dominant inheritance were considered, SCA36 would represent 21.3% of our series. One of the SCA36 index cases was a 69-year-old patient, whose parents had died at the age of 80 and 63 years, respectively, without having shown signs of the disease. Also, no additional affected individuals were recognized even after thorough family interview, suggesting that SCA36 should be considered also in the diagnosis of apparently sporadic ataxia cases. Two genes, PDYN and TGM6, within this same chromosomal region have been associated with SCA23 and SCA35, respectively (Bakalkin et al., 2010; Wang et al., 2010). We screened the coding region of these genes and did not detect mutations.
Our results confirm, in the Spanish population, the co-segregation of the GGCCTG hexanucleotide expansion in NOP56 with SCA, as well as the absence of expansions in normal individuals. Lack of co-segregation of the mutation with the disease and the presence in normal individuals in other SCAs, such as SCA31 (Ishikawa et al., 2011), challenge the interpretation of the pathogenic role of some expansions and strengthen the importance of our findings. We detected a total of 63 individuals with an SCA36 mutation, while over 400 people belong to any of the at risk family branches. These data have crucial implications for the establishment of diagnostic and follow-up protocols for the late-onset ataxias in the region of Galicia. The availability of a robust TP-PCR protocol, together with the presence of a characteristic associated haplotype, will facilitate the molecular diagnosis in this population. The question now remains whether SCA36 will turn out to be a frequent cause of ataxia in other regions of Spain and/or in Portugal, as well as in other Caucasian populations, especially those associated with traditional Spanish emigration.
From a clinical standpoint, the Spanish patients with SCA36 present with a late-onset spinocerebellar syndrome, with truncal ataxia as the foremost manifestation. Disease progression was slow, most of the patients remaining ambulant for 15–20 years after onset and longer. Appendicular ataxia, manifesting as dysmetria, uncoordinated alternating movements and overshoot, was less prominent than gait ataxia. A diverse combination and severity of eye movement abnormalities were also present in the patients. While no hearing impairment was reported in the Japanese SCA36 families (Kobayashi et al., 2011), a characteristic pattern of sensorineural deafness was detected in all Spanish patients examined, with audiometry showing a moderate to severe loss of ≥40 dB over 2500 Hz. Although hearing loss may be present in several ataxia types (Ikeda et al., 2011), the consistency of this feature in our patients suggests it may be a distinctive clinical aspect of the disease, at least in Spain, and should point to the possibility of SCA36 when present in ataxia cases. Similar to the cases reported by Kobayashi and collaborators (2011), numerous Galician patients with SCA36 had tongue fasciculations. Although these signs of denervation were more prominent as the disease progressed, functional consequences of lower motor disease on swallowing and speech were not significant. Our patients had relatively well preserved swallowing throughout the disease course, i.e. dysphagia was mild and not different from what is generally seen in other SCAs. Kobayahi et al. (2011) also noted the lack of significant dysphagia in their patients, in spite of the tongue fasciculations and atrophy. Also, further clinical or neurophysiological signs of lower motor neuron involvement were not evident in our cases. Mild upper motor neuron signs were generally present. Action myoclonus and myokymia in the facial muscles can be observed in some forms of olivopontocerebellar atrophy and in these cases enhanced long-latency facial reflexes can sometimes be demonstrated (Valls-Solé et al., 1994). We did not notice myokymia, twitching or other abnormal movements in perioral or other craniofacial muscles in the SCA36 cases, and only one patient had myoclonic jerks of the neck and trunk muscles (Table 1). Electrophysiological study of brainstem function will be useful to further detail the characterization of the SCA36 phenotype in the future. No significant sensory alteration was detectable upon clinical examination of most of the patients. Only very mild hypopallesthesia was present in one of the oldest individuals examined and was considered unspecific. Sensory nerve conduction studies of the sural nerve were normal, whereas lower limb somatosensory evoked potentials were abnormal in all individuals in whom they were examined, demonstrating affectation of the central sensory tracts. This may suggest that SCA36 could be another central distal axonopathy involving the sensory system (Thomas et al., 1984). Further somatosensory evoked potentials of additional patients, as well as electrophysiological evaluation of the pyramidal tracts will be needed to demonstrate this hypothesis. Cognition was normal in most patients, only two showing a mild impairment, with a frontal-subcortical pattern, and a mild degree of emotional lability.
A common characteristic in many neurodegenerative disorders associated with genomic expansions is clinical anticipation. Kobayashi et al. (2011) found no evidence of anticipation in 17 patients from nine families with a mean onset age of 52.8 years. We observed a tendency of the expanded SCA36 alleles to increase in size upon transmission (Table 2), more frequently in paternal transmission, as occurs in SCA10 (Matsuura et al., 2004), as well as in (CAG)n expansions, where a slight maternal contraction bias as opposed to a paternal expansion bias of the mutant has been reported (Aziz et al., 2011). There were several instances of an earlier onset and worse symptoms when compared with their affected parent, and the mean onset age was slightly lower in the fifth generation (52.4 years) than in the fourth generation (56.3 years) in Family 1, suggesting a tendency towards clinical anticipation. A significant correlation between allele size and onset age was not derived, however, from our data. These data should be taken cautiously, since both allele size and accurate age of disease onset are difficult to establish, as is usually the case with most late-onset neurodegenerative diseases where ascertainment biases are also likely. Therefore, more extensive and detailed studies are needed to fully address this question.
Patients with SCA36 show signs of both upper and lower motor neuron dysfunction. Furthermore, SCA and spastic paraplegia are two groups of neurodegenerative disorders with a significant degree of clinical and genetic overlap. Thus, we looked for expansions in 214 patients with pure or complicated forms of spastic paraplegia. No GGCCTG repeat expansions were seen in this group or in 234 healthy Galician controls. This is consistent with the report by Kobayashi et al. (2011) that no SCA36 mutations are found in patients with amyotrophic lateral sclerosis or dementia. Together, these results suggest that the pathogenic effects of the large expansions in NOP56 may be more specific on cerebellar function. Interestingly, a hexanucleotide repeat expansion in the first intron of C9ORF72 was recently reported as the most common genetic cause of frontotemporal dementia and amyotrophic lateral sclerosis, another neurological disorder with degeneration of motor neurons (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
The size of expanded alleles in the Spanish families ranged from ∼650 to 2500 repeats, a wider range than reported by Kobayashi et al. (2011) for the Japanese SCA36 expansions (1700–2300 repeats). On the other hand, the normal GGCCTG repeat size ranged from three to eight in 300 Japanese controls, while the normal size range in our population was also slightly broader (3–14), the nine-repeat allele being the most abundant. However, because it is not clear whether the primers used by Kobayashi et al. (2011) for amplification of the repeat also include the nearby indel, ranges of allele size may not be comparable. We then characterized the structure of non-expanded alleles by sequencing, and showed that non-expanded alleles in our populations consist of diverse combinations of an imperfect repeat (RGCCTG)-(GGCCTG)n-(CGCCTG)n, with or without a CGGGCG deletion 44 bp upstream of it (Fig. 3).
The SCA36 mutation lies within a common haplotype in all our patients, suggesting a founder origin, which is further supported by the fact that the largest kindreds (Families 1 and 2) originated from an isolated coastal area. We have traced back the origin of the Galician SCA36 expansion to ∼51 generations (∼1275 years ago), although these estimates partly depend on parameters that are difficult to determine, such as population growth rate and proportion of disease-bearing chromosomes ascertained in the study (Fig. 5). The results obtained with ESTIAGE and DMLE are consistent between them and with the fact that the age of the most recent common ancestor has to be equal to or less than the time in history at which the mutation arose (Rannala and Bertorelle, 2001). The nine SCA36 families reported in Eastern Japan also shared a common haplotype, which is not directly comparable with Galician haplotypes with the marker information available (Kobayashi et al., 2011). The haplotype shared by the Japanese patients is larger (1.8 Mb) than the Galician SCA36 haplotype (0.8 Mb), suggesting the former might have a more recent origin. It will be interesting to investigate whether the mutation found in Japanese patients and that in Spain have a common origin, such as is the case for the SCA3/Machado–Joseph disease CAG expansion, where a common original chromosome has been found in most families worldwide (Gaspar et al., 2001). Founder mutations have also been described for other SCAs and populations (Verbeek et al., 2004; Sequeiros et al., 2011). The Asian origin for an ancestral Machado–Joseph disease/SCA3 mutation has been shown (Martins et al., 2007); Portuguese dentatorubropallidoluysian atrophy families also share a common ancestral haplotype with Japanese patients (Martins et al., 2003). A similar relationship with Japan could apply to Galicia. Nevertheless, independent mutational events for the Galician and Japanese SCA36 expansions are also possible. It should also be emphasized that our region has traditionally been associated with emigration towards Latin America, especially during the 19th and 20th centuries. In fact, many family members of the kindreds in this study had reportedly emigrated to Argentina, Uruguay, Chile and Mexico. Thus, a proportion of SCA36 among ataxia cases in Central and South America is expected.
Many different mutation types have been identified in SCAs (www.scabase.eu). In addition to coding CAG repeats, non-coding triplet-repeat expansions, as well as larger intronic expansions and point mutations have been linked to the dominant ataxias (Schöls et al., 2004; Sequeiros et al., 2010, 2011). Large repeat expansions are thought to cause disease through haploinsufficiency of the repeat-harbouring gene, or through the generation of RNA species toxic for the cellular machinery. The NOP56 gene encodes a 56-kDa protein, which interacts with NOP1 and NOP58 to form the 60S ribosomal subunit. Alternatively spliced NOP56 isoforms have not been fully characterized. The GGCCTG repeat expansions in SCA36 lead to the formation of intranuclear RNA foci in patient’s cells, possibly disrupting the normal function of factors involved in transcription and splicing (Kobayashi et al., 2011). The presence of RNA foci has also been reported in other SCAs (Daughters et al., 2009; Sato et al., 2009), in the myotonic dystrophies, Huntington disease-like 2 (Rudnicki et al., 2007) and more recently a toxic effect on RNA metabolism has also been proposed for dementia–amyotrophic lateral sclerosis (Renton et al., 2011), further supporting a wider role of RNA-mediated mechanisms in neurodegenerative disorders, including those affecting cerebellar, motor neuron and cognitive functions.
The RNA-mediated mechanism of SCA36 may not be limited to a toxic gain of function. The NOP56 expanded repeat lies next to the microRNA gene MIR1292, which is downregulated in patient cells, thus suggesting that a defective regulatory function of mir1292 might be critical for SCA36 pathogenesis (Kobayashi et al., 2011). A combination of toxic gain of function and loss of function mechanisms are being hypothesized for several neurodegenerative ataxias (Zoghbi and Orr, 2009; Seixas et al., 2011). It is interesting that TMC2 is a gene located near NOP56. TMC1 causes dominant and recessive forms of hearing loss (Kurima et al., 2002). It could be hypothesized that the sensorineural hearing loss, almost constant in Galician patients with SCA36, may be due to dysfunction of TMC2.
In summary, our data add up to the increasing number of neurodegenerative syndromes associated with very large, non-coding repeat expansions and possible RNA-mediated pathogenesis. We show that SCA36 is not restricted to the eastern coast of Japan, but is also present in a European population. Furthermore, it appears to be the most frequent type of SCA in the Galician region of Northwest Spain. This description of 44 patients in 10 families contributes to the SCA36 clinical picture, consisting of a late-onset and slowly progressive, predominantly midline ataxia, with diverse eye movement abnormalities, sensorineural hearing loss and mild motor neuron signs, mainly in medullar territories. We dated the founder mutation responsible for this geographic cluster to ∼1275 years ago, indicating that the disease might have worldwide spread during the last centuries.
Funding
This work was supported by projects from the following agencies: Conselleria de Sanidade, Xunta de Galicia (PGIDIT05SAN26PR); Xunta de Galicia, INCITE plan (10PXIB9101280PR); Instituto de Salud Carlos III (CP05/00251); FCT grant (PTDC/SAU-GMG/098305/2008, Portugal, to I.S.); Miguel Servet contract (Instituto de Salud Carlos III, to M.J.S.); Lucas Labrada program, Consellería de Innovación, Xunta de Galicia (to M.G.-M.); ISCIII-SERGAS (to B.Q.) and postdoctoral scholarship from FCT grant (PTDC/SAU-GMG/098305/2008, to A.I.S.).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
The authors are indebted to the participating patients and their families, as well as to the Asociación Galega de Ataxias (AGA). We thank Drs M. Rossi (ENT), J. Otero-Costas (Neurophysiology) and F. Vázquez (Radiology) for their help with workup studies. Genotyping was carried out at the Santiago de Compostela node of the Spanish National Genotyping Center (CEGEN-ISCIII).
Glossary
Abbreviations
- SCA
spinocerebellar ataxia
- SNP
single nucleotide polymorphism
- TP-PCR
triplet repeat primed polymerase chain reaction
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Arias M, Arias-Rivas S, Blanco-Arias P, Dapena D, Vázquez F, Rossi M, et al. [SCA from the Costa da Morte: ‘A new SCA’. Description of the phenotype] Neurologia. 2008;23:628–9. [Google Scholar]
- Aziz NA, van Belzen MJ, Coops ID, Belfroid RD, Roos RA. Parent-of-origin differences of mutant HTT CAG repeat instability in Huntington’s disease. Eur J Med Genet. 2011;54:e413–e418. doi: 10.1016/j.ejmg.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Bakalkin G, Watanabe H, Jezierska J, Depoorter C, Verschuuren-Bemelmans C, Bazov I, et al. Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. Am J Hum Genet. 2010;87:593–603. doi: 10.1016/j.ajhg.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, et al. RNA gainof-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–62. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar C, Lopes-Cendes I, Hayes S, Goto J, Arvidsson K, Dias A, et al. Ancestral origins of the Machado-Joseph disease mutation: a worldwide haplotype study. Am J Hum Genet. 2001;68:523–8. doi: 10.1086/318184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin E, Tullio-Pelet A, Begeot F, Lyonnet S, Abel L. Estimating the age of rare disease mutations: the example of Triple-A syndrome. J Med Genet. 2004;41:445–9. doi: 10.1136/jmg.2003.017962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Nagai M, Kurata T, Yamashita T, Ohta Y, Nagotani S, et al. Comparisons of acoustic function in SCA31 and other forms of ataxias. Neurol Res. 2011;33:427–32. doi: 10.1179/1743132810Y.0000000011. [DOI] [PubMed] [Google Scholar]
- Infante J, Combarros O, Volpini V, Corral J, Llorca J, Berciano J. Autosomal dominant cerebellar ataxias in Spain: molecular and clinical correlations, prevalence estimation and survival analysis. Acta Neurol Scand. 2005;111:391–9. doi: 10.1111/j.1600-0404.2005.00400.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Dürr A, Klopstock T, Müller S, De Toffol B, Vidailhet M, et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology. 2011;77:1853–5. doi: 10.1212/WNL.0b013e3182377e3a. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of Intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–30. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2011;40:1–16. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30:277–84. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–7. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- Martins S, Calafell F, Gaspar C, Wong VC, Silveira I, Nicholson GA, et al. Asian origin for the worldwide-spread mutational event in Machado-Joseph disease. Arch Neurol. 2007;64:1502–8. doi: 10.1001/archneur.64.10.1502. [DOI] [PubMed] [Google Scholar]
- Martins S, Matamá T, Guimarães L, Vale J, Guimarães J, Ramos L, et al. Portuguese families with dentatorubropallidoluysian atrophy (DRPLA) share a common haplotype of Asian origin. Eur J Hum Genet. 2003;11:808–11. doi: 10.1038/sj.ejhg.5201054. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Fang P, Lin X, Khajavi M, Tsuji K, Rasmussen A, et al. Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am J Hum Genet. 2004;74:1216–24. doi: 10.1086/421526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nature Genet. 2000;26:191–4. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- Pujana MA, Corral J, Gratacòs M, Combarros O, Berciano J, Genís D, et al. Spinocerebellar ataxias in Spanish patients: genetic analysis of familial and sporadic cases. The Ataxia Study Group. Hum Genet. 1999;104:516–22. doi: 10.1007/s004390050997. [DOI] [PubMed] [Google Scholar]
- Rannala B, Bertorelle G. Using linked markers to infer the age of a mutation. Hum Mutat. 2001;18:87–100. doi: 10.1002/humu.1158. [DOI] [PubMed] [Google Scholar]
- Rannala B, Reeve JP. Joint Bayesian estimation of mutation location and age using linkage disequilibrium. Pac Symp Biocomput. 2003;8:526–34. doi: 10.1142/9789812776303_0049. [DOI] [PubMed] [Google Scholar]
- Reeve JP, Rannala B. DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics. 2002;18:894–95. doi: 10.1093/bioinformatics/18.6.894. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol. 2007;61:272–82. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–57. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, Pruitt JL, et al. Loss of junctophilin-3 contributes to Huntington’s disease-like 2 pathogenesis. Ann Neurol. 2011;71:245–57. doi: 10.1002/ana.22598. [DOI] [PubMed] [Google Scholar]
- Sequeiros J, Martins S, Silveira I. Epidemiology and population genetics of degenerative ataxias. In: Subramony SH, Dürr A, editors. Ataxic disorders. Handbook of clinical neurology. Vol. 103. 3rd series, Edinburgh: Elsevier; 2011. pp. 225–48. [DOI] [PubMed] [Google Scholar]
- Sequeiros J, Seneca S, Martindale J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur J Hum Genet. 2010;18:1188–95. doi: 10.1038/ejhg.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira I, Lopes-Cendes I, Kish S, Maciel P, Gaspar C, Coutinho P, et al. Frequency of spinocerebellar ataxia type 1, dentatorubropallidoluysian atrophy and Machado-Joseph disease mutations in a large group of spinocerebellar ataxia patients. Neurology. 1996;46:214–8. doi: 10.1212/wnl.46.1.214. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. The Staden sequence analysis package. Mol Biotechnol. 1996;5:233–41. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–9. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Liu C, Shen L, Dai H, Pan Q, Jing L, et al. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol. 2000;57:540–4. doi: 10.1001/archneur.57.4.540. [DOI] [PubMed] [Google Scholar]
- Thomas PK, Schaumburg HH, Spencer PS, Kaeser HE, Pallis CA, Rose FC, et al. Central distal axonopathy syndromes: newly recognized models of naturally occurring human degenerative disease. Ann Neurol. 1984;15:313–5. doi: 10.1002/ana.410150402. [DOI] [PubMed] [Google Scholar]
- Valls-Solé, Lou JS, Hallett M. Brainstem reflexes in patients with olivopontocerebellar atrophy. Muscle Nerve. 1994;17:1439–48. doi: 10.1002/mus.880171213. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek DS, Piersma SJ, Hennekam EF, Ippel EF, Pearson PL, Sinke RJ. Haplotype study in Dutch SCA3 and SCA6 families: evidence for common founder mutations. Eur J Hum Genet. 2004;12:441–6. doi: 10.1038/sj.ejhg.5201167. [DOI] [PubMed] [Google Scholar]
- Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–8. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, et al. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–6. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi YH, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia Type 1. J Biol Chem. 2009;284:7425–9. doi: 10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.