

Abstract
Assays for identification and quantification of host-cell proteins (HCPs) in biotherapeutic proteins over 5 orders of magnitude in concentration are presented. The HCP assays consist of two types: HCP identification using comprehensive online two-dimensional liquid chromatography coupled with high resolution mass spectrometry (2D-LC/MS), followed by high-throughput HCP quantification by liquid chromatography, multiple reaction monitoring (LC-MRM). The former is described as a “discovery” assay, the latter as a “monitoring” assay. Purified biotherapeutic proteins (e.g., monoclonal antibodies) were digested with trypsin after reduction and alkylation, and the digests were fractionated using reversed-phase (RP) chromatography at high pH (pH 10) by a step gradient in the first dimension, followed by a high-resolution separation at low pH (pH 2.5) in the second dimension. As peptides eluted from the second dimension, a quadrupole time-of-flight mass spectrometer was used to detect the peptides and their fragments simultaneously by alternating the collision cell energy between a low and an elevated energy (MSE methodology). The MSE data was used to identify and quantify the proteins in the mixture using a proven label-free quantification technique (“Hi3” method). The same data set was mined to subsequently develop target peptides and transitions for monitoring the concentration of selected HCPs on a triple quadrupole mass spectrometer in a high-throughput manner (20 min LC-MRM analysis). This analytical methodology was applied to the identification and quantification of low-abundance HCPs in six samples of PTG1, a recombinant chimeric anti-phosphotyrosine monoclonal antibody (mAb). Thirty three HCPs were identified in total from the PTG1 samples among which 21 HCP isoforms were selected for MRM monitoring. The absolute quantification of three selected HCPs was undertaken on two different LC-MRM platforms after spiking isotopically labeled peptides in the samples. Finally, the MRM quantitation results were compared with TOF-based quantification based on the Hi3 peptides, and the TOF and MRM data sets correlated reasonably well. The results show that the assays provide detailed valuable information to understand the relative contributions of purification schemes to the nature and concentrations of HCP impurities in biopharmaceutical samples, and the assays can be used as generic methods for HCP analysis in the biopharmaceutical industry.
Key words: host cell proteins, protein quantification, biotherapeutic proteins, mAbs, HCP
Introduction
A major portion of biopharmaceuticals today are produced by recombinant DNA-technology using a well-selected host cell system. Host cells express a large number of their own proteins that can easily contaminate the recombinant protein drug. Even after sophisticated purifications steps, low levels (1–100 ppm) of host cell proteins (HCPs) may still remain in the final purified biopharmaceutical. Because HCPs can sometimes elicit an immunogenic response, regulatory guidelines mandate that they are identified and quantified in order to protect patient safety.1
The presence of HCPs in protein drugs may determine whether the biopharmaceutical is accepted or not by the regulatory agencies. For example, in 2008 the European Medicines Agency approved a recombinant form of human somatropin only after the manufacturer added additional purification steps for removal of the HCPs responsible for immunogenic response in patients.2 The same agency rejected an interferon biosimilar in 2006 because of insufficient validation for immunogenicity testing.3 As the patents for the first generation of approved biopharmaceuticals have either expired or are about to expire, giving way to emerging biosimilar therapeutics, more biopharmaceutical companies have become interested in HCPs analysis in recent years.4
All analytical methods for measuring HCPs face significant challenges due to the wide dynamic range of the protein concentration (4 to 5 orders of magnitude). Analytical methods used for measuring HCP have not drastically changed over the past 10–15 y.5–7 The most popular methods are process-specific enzyme linked immunosorbent assays (ELISAs) and protein gel blots.8–11 Both methods require prior knowledge regarding the nature of HCP contaminants. In addition, process specific immunoassays8–11 are time consuming (e.g., 6 mo) and expensive to develop (>$100K), and may produce misleading results due to antibody cross-reactivity.9 ELISA assays are not easily adapted to fully evaluate biopharmaceutical products from different cell types and purification schemes. The generic ELISA kits that are commercially available for monitoring HCPs are less specific than the process specific immunoassays, and do not offer complete coverage for all the HCPs present in a sample. Other techniques such as two-dimensional gel electrophoresis coupled to fluorescent staining12,13 is only semi-quantitative, has a limited dynamic range (3 to 4 orders of magnitude), and requires additional techniques (e.g., mass spectrometry) for HCP identification.
As a universal detection technique, mass spectrometry (MS) has some advantages over the current HCP methods to identify all possible existing HCPs in a biopharmaceutical sample. When coupled to chromatographic separations, MS is routinely used for characterization of therapeutic proteins14–17 and in proteomics and biomarker discovery arenas for identification of low-abundance proteins across a dynamic range of 3–4 orders of magnitude in a variety of samples.18,19 The combination of MS with two-dimensional gel electrophoresis has recently been applied for the identification of host-cell proteins co-purified with recombinant monoclonal antibodies20 and with apolipoprotein A–I.21 In addition, two recent reports describe the use of SELDI-TOF MS22,23 and SDS-PAGE followed by 1D LC/MS/MS23 for screening22 and identification of HCPs23 from purified recombinant proteins. However, there are no known literature reports demonstrating the utility of LC/MS for comprehensive HCP analysis in biopharmaceuticals.
One dimensional chromatographic separation typically has a limited dynamic range of about 3 orders of magnitude.24 Multidimensional chromatographic separations, however, permit the first dimension column to be overloaded, allowing the overall dynamic range of the analysis to be increased to reach the concentration range of 1–100 ppm for the detection of HCPs. For peptide separations, widely practiced two dimensional chromatography (2D-LC) separation schemes are based on the coupling of strong cation exchange (SCX) chromatography in the first dimension with low-pH reversed-phase (RP) 25 separation in the second dimension. In recent years, a different 2D-LC methodology has been developed, based on the RP separation under basic pH (pH 10) conditions in the first dimension and a low-pH RP separation in the second dimension.26–39 In all reported work using capillary chromatography (e.g., 300 µm ID columns),26–36 the first LC dimension was operated in an off-line mode using fraction collection, followed by excess organic solvent evaporation or sample concentration so that the samples could be re-injected onto the 2nd dimension RP column. In comparison with the off-line fractionation approach, the online (automated) fractionation approach recently reported for nanoscale separations37 appears to be a better alternative because it has the benefits of higher throughput, less sample loss and better reproducibility. Recently, online 2D-LC configurations using high-pH RP/low-pH RP (or vice versa) for peptide fractionation on an analytical scale columns (2.1 or 4.6 mm ID) have also been reported in references 38 and 39. While these reports confirm the advantages of online comprehensive multidimensional chromatography, their utility for the analysis of complex protein samples is limited due to the larger amount of sample required for LC/MS analysis.
In this work, we introduce an online two dimensional capillary scale LC setup (0.3 mm ID) for comprehensive peptide separations using high-pH RP/low-pH RP. We evaluated the chromatography and MS performance of this setup for analysis of complex peptide mixtures (protein digests), and applied this method to the analysis of host-cell protein impurities in monoclonal antibody (mAb) preparations. Subsequently, a fast, quantitative LC-MRM assay was developed for a flexible, rapid, high-throughput monitoring and for the quantification of HCPs from various mAb samples derived from different cell lines and purification protocols. The final objective of the current study was to develop a generic method for the identification and quantification of HCPs in any biopharmaceutical.
Results
Chromatographic performance of the high-pH/low-pH 2D-LC system for peptide separation.
Figure 1 shows the fluidic configuration of the online setup for 2D-LC fractionation of peptide samples. During the sample loading step (Fig. 1A), peptide samples are loaded and retained on a reversed-phase XBridge column designed to withstand high pH mobile-phase conditions. After the sample loading step (15 min), the first fraction of peptides is eluted (Fig. 1B) from the XBridge column by slightly increasing the ACN percentage of the Eluent B (pure ACN) of the first chromatographic dimension. The elution step takes 15 min and the eluent (containing peptides) is automatically diluted by 10-fold with 0.1% TFA (pH 2.1) aqueous solution. This online dilution reduces the organic solvent percentage in the eluent and decreases the pH of the mobile phase to about 2.2, such that all the peptides eluted from the first dimension can be effectively retained again on the trap column. At the end of the trapping step as shown in Figure 1C, peptides retained on the trap column are back-flushed onto the analytical column where a high-resolution RP separation is undertaken at pH 2.3. The low-pH high-resolution separation is performed at relatively high temperature (65°C) to shorten the analysis time in the second dimension and to improve the chromatographic peak shapes of proline/proline containing peptides.40,41 Although the separation at low-pH was performed under sub-optimal conditions (12 µL/min flow rate and 65°C column temperature), the data provided in Figure S1 demonstrates that the average peak capacity (PC ∼ 160) decreased only about 15% in comparison with the values from the normal separation conditions (4 µl/min flow rate and 35°C column temperature).
Figure 1.

Fluidic configuration of the 2-dimensional chromatography with online dilution: (A) sample loading; (B) peptide fractionation using the first chromatographic dimension (high pH reversed-phase), and peptide trapping; (C) peptide separation in the second dimension (low pH reversed-phase).
A critical chromatographic parameter in multidimensional chromatographic separations is the reproducibility of peptide fractionation during an extended period of operation. This is illustrated by the extracted mass chromatograms of the T49/50 peptide from BSA (DAIPENLPPLTADFAEDKDVCK, monoisotopic mass [MH3]3+: 820.06, Fig. 2A) and the T43 peptide from ENL protein (VNQIGTLSESIK, monoisotopic mass of [MH2]2+: 644.86, Fig. 2B) from the second dimension (low-pH) separations in four consecutive injections (experiments). In each experiment, a 5-step fractionation using 10.8, 12.4, 15.4, 18.6 and 50% Eluent B (100% ACN) was performed in the first dimension. T49/50 peptide from BSA was detected only in Fraction 3 and T43 ENL peptide was eluted only in Fraction 4, demonstrating excellent reproducibility of the first dimension fractionation. The retention time reproducibility (around 0.1% RSD) for the same peptides over 48 h of separation additionally confirms the stability of the 2D-LC system.
Figure 2.

Reproducibility of the 2D-LC setup for four consecutive experiments: (A) extracted mass chromatograms of T49/50 peptide from BSA (DAIPENLPPLTADFAEDKDV(C)K, [MH3]3+ = 820.06) which eluted only in Fraction 3 of 5 step elutions (with 15.4% ACN); (B) extracted mass chromatograms of T43 peptide from ENL (VNQIGTLSESIK, [MH2]2+ = 644.86) eluted only in Fraction 4 out of 5 (using 18.6% ACN). All mass chromatograms were generated using an extraction mass window of 0.1 Da around the corresponding monoisotopic peaks. Second dimension chromatography runs were performed at 12 µL/min using a 30 min gradient (7–35% ACN, 0.1% FA). The amounts of digests loaded on column were 20 fmoles of ENL and 100 fmoles of BSA.
The ability of the 2D-LC system to maintain good chromatographic performance in the 2nd dimension separation, independent of the number of fractionation steps in the first dimension, was investigated in the subsequent experiment. Figure 3 displays the extracted mass chromatograms of the ENL T43 peptide generated from the second dimension separations under four fractionation schemes: “simulated 1D” (a single one-step elution from 10.8 to 50% of Eluent B), 3-step, 5-step and 10-step fractionations, respectively. The retention time for the T43 peptide over the different operation schemes was highly reproducible (0.15% RSD). The 10-step fractionation experiment revealed an important finding: a peptide can elute completely in a single fraction (fraction 5 of 10) even though a relatively narrow step-gradient (1.9% B) was employed. As shown in Figure 4A, no T43 peptide could be detected in the previous or following fractions. The same performance was seen for the T26 peptide from ADH (Fig. 4B) despite a 100-fold higher loading of ADH digest on the column. The data indicates that high-pH fractionation has greatly improved the performance of peptide separation and overcame one of the major limitations often observed in SCX fractionation where the same peptide can be split into multiple fractions.
Figure 3.

Chromatographic performance (e.g., RT reproducibility and peak width) is maintained during 1st dimensional fractionation: mass chromatograms of ENL T43 peptide obtained under four fractionation conditions: (A) “simulated” 1D run using a single elution step (from 10.8 to 50% ACN); (B) fraction 2 out of 3 (from 10.8 to 18.6% ACN); (C) fraction 4 out of 5 (from 15.4 to 18.6% ACN); (D) fraction 5 out of 10 (from 15.4 to 16.7% ACN). The data was acquired in continuum mode and all mass chromatograms used an extraction window of 0.1 Da around the corresponding monoisotopic peak. All separations used a 30 min gradient (7–35% ACN, 0.1% FA).
Figure 4.

Performance of high-pH peptide fractionation: (A) mass chromatograms of ENL T43 peptide recorded for 3 consecutive fractions during a 10-step 2D fractionation experiment: fraction 4/10 corresponds to a step elution from 14.0 to 15.4% ACN; fraction 5/10 was eluted from 15.4 to 16.7% ACN; fraction 6/10 (16.7 to 18.6% ACN). (B) mass chromatograms of ADH T26 peptide (EALDFFAR, [MH]1+ = 968.48) recorded for 3 consecutive fractions during a 10-step 2D fractionation experiment: fraction 5/10 corresponds to a step elution from 15.4 to 16.7% ACN; fraction 6/10 was eluted from 16.7 to 18.6% ACN; fraction 7/10 (18.6 to 20.4% ACN). The data was acquired in continuum mode and all mass chromatograms used an extraction window of 0.1 Da around the corresponding monoisotopic peaks. All separations were performed using a 30 min gradient (7–35% ACN, 0.1% FA). The amount loaded on-column was 20 fmoles ENL and 2 picomoles ADH digest.
The utility of multiple fractionations for the identification of low-abundance peptides from a complex mixture was further investigated using Sample II (See experimental description). The presence of the E. coli lysate digest in a MIX-4 samples creates a useful biological background for probing the role of fractionation in differentiating specific peptide signals. The four fractionation schemes discussed above (comprising 1, 3, 5 and 10-step fractionation in the first dimension) were undertaken, and the ESI-MS signal produced by the ENL T43 peptide was used as a typical indicator to demonstrate the role of multiple-step fractionation and the performance of the 2D-LC system. The ESI-MS spectra of ENL T43 peptide, summed across the entire chromatographic peak width (∼10 sec wide peak), are shown in Figure 5A–E. As shown in Figure 5A and B, under no or with few fractionations (a single step or a 3-step fractionation in the first dimension), the monoisotopic peak of the spiked T43 peptide (m/z: 644.86) is completely buried under the isotopic distributions of other more intense co-eluting peptides and cannot be reliably distinguished. However, the monoistopic peak of the T43 peptide starts to emerge from the peptide background with the 5-step fractionation (Fig. 5C), and is clearly resolved from the background signals with 10-step fractionation (Fig. 5D). Interestingly, with the 10-step 2D-LC separation, the ion counts for the summed ESI-MS signal intensity of T43 peptide in the presence of E. coli digest background is about 85% of the summed ESI-MS intensity of the same peptide from a purified sample containing just MIX-4 proteins (Fig. 5E).
Figure 5.

ESI-MS spectra of ENL T43 peptide in a complex peptide background produced by spiking the ENL digest in an E. coli lysate digest. Each spectrum is composed of 10 combined scans across the entire chromatographic peak-width of T43: (A) “simulated” 1D run using a single elution step (from 10.8 to 50% ACN); (B) fraction 2/3 (from 10.8 to 18.6% ACN); (C) fraction 4/5 (from 15.4 to 18.6% ACN); (D) fraction 6/10 (from 16.7 to 18.6% ACN); (E) fraction 6/10 from the ENL digest (no E. coli digest, same 2D fractionation protocol). All separations employed a 30 min gradient (7–35% ACN, 0.1% FA). The amount of ENL digest loaded on column was 20 fmoles for all experiments.
Identification and quantification of HCPs from a monoclonal antibody.
Six PTG1 mAb samples were analyzed using the 2D-LC/MSE method to identify the HCPs present in each sample, and to understand the contribution of different cell lines and purification schemes to the final HCP composition in biopharmaceuticals. Tryptic digests from each of the PTG1 samples were fractionated using only the 10-step fractionation protocol, and MSE data were collected for each fraction during the second dimension separations. The 2D-LC/MSE experiments were performed in triplicate for each PTG1 sample.
Table 1 lists 33 HCPs that were identified in total from the six PTG1 samples. Detailed information regarding the sample and HCP composition is summarized in Table S3. Each of the HCPs in Table 1 was identified in at least 2 of 3 replicates. In addition, four of the five spiked proteins (LA, PHO, ADH and BSA) were identified in all mAb digests. ENL, which has the lowest concentration among all the spiked proteins, was not identified in any PTG1 sample, but was identified in Sample I.
Table 1.
HCPs identified across six PTG1 preparations.
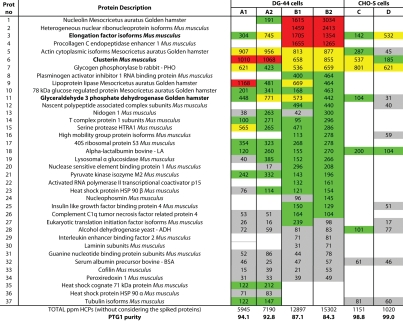 |
Host-cell proteins (HCPs) identified in six PTG1 preparations and their corresponding concentrations (ppm, expressed in ng HCP/mg total protein) The protein concentrations were calculated using top three best responding peptides in ESI-MS [38]. All samples were purified on a ProSep-vA column (EMD Millipore). Samples A1/A2 and B1/B2 were biological replicates, grown in a DG-44 CHO cell line. Samples C/D were expressed in a CHO-S cell line. The MRM assays were developed for three proteins highlighted in bold.
It has been reported that the concentration of a protein in a complex mixture can be estimated using the added ESI-MS signal of the top three best responding peptide precursors42,43 (the so-called Hi3 method). The presence of the spiked proteins in all six samples allows us to use the Hi3 method to quantify the concentration of each HCP discovered. For this purpose, PHO was selected as an internal reference, of which a known amount of 400 fmoles was loaded on-column in every 2D-LC experiment. The quantification method calculates the molar amounts loaded on column for all identified proteins (including the spiked ones). Based on the average molecular weight of each protein, the on-column HCP amount (in nanograms) can be readily calculated. This value can then be used to back-calculate the protein concentration in the original sample, after taking into account the volume changes during the sample preparation. Finally, the HCP concentration in each PTG1 sample is expressed in ppm (as ng of HCP for every mg of total protein) based on the total protein concentrations (Bradford assay results). The HCPs listed in Table 1 were sorted in the decreasing order of their ppm concentrations measured in Sample B2, which has the highest number of HCPs and the greatest total HCP concentration. To compare the HCP distribution from different cell lines or purification methods, the HCPs were grouped into four concentration categories for visual clarity: high abundance (highlighted in red, with concentrations >1,000 ppm), medium abundance (yellow, concentrations: 500–1,000 ppm), low abundance (green concentrations: 100–500 ppm) and very low-abundance (gray, under 100 ppm). As shown in Table 1, higher purity samples (>98.8% purity) were obtained for PTG1 expressed from CHO-S cell line regardless of the purification column/protocol used. The purity of the mAb expressed in the DG-44 cell line varied more between the two purification procedures (7–8%), than between biological replicates (2–3%). The data clearly indicates that for these protocols the final composition/concentration of HCPs depends more on the type of cell lines used for mAb production.
LC-MRM assay for monitoring HCPs across multiple mAb preparations.
Quantitative LC-MRM analysis is routinely applied to the measurement of specific peptides in complex mixtures such as tryptic digests of plasma or urine, and is an established technique in bioanalysis. In this case, a specific tryptic peptide is selected as a stoichiometric representative of the protein from which it is cleaved to provide a measure of protein concentration. When absolute quantification is needed, the measurement is performed against a spiked internal standard (a synthetic stable isotope-labeled peptide). In principle, the execution of this type of mass spectrometric experiment only requires the knowledge of the precursor m/z and a fragment m/z (MRM transition) as well as the ability to synthesize the stable isotope-labeled analog of the peptide. Thus, MRM experiments are widely used to obtain the maximum sensitivity for detection of target compounds. However, the development of a set of MRM transitions for a number of proteins of interests has always been a challenging process and usually takes many iterative steps.
One of the benefits using the LC-MSE approach for protein identification44–46 is that for every identified protein, a large number of the best ionized peptides and the corresponding fragment ions are simultaneously acquired. This information provides a solid experimental foundation to design a sensitive MRM experiment because, unlike data dependent acquisition, the MSE data acquisition method acquires all ions all the time and therefore good MRM transitions are obtained in the first ‘discovery’ step with no need to re-run the samples. In the case of the HCP assay described here, the accurate mass and sequence of all the responding peptides identified and their corresponding fragments are contained in the LC-MSE data set acquired during the discovery process. This information greatly facilitates the design of an MRM experiment to specifically quantify any HCP identified in the sample of interest, thus providing a rapid method to monitor the HCP in a high-throughput manner. Any ions that may potentially interfere in an MRM experiment are also measured in the MSE data and this information can also be used in the design of the assay.
The reproducibility of the MRM assay is presented in Figure S2. The results are obtained from the MRM analysis of all six PTG1 samples, each with 5 replicate injections. The average RSD percentage of the peak area for 50 transitions was 13.3%. The sensitivity of the MRM assay was probed by a separate MRM experiment, designed to monitor the spiked protein standards with two peptides for each standard and two transitions per peptide (16 MRMs in total). The MRM assay was able to detect the lowest spiked protein (ENL, 20 fmoles on-column) with a calculated concentration of 4–8 ppm in the six PTG1 samples, as shown in Figure 6A. However, only one transition (644.9→947.5) for the selected ENL T43 peptide could be used for quantification due to the presence of a significant interference for the other transition (644.9→834.4, Fig. 6B).
Figure 6.

Example of MRM interference: (A) transition 644.9→947.5 of ENL T43 peptide provides a _clean_channel for quantification of this peptide in the PTG1 digest; (B) another transition from the same peptide is obscured by a very strong interference from the sample matrix.
In addition to HCP monitoring experiments, samples spiked with 13C15N-isotopically labeled peptides were also analyzed using the constructed MRM methods for the absolute quantification of three selected HCPs (clusterin, elongation factor 1-α and glyceraldehyde-3-phosphate dehydrogenase). The isotopically labeled peptide analogs (one peptide for each HCP) were spiked in the six PTG1 samples before the samples were digested with trypsin (see the Experimental Section). The digests were analyzed using two instrument setups (platforms) to compare the accuracy and reproducibility of the MRM approach on two chromatographic scales: analytical scale chromatography (2.1 mm column ID) and capillary scale chromatography using a microfluidic device containing a separation channel with an internal diameter of 150 µm.
The experimental conditions for the MRM assays involving isotopically labeled peptides are given in Table S2, and the MRM chromatograms recorded for the sample-derived and 13C15N-isotopically labeled peptides for each HCP are shown in Figure 7. For each sample, a replicate of 5 MRM assays were performed on both LC-MRM platforms. Figure 8 shows a comparison between the HCP quantification results obtained on the HCP Discovery platform (QTOF instrument) and the LC-MRM platforms (tandem quadrupoles).
Figure 7.



MRM chromatograms recorded for three selected HCPs measured in Sample B1. Each part displays the best responding transitions for the native and the corresponding 13C15N-isotopically labeled peptide belonging to (A and D) clusterin, (B and E) elongation factor 1α and (C and F) glyceraldehyde 3-phosphate dehydrogenase. MRMs displayed in (A–C) were recorded using the analytical scale chromatography setup (2.1 mm column ID), and the chromatograms presented in (D–F) were obtained on the prototype TRIZAIC nanoTile microfluidic device (150 µm channel ID).
Figure 8.

Comparison of HCP quantification between MSE and MRM methods: TOF-based quantification (MSE)is based on the Hi3 method and MRM quantification is based on the peak area from the signal of spiked 13C15N-isotopically labeled peptides with a known concentration. Protein concentrations (ppm) measured in six mAb preparations are shown for (A) clusterin, (B) elongation factor 1α and (C) glyceraldehyde-3-phosphate dehydrogenase.
Discussion
Different strategies have been evaluated by researchers to design 2-dimensional LC systems for the analysis of complex protein digests with the aim to maximize the separation of all peptides. The choice of the two separation columns for the creation of a practical 2D-LC system, regardless of system configuration, is largely dependent on the orthogonality of the separation selectivity provided by each column. One of the historically common 2D strategies is based on the coupling of a strong cation-exchange (SCX) column with a reversed-phase (RP) column.25 Peptide fractions are eluted from the first-dimensional SCX column by applying a series of step gradients of salt solutions with increasing ionic strengths. At each step, peptides are subsequently separated on the 2nd dimension RP column using a linear acetonitrile gradient. In principle, the retention in SCX is driven by the analyte charge. Because the majority of tryptic peptides in a protein digest are doubly and triply charged at the pH used for the SCX separation, the distribution of peptides fractions is bimodal, with 2+ and 3+ charged peptides eluting in clusters. As a result, significant splitting of high-abundance peptides across multiple fractions has been reported in reference 26. In addition, SCX separation tends to suffer from significant peptide losses and poor reproducibility in separation.26
It has previously been demonstrated that pH can significantly alter the selectivity of peptide separations in reversed-phase chromatography.26,27,30,31 This observation prompted much interest in coupling two RP columns, operated at two pH extremes (pH 10 and pH 2.5), as a 2D chromatographic system for peptide separation, using online or off-line configurations.26–39 Although the coupling of high-pH RP/low-pH reversed-phase separations was shown to be less orthogonal than the classical SCX/RP multidimensional system for the separation of complex peptide mixtures in proteomic experiments,30 the separation resolution offered by the high-pH RP in the first chromatographic dimension is far superior to the SCX separation. RP separation elutes peptides almost equally over the entire retention window (trapezoidal distribution of peptides) allowing for a greater spread of peptides across the same number of fractions.30 Taken together, the advantages offered by a 2D-LC system based on high-pH/low-pH RP separation translate into better chromatographic performance. In the case of the HCP assay, these chromatographic advantages can provide more HCP identifications or better sequence coverage for low abundance (10–1,000 ppm) HCPs.
One of the efficiency benefits of the RP/RP 2D setup over the SCX/RP approach is the significant improvement in peptide resolution in the first dimension of fractionation. Figure 4 highlights the capability of the high-pH fractionation for isolating a low-abundance ENL peptide (T43) as well as a high-abundance ADH peptide (T26). Despite the great abundance difference between the two peptides (100-fold), both peptides are “digitally” eluted in a single fraction with a very narrow elution window (2% of ACN change). This data clearly demonstrates the potential of high-pH RP/low-pH RP chromatography for enrichment (heart-cutting) of targeted peptides from complex samples.
During the analysis of complex peptide samples, it is often observed that the isotopic distribution of a low-abundance peptide of interest is frequently obscured by the isotopic pattern of higher abundance ions due to the overlapping of multiple isotopic distributions, making the identification of low abundance peptides extremely difficult. The ability to identify such low abundance peptides is greatly enhanced by removing the interfering peptides to reduce significantly the chemical background around the analyte signal. Figure 5 illustrates the utility of the high-pH/low-pH approach in differentiating a low abundance peptide from complex background signals. In this example, an ENL digest was spiked at a concentration of 50 ppm in a complex protein digest sample (1 ng ENL T43 in 20 µg E. coli lysate digest loaded on column), and the mixture was analyzed by the 2D-LC setup in four separate experiments. The difference between the four experiments appears in the number of fractionation step undertaken during the first dimension separation. Four different fractionation schemes, with 1, 3, 5 and 10-steps, were employed. As shown in Figure 5, the isotope pattern of the peptide of interest (ENL T43) became recognizable only after the 5-step fractionation, although its isotopic distribution was distorted by another interfering species. With further fractionation (10-step fractionation), the experiment produced a rather clean MS spectrum of the spiked peptide. The intensity of the MS peak is very close to the intensity of the same amount of pure peptide from an analysis recorded in the absence of the E. coli background. This observation suggests that ion suppression caused by the co-eluting higher abundance peptides did not play a significant role in the case of T43 peptide. Instead, the interference from other ion species that have close m/z results in the failure of detecting this low abundance peptide. Overall, a 10- to 20-fold improvement to the dynamic range of a peptide assay can be achieved when a 2D-RP/RP LC system is used for a comprehensive separation. The data presented above illustrates that the 2D-LC system can adequately address the needs for the inherently wide dynamic range (105) encountered in HCP analysis. As shown in Figure 5, the final results depend on the number of fractionation steps involved.
Two operational modes involved in the applications of multidimensional liquid chromatography are often described as heart-cutting and comprehensive chromatography. Heart-cutting refers to the isolation of one or multiple fractions of the analyte(s) of interest from the first separation column and conducting the analysis typically on a subset of the isolated fractions on the second dimension separation column. For the complete identification of HCPs in a biopharmaceutical sample, comprehensive multidimensional chromatography might be necessary, where every fraction from the first dimension is transferred to the second dimension for analysis, and a single sample injection might take 10–12 h to finish. This experimental procedure relies on the system stability and reproducibility. The chromatographic performance of the 2D-LC setup with respect to the retention time, peak width, peak shape and MS signal intensities for selected peptide were investigated in this study. The data in Figure 3 demonstrates that a highly reproducible chromatographic performance can be maintained, regardless of the number of fractionation steps employed in the first dimension. The stability of the setup over an extended period of time (48 h, 4 consecutive experiments) is demonstrated by Figure 2, where the mass chromatograms for two peptides from the MIX-4 digest (Sample I) are displayed. These results directly demonstrate that the high-pH XBridge column can retain peptides over a long fractionation experiment (6–10 h) without measurable sample losses and the multidimensional chromatography setup is capable of generating highly reproducible chromatographic separations.
The identification and quantification of low-abundance HCPs in biopharmaceuticals requires a sensitive and specific analytical technique that is able to detect these protein impurities at very low concentration levels (10–1,000 ppm). Methods with poor sensitivity are of little value in HCP assay because of stringent product quality and safety requirements by the regulatory agencies. In addition, an assay with high specificity is necessary not only to identify individual HCPs, but also to allow differentiation of HCPs from other process-related contaminants.
When coupled with LC separations, MS has been widely used in many proteomic experiments to identify proteins from complex mixtures. In a typical shotgun proteomics experiment, peptides from a complex protein digest are separated by LC, introduced into a mass spectrometer and fragmented. The parent proteins are identified via searching the peptide fragments against a protein database. In this approach, each peptide precursor ion is individually selected in the mass spectrometer for fragmentation, and is often referred to as data-dependent acquisition (‘DDA’). The rate at which a mass spectrometer can perform the fragmentation (i.e., switch between MS and MS/MS) determines the sampling depth and the dynamic range of the analysis. Because the concentration of the biopharmaceutical molecule is at least three orders of magnitude higher than the HCPs, the signals of the peptides from the biotherapeutic protein dominate throughout the LC run. As a result, in many situations, peptides from HCPs co-elute with multiple peptides that have much higher concentration. This situation quickly overwhelms the MS/MS acquisition rate of even the fastest instruments, because peptide precursors are selected for MS/MS fragmentation based on their intensities. Since the low intensity ions are those of interest for HCP identification, the inability of data-dependent techniques results in preferential fragmentation of high-abundance peptides (coming from the biopharmaceutical itself), with little or no chance for the fragmentation of HCP peptides. In a data-dependent approach, the MS instrument is greatly biased toward uninformative peptide fragmentation of the biopharmaceutical itself, rather than focusing on the low abundant HCP peptides.
Multiplexed data acquisition method (MSE)has proven to be advantageous to provide in-depth signal sampling44–46 because the fragmentation process does not involve any precursor selection. All the peptide precursor ions, whether they have high or low intensity, have an equal opportunity to be fragmented. This technique provides an efficient way to sample the low intensity peptide precursors when there is a wide dynamic range distribution. For the identification of HCPs in biopharmaceuticals, where low abundance proteins are the primary subject of interest, the use of the multiplexed MS method seems to be a sensible choice. Therefore, when coupled with 2D separations, MSE provides the specificity needed for HCP analysis. Further detailed description of the MSE methodology can be found in previous publications.44–46
To test the analytical capability of the 2D-LC MSE technology for identification of low-abundance HCPs in biopharmaceuticals, five proteins standards (originating from species other than the host) were spiked in the PTG1 preparations before tryptic digestion to probe the dynamic range of the assay. From the analysis of six Protein A purified mAb (PTG1) samples, HCP concentrations as low as 50 ppm can be confidently identified routinely by the approach (see Table 1). HCPs with a concentration lower than 50 ppm can be identified if the sample digest is less complex, such as when the MW of the biopharmaceutical is smaller, or when fewer high abundance HCPs are present.
The protein database used for identification of CHO proteins contains a significant number of entries of mouse proteins (95%). The genomic sequence information for the CHO-K1 cell line became publicly available only recently.47 By relying on the homology between mouse and hamster, several MS-based proteomics approaches have been able to overcome this apparent limitation and proved that relevant protein changes during antibody expression in CHO host cells can be detected.48,49 To our knowledge, there are no MS-based applications reported in the scientific literature that describe the identification and monitoring of HCPs during purification of biopharmaceutical products. As shown in Table 1, using the mouse/hamster homology, we were able to identify a total of 33 HCPs across six PTG1 preparations, of which five HCPs are authentic hamster proteins. The HCP concentrations measured varied widely from 16 to 3,034 ppm (ng HCP/mg total protein), and four out of five spiked proteins were identified in all samples (2 out of 3 replicates). Interestingly, only four high-abundance HCPs were found in common across all PTG1 preparations (see Tables 1 and S3). Most of the HCPs identified in this study are high-abundance CHO proteins and ten of them were shown to be differentially expressed in CHO cells used for mAb production.48,49
The six PTG1 mAb samples analyzed in the current study were produced by a single-step purification process, and they do not necessarily represent highly purified biotherapeutic products. In addition, the single-step purification protocol was not fully optimized to provide the highest purity of PTG1. Therefore, a relatively high number of CHO proteins (33) were co-purified (see Table 1). However, the samples served as good models to test the capability of the 2D-LC/MSE assay in dealing with complex samples. In addition, since Protein A is normally included as the initial step in a sophisticated purification scheme of a biopharmaceutical (mAb) product, this assay can yield a comprehensive list of HCPs in the early stage of purification (while HCP concentrations are high) thereby providing vital information to the design of downstream purification processes. More importantly, because the identities of each individual HCPs are known, these proteins can be effectively monitored in a more sensitive and high-throughput manner.
Compared with other methods for HCP analysis, the 2D-LC/MSE assay has several advantages. For examples, the method can be applied to any protein biopharmaceutical sample and provides the concentration and the identity of individual HCPs. The concentration measurement is also available from a calculation that does not involve the synthesis of internal protein standards. Additionally, the development of a method is far shorter than many other techniques that involve immunoassay steps. However, the assay is not without limitations. With a total run time of 10–12 h per sample (one injection, 10 fractions), the assay is not practical for analyzing multiple pharmaceutical preparations simultaneously. Therefore additional effort was invested in developing a sensitive and high-throughput MRM assay to address the limitations of the HCP discovery method. MRM methods represent a rational, cost-effective strategy and have been widely applied to process development and routine quality control.
An MRM assay is a targeted quantitative method that requires knowledge of protein identity. This information is readily available from the 2D-LC/MSE assay. In this sense, the MRM assay is the natural extension to the 2D-LC/MSE assay and an essential component of the entire workflow for HCP analysis. It paves the way for high-throughput HCP monitoring during later stages of biopharmaceutical purification. The developed LC-MRM assay allowed us to monitor a list of 21 HCPs within 20 min, and generated highly reproducible measurements with an average peak area RSD of 13.3% (Fig. S2). However, as exemplified by the MRM chromatograms from Figure 6, MRM interferences caused by higher abundance peptide can sometime pose a challenge when complex peptide mixtures are analyzed with a fast gradient (15 min) on a single chromatographic column. This problem can be alleviated by using at least two peptides per protein and two transitions per peptide.
With the use of isotope labeled peptides, the MRM assay can be used for absolute quantification of HCPs. The MRM assays developed for three abundant HCPs out of the four proteins found in all six PTG1 preparations (see Table S3) were tested on two different LC-MRM platforms for the high-throughput quantification. For each protein, a 13C15N-isotopically labeled peptide was also spiked in each sample as an internal standard. Representative MRM chromatograms recorded on an analytical scale column (2.1 mm × 150 mm) and a nanoscale micro-column (150 µm × 50 mm) are shown in Figure 7. Despite the difference in chromatographic scales, the quantification results obtained on the two LC-MRM platforms are in very good agreement (<5% difference in the absolute HCP concentrations measured), as illustrated by Figure 8. The nanoscale platform is able to deliver the same quantitative information as the analytical scale platform, while only using 1/50 of the sample. Although the amount of sample available for HCP analysis is generally not limited, there are other applications (e.g., clinical proteomics, biomarker validation, PK/PD studies for monitoring therapeutic proteins in biological fluids) where the microfluidic MRM platform would bring greater benefits. Additionally, microfluidic platforms are potentially easier to automate or simplify for less experienced users.
The quantification results for three HCPs obtained from the MSE method and the LC-MRM absolute quantification methods are compared in Figure 8. The MSE results were calculated using the Hi3 best responding peptides for each protein and the LC-MRM quantification results were generated using isotopically labeled peptide analogs. The results agree with each other within 25% of the measured values. The Hi3 quantification methodology is likely to be responsible for the discrepancy between these methods because the accuracy of this method is reported to be in the range of 20–40%.38,39 In almost all measurements, the Hi3 method slightly overestimated the HCP concentrations. One explanation for this observation is that that the Hi3 method combines all potential HCP isoforms into one measurement, whereas the MRM method is isoform-specific. Further comparisons between the Hi3 method and the MRM method for larger data sets should help to validate these findings.
In summary, an online comprehensive two-dimensional LC/MS assay for identification and quantification of host-cell proteins (HCPs) in biotherapeutic proteins over 5 orders of magnitude was developed. The 2D-LC setup used high-pH RP fractionation followed by low-pH RP separation to achieve the separation of protein digests, followed by peptide detection by the MSE approach. The application of this assay to six mAb preparations allowed us to identify 33 HCPs with the lowest concentration of 16 ppm. Subsequently, a high-throughput LC-MRM assay was developed based on the identification information for rapid HCP monitoring. The performance of the MRM assay was evaluated by analyzing 21 HCPs in 20 min with an average peak area RSD of 13.3%. Absolute quantification using 13C15N-labeled peptides analogs as internal standards for three selected HCPs was performed on two LC-MRM platforms. The results were compared with TOF-based, 2D-LC/MSE quantification method using the Hi3 peptide method. The TOF and MRM data sets correlated well, providing strong evidence for the validity of each method, as well as the consistency of the entire workflow. Because the HCP assay provides not only the total concentration of all the HCPs, but also the identity and the concentration of each HCP, it allows us to correlate the contribution of different purification schemes and cell lines to the nature and concentration of HCP impurities present in biopharmaceuticals. This in-depth characterization of biotherapeutic products will greatly facilitate the development of purification methods for both process development and final product quality control. The HCP assay has a great potential to become a universal method for HCP analysis in biopharmaceutical industry.
Materials and Methods
Chemicals.
Ammonium bicarbonate, ammonium hydroxide (28% w/w), bovine α-lactalbumin (LA), bovine serum albumin (BSA), dithiothreitol (DTT), Glu1-fibrinopeptide B (GFP), iodoacetamide (IAM), rabbit glycogen phosphorylase b (PHO), yeast alcohol dehydrogenase (ADH) and yeast enolase (ENL) were purchased from Sigma Chemical Co., (St. Louis, MO USA). HPLC grade acetonitrile (ACN) and trifluoroacetic acid (TFA) were obtained from Thermo Fisher Scientific (Rockford, IL USA). MS-grade formic acid (FA) was acquired from Sigma Chemical Co., A Milli-Q Elix-3 purification system (Millipore, Bedford, MA USA) was used for preparing the deionized (DI) water (18 Mωcm) required for all experiments.
Sample preparation.
Preparation of ammonium formate (NH4FA, pH 10) solution. A stock solution of ammonium formate (200 mM, pH 10) was prepared by mixing 6.95 mL of 28% (w/w) ammonium hydroxide with 450 mL of DI water. Then 0.81 mL of formic acid was added to the solution. The pH of the stock solution was adjusted to 10 with FA, and the final volume was brought to 500 mL. The stock solution was diluted (1:10, v/v) using DI water to yield a 20 mM NH4FA solution for sample preparation and 2D-LC separations.
Protein digest standard mixtures.
MIX-4 protein digest standards (Sample I). A simple protein digest mixture was created by mixing four individual protein digest standards at an appropriate ratio. Stock solutions of individual MassPREP™ protein digests of yeast alcohol dehydrogenase (ADH), rabbit phosphorylase b (PHO), bovine serum albumin (BSA) and yeast enolase (ENL) (all from Waters Corp.) in 20 mM ammonium formate, pH 10, were mixed to achieve the final concentrations of 20 nM ADH, 4 nM PHO, 1 nM BSA and 0.2 nM ENL respectively.
MIX-4 protein digests standards in an E. coli lysate digest solution (Sample II). The second MIX-4 protein digest was prepared by mixing the same sets of protein digest standards at the same concentration levels (20-0.2 nM) except that the four protein digests were spiked into a complex peptide background of an E. coli lysate digest (Waters Corp.,), which was prepared by dissolving 100 µg of E. coli lysate digest in 0.5 mL of 20 mM ammonium formate (pH 10).
HCP samples. A chimeric anti-phosphotyrosine IgG1 monoclonal antibody (PTG1 mAb) was expressed in two different Chinese hamster ovary (CHO) cell lines (DG-44 CHO and CHO-S). Samples A1, B1, A2 and B2, were collected from cell line DG-44 CHO, and samples C and D, were from CHO-S cell lines. All samples were clarified using a Millistak+ D0HC pod filter for primary clarification, a Millistak+ A1HC pod filter for secondary clarification and an Express membrane for sterile filtration (all from EMD Millipore, Bedford, MA). Samples were then purified using Protein A chromatography on a ProSep-vA column (Millipore, Bedford, MA). Samples A1, B1 and C were purified using the recommended manufacturer's protocol, while for samples A2, B2 and D different washing and elution conditions were used to maximize differences in purification performance. The effect of cell viability was investigated for two sample pairs (A1/A2 and B1/B2) with samples A2/B2 being cultured at lower cell viability. Total protein concentrations (expressed in mg/mL) were measured for each PTG1 sample using a Bradford assay. Five protein standards (LA, PHO, ADH, BSA and ENL) were spiked in 250 µL of each of the PTG1 (5–10 mg/mL) samples at fixed concentrations ranging from 20 to 1,000 ppm. The spiked-in proteins were used for quantification of HCPs and for probing the dynamic range of the 2D-LC/MSE assay. The resulting protein mixture was denatured with 0.1% RapiGest (Waters Corp.,) for 15 min at 60°C, reduced with 10 mM DTT for 30 min at 60°C, alkylated with 20 mM IAM for 30 min (at RT) and enzymatically digested overnight (37°C) with porcine trypsin (Promega, Madison, WI) using a 20:1 (w/w, protein:enzyme) ratio. After digestion, the RapiGest surfactant was decomposed by adding 5 µL of pure TFA and the samples were incubated for 30 min at 37°C, and centrifuged (10 min at 10,000 rpm) to remove the insoluble component of the degraded RapiGest. After adjusting the pH of the supernatant solution to pH 10 using 28% (w/w) ammonium hydroxide (pH 11), the digestion volume was brought to 1 mL using 20 mM ammonium formate (pH 10). The digestion protocol was designed to produce the same volume of peptide digest for each sample (1 mL), irrespective of the initial mAb concentration. Because the injection volume for each sample was kept the same for each 2D-LC/MSE experiment (100 µL), the amount of the spiked proteins loaded on-column was constant for every injection: 4,000 fmoles LA, 1,000 fmoles ADH, 400 fmoles PHO, 100 fmoles BSA and 20 fmoles ENL.
Internal standards for MRM analysis.
For absolute protein quantification by the MRM approach, three 13C15N-isotopically labeled peptides (Sigma Aldrich, St. Louis, MO) were spiked into 250 µL of PTG1 sample before the digestion. The final concentration for all three isotopically labeled peptides spiked in the PTG1 digest was 20 nM.
2D-LC separation setup and methods.
A nanoACQUITY™ UPLC£ system (Waters Corporation, Milford, MA) equipped with online 2D-LC technology was developed to perform the peptide separations. A schematic diagram illustrating the operation of the 2D-LC system is presented in Figure 1. The first chromatographic dimension performs peptide fractionation under basic (pH 10) conditions on a 1.0 mm × 50 mm XBridge C18 (5 µm particles) reversed-phase column (Waters Corp.,) at a flow rate of 10 µL/min. Eluent A was 20 mM ammonium formate in water (pH 10), and eluent B was pure ACN. A trap column (2.7 µL internal volume) packed with Symmetry C18 (5 µm, Waters Corp.) was used to trap peptides eluted from the first dimension. Peptide fractions were eluted in step gradients from the first dimension column and mixed online with 100 µL/min of 0.1% TFA solution (1:10 dilution) before being trapped on the trapping column (Fig. 1B). The 10-fold online dilution reduces the organic content and the pH of the mobile phase so that peptides can be effectively retained on the trap column before the second dimension separation. The mobile phases for the second chromatographic dimension (low pH RP) were 0.1% FA in water (mobile phase A) and 0.1% FA in ACN (mobile phase B). The second dimension column is a 0.3 mm × 150 mm C18 column (BEH300, 1.7 µm particles, Waters Corp.). The flow rate for the second dimension separation was set at 12 µL/min and the column was maintained at 65°C. A 30 min gradient from 7–35% B was used for peptide separation in the 2nd dimension separation. The column was washed using 90% B for 5 min and re-equilibrated at 7% B for 10 min before returning to the next step of fractionation.
The step elution gradients for the first dimension were optimized such that approximately the same amount of peptides was eluted off at each step. Throughout the study, four 2D-LC methods were employed: (1) simulated 1D (i.e., 1-step fractionation): all the peptides loaded onto the first dimension column were eluted from the XBridge (high pH) column with 50% B; (2) 3-step fractionation: peptides were eluted from the first dimension column using three successive elution steps, with 10.8, 18.6 and 50% B, respectively; (3) 5-step fractionation: five fractions were eluted with 10.8, 12.4, 15.4, 18.6 and 50% B; (4) 10 step fractionation: ten fractions were eluted using 10.8, 12.4, 14.0, 15.4, 16.7, 18.6, 20.4, 25.0, 30.0 and 50% B. The fractionation process started right after the completion of sample loading (15 min at 10 µL/min with 3% B), and each elution step took 15 min (using a flow rate of 10 µL/min) to complete.
Mass spectrometry.
HCP discovery platform. A multiplexed data acquisition method (MSE) was employed for the mass spectrometric analysis. The LC/MSE data was acquired using a quadrupole time-of-flight mass spectrometer (SYNAPT HDMS System, Waters Corp., Milford, MA) equipped with the standard electrospray ionization (ESI) probe fitted with a small bore (45 µm ID) stainless steel capillary (Waters Corp., p/n M956357DC4-S). For all measurements, the mass spectrometer was operated in positive ESI ion mode with a typical resolving power of 10,000 FWHM. Data were acquired in continuum mode over m/z range of 50–1,990, using a capillary voltage of 2.6 kV, a source temperature of 90°C and a cone voltage of 35 V. The desolvation temperature was set to 300°C and the desolvation gas flow rate was 500 L/hour.
The LC/MSE data was collected by alternating the collision energy of the MS instrument between low energy (MS) and elevated energy (MSE) without precursor selection. The spectral acquisition time at each energy setting was 0.5 sec such that one spectrum of MS and MSE data was acquired every second. In the low energy MS mode the data was collected at a constant collision energy of 5 eV, while in the MSE mode the collision energy was ramped from 15 to 35 eV. The collision energy of the transfer cell was correspondingly alternated between 4 eV (low energy MS acquisition) and 10 eV (ramped MSE acquisition). A solution of 0.2 µM Glu1-fibrinopeptide B (GFP) in 50% acetonitrile with 0.1% FA was used as a lock-mass solution. The solution was delivered at a flow rate of 3 µL/min using an auxiliary pump of the nanoACQUITY 2D-LC system. The lock-mass data was sampled every 4 min using 0.5 sec scans over the same mass range.
LC-MRM analysis using a 2.1 mm analytical column. LC-MRM assays were performed on an ACQUITY™ UPLC system coupled to a Xevo TQ MS tandem quadrupole mass spectrometer (Waters Corp., Milford, MA USA). UPLC separations were performed on a 2.1 × 150 mm BEH300 C18 column packed with 1.7 µm particles (Waters Corp.). The column temperature was kept at 35°C. Mobile phase A was 0.1% FA in DI water, and mobile phase B was 0.1% FA in ACN. Fifty microliters of the digest samples were injected onto the analytical column using a linear gradient from 0 to 35% B over 15 min at a flow rate of 300 µL/min. The ESI-MS conditions are as follows: electrospray capillary voltage 3.5 kV, cone voltage 35 V, source temperature 90°C, desolvation temperature 400°C and desolvation gas flow 800 L/hour. All MRM measurements were acquired at unit mass resolution (0.75 Da FWHM) for both MS1 and MS2 using dwell times ranging from 10–40 ms. Collision energies were assigned according to the formula CE = 0.034 × m/z + 3.3,50 regardless of precursor charge state.
Twenty one HCPs (unique protein isoforms) identified in the six PTG1 samples were selected to demonstrate the development of LC-MRM methods for rapid monitoring of target HCPs. These HCPs represent proteins from each abundance category and span a broad range of concentration (from 16 ppm for eukaryotic translation initiation factor 5A-1 to 3,034 ppm for nucleolin). The selection of representative peptides for MRM assays starts from the MSE data set from which the HCPs were identified. The VerifyE software (Waters Corp.,) was used to automatically filter the MSE data set based on a number of simple rules: (1) identification of four peptides for each HCP, having the best precursor ion intensity in ESI-MS; (2) identification of four transitions per peptide; (3) exclusion of peptides containing modified residues such as Cys carbamidomethylation, Met oxidation and N/Q deamidation, although tryptic miscleavages were allowed. For the 21 HCPs, VerifyE generated 304 MRM transitions for further optimization.
Although all the transitions from a VerifyE process are based on experimental data, the ability to detect a peptide in MRM is potentially affected by a higher abundance peptide that shares the same transition and elutes at the same retention time. This observation is common in complex samples with a wide dynamic range, especially when the MRM analysis is done in a fast chromatographic run. To improve the selection of MRM transitions for the HCPs and find the best transitions for each peptide, all 304 transition candidates were tested to validate the detectability under typical chromatographic conditions used for a high-throughput assay (15 min gradient separation). The transitions were split into three scouting runs for validation (approximately 100 transitions per run) and each transition was monitored throughout the entire LC gradient with no retention time windows specified. The final MRM method was constructed after analyzing the MRM chromatograms from the three scouting runs, and it contained 50 MRMs from 25 peptides (2 transitions per peptide) covering 21 HCPs. These transitions were programmed into six scheduled retention time windows (2 min each) for the monitoring of HCPs during the 15 min assay. The experimental conditions for the MRM experiment (peptide sequences/transitions, collision energy and dwell times) are presented in Table S1. The absolute quantification for three selected HCPs after spiking three 13C15N-isotopically labeled peptides (one peptide for each HCP) was also performed for all six PTG1 preparations using the same setup and method described above.
LC-MRM analysis using a novel microfluidic device. The absolute quantification for the selected HCPs in all six PTG1 preparations was also acquired using a prototype microfludic device (a ‘Trizaic’ tile containing a 150 µm × 50 mm channel, packed with BEH 1.7 µm C18 particles, Waters Corp.,) coupled to a Xevo TQ-S tandem quadrupole (Waters Corp.). For the same samples analyzed on the microfludic device, the injection volumes were scaled down to match the chromatographic dimension of the separation channel. Only 1 µL of sample was injected on the microfluidic device compared with 50 µL of sample injected on a 2.1 mm ID column. The LC separations were performed at 45°C, using a flow rate of 3 µL/min and a linear gradient from 3–40% B over 7 min. The mobile phase A was 0.1% FA in DI water, and mobile phase B was 0.1% FA in ACN. The ESI-MS conditions were: electrospray capillary voltage 4.0 kV, cone voltage 40 V, source temperature 150°C, nanoflow nebulizing gas pressure 0.5 bar. Measurements were acquired at unit mass resolution (0.75 Da FWHM) for both MS1 and MS2 and the detailed MRM experimental parameters are provided in Table S2.
Data processing. The LC/MSE data was processed using PLGS 2.4 software (Waters Corp.,) 51,52 for HCP identification. For each PTG1 sample, all the MSE data from each fractionation step was digitally combined into a single file using PLGS software.51,52 The low-energy and high-energy (MSE) data were background subtracted, de-isotoped and charge-state reduced to the corresponding monoisotopic peaks. Each monoisotopic peak was then lock-mass corrected to yield the accurate mass measurement. Fragment ions and their corresponding precursor ions were automatically aligned (grouped) together based on the retention time profiles of the ions.51,52 Processed spectra were searched against a custom protein database which was compiled from 12,943 Swiss Prot mouse protein sequences, 654 Golden hamster proteins (from Swiss Prot database), the sequences of five spiked proteins (LA, ADH, PHO, BSA, ENL), the sequence of porcine trypsin, protein A (S. aureus) sequence, and the heavy/light chain sequences of PTG1. The final custom database also included an equal number of entries of randomized (decoy) sequences (one random sequence for each true sequence), containing a total of 27,212 entries in the database. The decoy strategy was used to control the false positive rate in HCP identification. The search was limited to tryptic peptides with one potential missed cleavage. The mass tolerance allowed for the low-energy precursor ions was 20 ppm, while the mass tolerance of elevated-energy fragment ions was set to 50 ppm.
PLGS search results (*.csv files) were exported to VerifyE software to generate transitions for the MRM assay.
All MRM chromatograms (five replicates per sample) were integrated using the TargetLynx application manager from MassLynx 4.1 (Waters Corp.,), and the results containing MRM transitions, chromatographic retention times and peak areas were exported to Excel spreadsheet (Microsoft, Redmond, WA) for additional data analysis.
Acknowledgments
The authors would like to thank Dr. Steve Cohen for his contribution in the early stage of this project. The authors would like to thank Karen Haas and Leslie Bartholomew from Waters Corp., for preparing Figure 1 of the manuscript. The following colleagues from EMD Millipore are acknowledged: Patricia Kumpey and Brett Belongia for the cell culture work, Scott Bliss for the clarification work and Ajish Potty for the Protein A purification work.
Supplementary Material
References
- 1.ICH Harmonised Tripartite Guideline, author. Specifications: Test procedures and acceptance criteria for biotechnological/biological products Q6B. 1999 [Google Scholar]
- 2.European Medicines Agency, author. Omnitrope: Scientific Discussion 2006. http://www.emea.europa.eu/humandocs/PDFs/EPAR/Omnitrope/060706en6.pdf.
- 3.European Medicines Agency, author. Alpheon: Scientific Discussion 2006. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/000585/WC500017451.pdf.
- 4.Schellekens H. Biosimilar therapeutics—what do we need to consider? NDT Plus. 2009;2:27–36. doi: 10.1093/ndtplus/sfn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman K. Strategies for host cell protein analysis. Biopharm. 2000;13:38–45. [Google Scholar]
- 6.Champion K, Madden H, Dougherty J, Shacter E. Defining your product profile and maintaining control over it, Part 2: Challenges of monitoring host cell protein impurities. BioProcess Intl. 2005;3:52–57. [Google Scholar]
- 7.Wang X, Hunter AK, Mozier NM. Host cell proteins in biologics development: identification, quantitation and risk assessment. Biotechnol Bioeng. 2009;103:446–458. doi: 10.1002/bit.22304. [DOI] [PubMed] [Google Scholar]
- 8.Rathore AS, Sobacke SE, Kocot TJ, Morgan DR, Dufield RL, Mozier NM. Analysis for residual host cell proteins and DNA in process streams of a recombinant protein product expressed in Escherichia coli cells. J Pharm Biomed Anal. 2003;32:1199–1211. doi: 10.1016/S0731-7085(03)00157-2. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Schomogy T, Wells K, Mozier NM. Improved HCP quantitation by minimizing antibody cross-reactivity to target proteins. BioProcess Intl. 2010;8:18–23. [Google Scholar]
- 10.Savino E, Hu B, Sellers J, Sobjak A, Majewski N, Fenton S, et al. Development of an in-house, process-specific ELISA for detecting HCP in a therapeutic antibody, Part 1. BioProcess Intl. 2011;9:38–47. [Google Scholar]
- 11.Savino E, Hu B, Sellers J, Sobjak A, Majewski N, Fenton S, et al. Development of an in-house, process-specific ELISA for detecting HCP in a therapeutic antibody, Part 2. BioProcess Intl. 2011;9:68–77. [Google Scholar]
- 12.Hayduk EJ, Choe LH, Lee KH. A two-dimensional electrophoresis map of Chinese hamster ovary cell proteins based on fluorescence staining. Electrophoresis. 2004;25:2545–2556. doi: 10.1002/elps.200406010. [DOI] [PubMed] [Google Scholar]
- 13.Jin M, Szapiel N, Zhang J, Hickey J, Ghose S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol Bioeng. 2010;105:306–316. doi: 10.1002/bit.22532. [DOI] [PubMed] [Google Scholar]
- 14.Srebalus Barnes CA, Lim A. Applications of mass spectrometry for the structural characterization of recombinant protein pharmaceuticals. Mass Spectrom Rev. 2007;26:370–388. doi: 10.1002/mas.20129. [DOI] [PubMed] [Google Scholar]
- 15.Chen G, Pramanik BH. LC-MS for protein characterization: Current capabilities and future trends. Expert Rev Proteomics. 2008;5:435–444. doi: 10.1586/14789450.5.3.435. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Warrack BM, Goodenough AK, Wei H, Wang-Iverson DB, Tymiak AA. Characterization of protein therapeutics by mass spectrometry: Recent developments and future directions. Drug Discov Today. 2011;16:58–64. doi: 10.1016/j.drudis.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Gross ML, Chen G, Pramanik BN, editors. Protein and peptide mass spectrometry in drug discovery. Hoboken, NJ USA: John Wiley and Sons, Inc; 2011. [Google Scholar]
- 18.Dowling P, Meleady P, Henry M, Clynes P. Recent advances in clinical proteomics using mass spectrometry. Bioanalysis. 2010;2:1609–1615. doi: 10.4155/bio.10.69. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Pramanik BN. Application of LC/MS to proteomics studies: Current status and future prospects. Drug Discov Today. 2009;14:465–471. doi: 10.1016/j.drudis.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Antonioli P, Fortis F, Guerrier L, Rinalducci S, Zolla L, Righetti PG, et al. Capturing and amplifying impurities from purified recombinant monoclonal antibodies via peptide library beads: A proteomic study. Proteomics. 2007;7:1624–1633. doi: 10.1002/pmic.200600778. [DOI] [PubMed] [Google Scholar]
- 21.Hunter AK, Wang X, Suda EJ, Herber JT, Shell RE, Thomas KE, et al. Separation of product associating E. coli host cell proteins OppA and DppA from recombinant apolipoprotein A-I in an industrial HIC unit operation. Biotechnol Prog. 2009;25:446–453. doi: 10.1002/btpr.106. [DOI] [PubMed] [Google Scholar]
- 22.Wierling PS, Bogumil R, Grunhagen EK, Hubbuck J. High-throughput screening of packed-bed chromatography coupled with SELDI-TOF MS analysis: monoclonal antibodies versus host cell protein. Biotechnol Bioeng. 2007;98:440–450. doi: 10.1002/bit.21399. [DOI] [PubMed] [Google Scholar]
- 23.Fortis F, Guerrier L, Areces LB, Antonioli P, Hayes T, Carrick K, et al. A new approach for the detection and identification of protein impurities using combinatorial solid phase ligand libraries. J Proteome Res. 2006;5:2577–2585. doi: 10.1021/pr060090s. [DOI] [PubMed] [Google Scholar]
- 24.Snyder L, Kirkland J, Dolan J. Basic concepts and the control of separation in: Introduction to modern liquid chromatography. Vol. 3. Hoboken, NJ USA: John Wiley & Sons; 2010. p. 73. [Google Scholar]
- 25.Washburn MP, Wolters DA, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 26.Gilar M, Olivova P, Daly AE, Gebler JC. Orthogonality in separation in two-dimensional liquid chromatography. Anal Chem. 2005;77:6426–6434. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 27.Gilar M, Olivova P, Daly AE, Gebler JC. Two-dimensional separation of peptides using a RP-RP-HPLC system with different pH in the first and second separation dimensions. J Sep Sci. 2005;28:1694–1703. doi: 10.1002/jssc.200500116. [DOI] [PubMed] [Google Scholar]
- 28.Toll H, Oberacher H, Swart R, Huber C. Separation, detection and identification of peptides by ion-pair reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry at high and low pH. J Chromatogr A. 2005;1079:274–286. doi: 10.1016/j.chroma.2005.03.121. [DOI] [PubMed] [Google Scholar]
- 29.Dowell JA, Frost DC, Zhang J, Li L. Comparison of two-dimensional fractionation techniques for shutgon proteomics. Anal Chem. 2008;80:6715–6723. doi: 10.1021/ac8007994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delmotte N, Lasaosa M, Tholey A, Heinzle E, Huber CG. Two-dimensional reversed-phase x ion-pair reversed-phase HPLC: An alternative approach to high-resolution peptide separation for shotgun proteome analysis. J Proteome Res. 2007;6:4363–4373. doi: 10.1021/pr070424t. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura T, Kuromitsu J, Oda Y. Evaluation of comprehensive multidimensional separations using reversed-phase, reversed-phase liquid chromatography/mass spectrometry for shotgun proteomics. J Proteome Res. 2008;7:1007–1011. doi: 10.1021/pr7005878. [DOI] [PubMed] [Google Scholar]
- 32.Gilar M, Olivova P, Chakrabory A, Jaworski A, Geromanos S, Gebler JC. Comparison of 1-D and 2-D LC MS/MS methods for proteomic analysis of human serum. Electrophoresis. 2009;30:1157–1167. doi: 10.1002/elps.200800630. [DOI] [PubMed] [Google Scholar]
- 33.Song C, Ye M, Han G, Jiang X, Wang F, Yu Z, et al. Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphopeptides. Anal Chem. 2010;82:53–56. doi: 10.1021/ac9023044. [DOI] [PubMed] [Google Scholar]
- 34.Calipo L, Capriotti SL, Cavaliere C, Gubbioti R, Samperi R, Lagana A. Evaluation of different two-dimensional chromatographic techniques for proteomic analysis of mouse cardiac tissue. Biomed Chrom. 2010;5:594–599. doi: 10.1002/bmc.1487. [DOI] [PubMed] [Google Scholar]
- 35.Zhou F, Cardozza JD, Ficarro SB, Adelmant G, Lazaro JB, Marto J. Online nanoflow RP-RP-MS reveals dynamics of multicomponent Ku complex in response to DNA damage. J Proteome Res. 2010;9:6242–6255. doi: 10.1021/pr1004696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Yang F, Gritsenko M, Wang Y, Clauss T, Liu T, et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stapels M, Fadgen K. A reproducible online 2D reversed phase-reversed phase high-low pH method for qualitative and quantitative proteomics. Curr Trends Mass Spectrom. 2009:14–15. [Google Scholar]
- 38.Donato P, Cacciola F, Sommella E, Fanali C, Dugo L, Dacha M, et al. Online comprehensive RPLC × RPLC with mass spectrometry detection for the analysis of proteomce samples. Anal Chem. 2011;83:2485–2491. doi: 10.1021/ac102656b. [DOI] [PubMed] [Google Scholar]
- 39.François I, Cabooter D, Sandra K, Lynen F, Desmet G, Sandra P. Tryptic digest analysis by comprehensive reversed phase x two reversed phase chromatography (RP-LC × 2RP-LC) at different pHs. J Sep Sci. 2009;32:1137–1144. doi: 10.1002/jssc.200800578. [DOI] [PubMed] [Google Scholar]
- 40.Griffiths SW, Cooney C. Development of a peptide mapping procedure to identify and quantify methionine oxidation in recombinant human alpha1-antitrypsin. J Chromatogr A. 2002;942:133–143. doi: 10.1016/S0021-9673(01)01350-4. [DOI] [PubMed] [Google Scholar]
- 41.Xie H, Gilar M. Improving LC peak shape of proline-rich peptides within an antibody peptide map. Waters Application 720003063en. 2009 [Google Scholar]
- 42.Silva J, Gorenstein M, Li GJ, Vissers P, Geromanos S. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;1:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Silva JC, Denny R, Dorschel C, Gorenstein M, Li G, Richardson K, et al. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol Cell Proteomics. 2006;5:589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Bateman RH, Carruthers R, Hoyes J, Jones C, Langridge J, Millar A, et al. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass spectrometer for studying protein phosphorylation. J Am Soc Mass Spectrom. 2002;13:792–803. doi: 10.1016/S1044-0305(02)00420-8. [DOI] [PubMed] [Google Scholar]
- 45.Purvine S, Eppel J, Yi E, Goodlett D. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3:847–850. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 46.Silva JC, Denny R, Dorschel A, Gorenstein M, Kass I, Li GJ. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 47.Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, et al. The genomic sequence of the chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carlage T, Hincapie M, Zang L, Lyubarskaya E, Madden H, Mhatre R, et al. Proteomic profiling of a high-producing chinese hamster ovary cell line. Anal Chem. 2009;81:7357–7362. doi: 10.1021/ac900792z. [DOI] [PubMed] [Google Scholar]
- 49.Kuystermans D, Dunn M, Al-Rubeai M. A proteomic study of cMyc improvement of CHO culture. BMC Biotechnol. 2010;10:1–13. doi: 10.1186/1472-6750-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maclean B, Tomazela D, Abbatiello S, Zhang S, Whiteaker J, Paulovich M, et al. Effect of collision energy optimization on the measurements of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 2010;82:10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geromanos SJ, Vissers J, Silva J, Dorschel C, Li GZ, Gorenstein M, et al. The detection, correlation and comparison of peptide precursors and product ions from data independent LC-MS with data dependent LC-MS/MS. Proteomics. 2009;9:1683–1695. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 52.Li GZ, Vissers J, Silva J, Golick D, Gorenstein M, Geromanos SJ. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mictures. Proteomics. 2009;9:1696–1719. doi: 10.1002/pmic.200800564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.