

Abstract
It is widely recognized that amyloid formation sensitively responds to conditions set by myriad cellular solutes. These cosolutes include two important classes: macromolecular crowders and compatible osmolytes. We have recently found that addition of macromolecular PEG only slightly affects fibril formation of a model peptide in vitro. Polyol osmolytes, in contrast, lengthen the lag time for aggregation, and lead to larger fibril mass at equilibrium. To further hypothesize on the molecular underpinnings of the disparate effect of the two cosolute classes, we have further analyzed the experiments using an available kinetic mechanism describing fibril aggregation. Model calculations suggest that all cosolutes similarly lengthen the time required for nucleation, possibly due to their excluded volume effect. However, PEGs may in addition promote fibril fragmentation, leading to lag times that are overall almost unvaried. Moreover, polyols effectively slow the monomer-fibril detachment rates, thereby favoring additional fibril formation. Our analysis provides first hints that cosolutes act not only by changing association or dissociation rates, but potentially also by directing the formation of fibrils of varied morphologies with different mechanical properties. Although additional experiments are needed to unambiguously resolve the action of excluded cosolutes on amyloid formation, it is becoming clear that these compounds are important to consider in the search for ways to modulate fibril formation.
Keywords: amyloid fibrils, protein aggregation kinetics, macromolecular crowding, osmolytes, protein stabilization, fibril breakage, osmotic pressure
Introduction
Associated with a variety of devastating pathologies, amyloids are formed through the self-association of alternatively folded proteins or peptides.1 In diseases ranging from Alzheimer to rheumatoid arthritis, these insoluble, β-sheet type, fibrillar aggregates are found to abnormally precipitate in affected cells and tissues.2–4 While the precise mechanism of amyloid aggregation is not fully resolved, solution conditions are found to play a crucial role. In fact, it has been realized that the kinetics and even the preferred fibril polymorph for a particular peptide can be determined simply by exposure to different growth conditions such as temperature, salt type and concentration, pH and buffers and hydrostatic pressure.5–7 To fully appreciate the impact of the cellular milieu on the fibrillation process requires scrutiny of even more complex, yet often neglected, solution conditions.
Cells are highly dense environments, crowded not only by many macromolecules such as proteins, nucleic acids and lipid membranes, but also by molecularly smaller cosolutes. Acting through excluded volume (steric) interactions, large macromolecules often stabilize proteins by reducing the free volume they allow the unfolded (denatured) state.8,9 Osmolytes, in contrast, are a prominent class of smaller cosolutes that are accumulated by cells to counteract external stresses. These solutes include sugars and polyols, urea and its derivatives and modified amino acids.10–12 While osmolytes are present at relatively high, even molar, concentrations, many are considered “compatible” as they tend to stabilize the native states of proteins. This effect is achieved because of the preferential exclusion of osmolytes from the protein interface that stabilizes protein states with smaller accessible surface area.11,13–19 Taken together, this wide range of cellular cosolutes presents a highly elaborate environment for the processes that lead to amyloid formation.
Macromolecular crowders and stabilizing osmolytes have been found to profoundly impact the process of amyloid formation.20–23 While both these types of solutes are preferentially excluded from the surfaces of many proteins, thereby stabilizing their folded state, they affect the aggregation process in very different ways. For example, certain inositol osmolytes have been shown to effectively slow and even dissolve Aβ fibril aggregation in vivo as well as in vitro.24 In contrast, the soluble polymer polyethelene glycol (PEG), often used to model macromolecular crowding in vitro, has been found to promote faster aggregation in a variety of amyloid forming proteins.21,25,26 This diverse action may come as no surprise, because osmolytes are chemically quite different from macromolecular crowders. And yet, this observation requires us to search for distinct mechanisms of cosolute action on aggregation kinetics that must also depend on cosolute type and not only on its concentration.
We have been following the amyloid aggregation of a model peptide to gauge the impact of various cosolutes, both large polymer “crowders” and smaller osmolytes, on the kinetic process. In the following sections we describe the primary results of our recently reported study, clearly indicating at least two distinct mechanisms of action associated with the two classes of cosolute.20 We then analyze the temporal evolution of aggregation in terms of a possible kinetic scheme, based on a mechanism used to describe amyloid nucleation and subsequent fibril elongation processes.27 This analysis allows us to further hypothesize on the molecular underpinnings of the disparate action of the two chemically distinct cosolute families. Interestingly, our findings hint that while for our model peptide the nucleation rate remains a function of cosolute concentration alone, different cosolute families (specifically, polyols and PEGs) may modulate fibril brittleness in diverse ways. In addition, we find that the rate of monomer fibrillation may also be modulated by cosolute chemical identity: polyols act to decrease the rate of monomer detachment, while PEGs hardly change it. Finally, we comment on the possible implications of our findings to future attempts to control fibril kinetics.
Different Cosolutes Distinctively Affect Aggregation Kinetics
Recently, we have tested the effect of different cosolutes on the folding and aggregation process of a designed, synthetic 16 amino-acid peptide.14,20,28 The cosolutes included polyols and PEGs of different molecular weights. The aggregation process was followed using several experimental techniques, including circular dichroism, electron microscopy and ThT fluorescence. Typical fluorescence assay results, recorded over the course of ∼40 h, are shown in Figure 1A. These kinetic curves were analyzed to derive typical rate constants in terms of the two standard stages of a nucleation-aggregation process.29 First, the nucleation lag time, τlag, describes the time it takes to form a critical amount of aggregating nuclei. Second, the elongation time, τel, is inversely related to the rate of fibril elongation once the nuclei form. In addition, we followed the fluorescence signal at maximal emission, Fmax. Control experiment showed that Fmax varied only by up to 30% in the presence of high concentrations of the different cosolutes, allowing us to use it as a measure of the amount of monomer that has undergone fibrillation when the system has reached steady-state.
Figure 1.

ThT fluorescence assay of peptide aggregation and corresponding typical time scales in the aggregation process. (A) Fluorescence intensity vs. time in aqueous solution (orange hexagons) and in the presence of various cosolutes: sorbitol 1.66 m (black squares); glycerol 2.37 m (red circles); PEG4000 0.03 m (green upward pointing triangles); PEG400 0.48 m (blue downward pointing triangles); TEG 0.83 m (purple diamonds). (B) Nucleation lag time, τlag, as a function of the molality of cosolute monomers. (C) Peak fluorescence emission, Fmax, as a function of cosolute molality. (D) Elongation time, τel, as a function of cosolute molality. The dotted lines are guides for the eye, and error bars represent standard deviations from an average of 4 kinetic curves.
The changes in the rate constants with concentrations of cosolutes (Ccs) highlighted differences in aggregation mechanisms. While addition of PEGs shows little or no effect on the nucleation constant τlag, this typical time showed a marked, concentration dependent increase in the presence of both sorbitol and glycerol (see Fig. 1B). The peak fluorescence emission Fmax follows a similar trend, showing very little change upon PEG addition, but a concentration dependant increase in the presence of polyols (Fig. 1C). Moreover, at the same concentration, sorbitol has a stronger effect on aggregation than that of glycerol, suggesting a dependence on cosolute size. Surprisingly, there was little change in the elongation rate τel regardless of cosolute identity or concentration (Fig. 1D). This is even more unexpected considering that solution viscosity changes considerably at high cosolute concentrations. However, previous analysis showed that even at very high cosolute concentrations viscosity does not correlate with either τel or τlag, indicating that the changes in kinetics cannot be attributed solely to changes in viscosity.20,21
These results led us to conclude that changes in the mechanism of amyloid aggregation depend not only on the concentration, but also on the specific chemical identity of the cosolute added. It was apparent that the polyols and the PEGs have disparate impacts on the aggregation processes, and that they differently affect each phase of fibril formation.
Kinetic Model Helps Dissect the Aggregation Process
In order to gain further insight into how different stages in the aggregation are modulated by the action of the two cosolute classes, we used a kinetic model to fit the experimental results. Kinetic models can afford further insights into the different possible processes that occur during aggregation. While it is exceedingly difficult to prove specific suggested mechanisms for the process of amyloid formation because of limited information on the process, a wide variety of models have been proposed.29 We focus, therefore, on models that could provide clues to the reasons for the prolonged nucleation time, as well as the increased propensity of the peptide to aggregate in the presence of polyols, but not with PEGs. One kinetic model that satisfies these requirements has been recently suggested by Knowles et al.27 Specifically, in addition to the common nucleation and elongation terms, the model also includes a term describing possible fibril breakage leading to secondary nucleation. It has been shown that this process can significantly contribute to amyloid nucleation and may have major implications to the progression of disease linked with amyloid aggregation.30–32 We slightly modified the kinetic scheme by adding a term describing the dissociation of monomers from formed fibrils; the full kinetic mechanism we used is shown schematically in Figure 2A. This additional term was added to accommodate our experimental observation that, when different cosolutes are present, equal starting monomer concentrations can result in different fibril mass concentrations at steady-state (Fig. 1A). The monomer dissociation step should be distinct from fibril breakage, as it allows reversible monomer detachment from fibrils, with its own typical timescale. This modified scheme resulted, as expected, in kinetic curves that did not reach a state of full aggregation.
Figure 2.
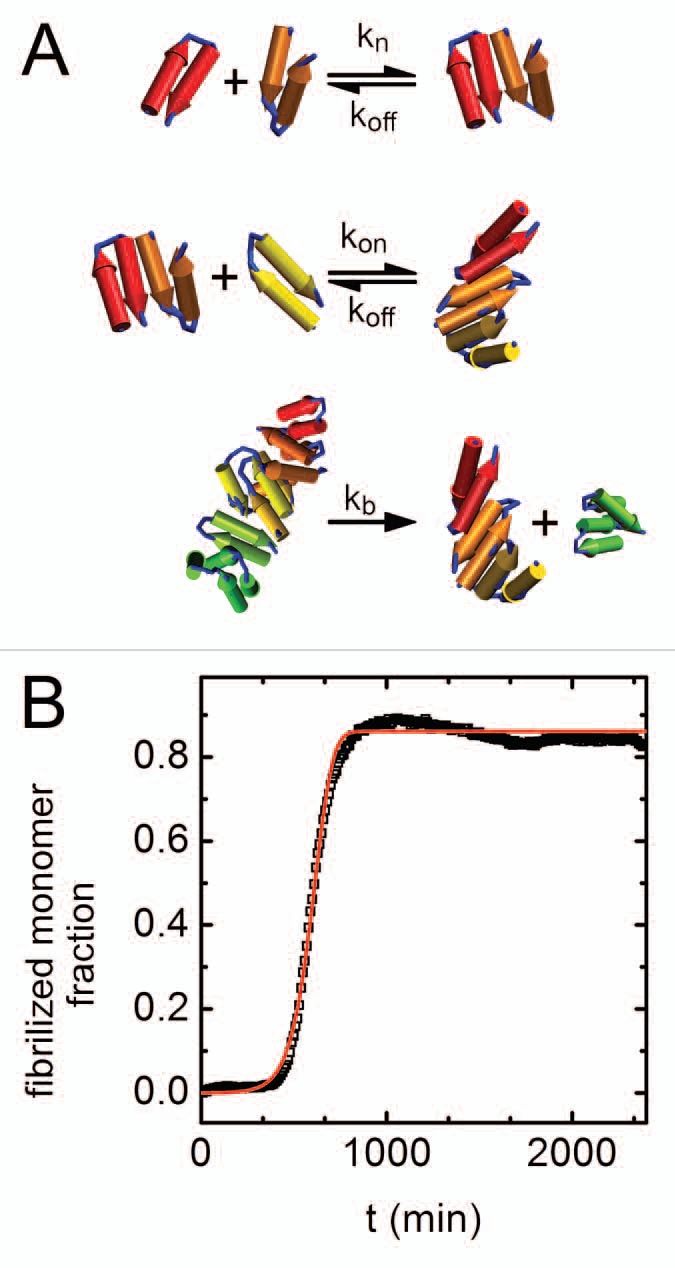
Kinetics of amyloid aggregation. (A) A schematic representation of the steps in the kinetic scheme used to fit the data. The mechanism involves reversible nucleation, reversible monomer-fibril association, and fibril fragmentation steps. (B) Fibrilized monomer fraction as a function of time (black squares) with an overlaid optimized fit to the kinetic model (red line). The fibrilized monomer fraction relates to the fluorescence signal (Fig. 1A) by selecting the highest fluorescence signal in the entire data set (found for the highest concentration of sorbitol) and assuming that value is the fluorescence signal for a fully fibrilized sample.
Two simplified equations can be used to describe the aggregation process in terms of P(t), the concentration of formed fibril at time t, and M(t), the monomer concentration that has been fibrilized:
In these equations m(t) is the free monomer concentration; we determine m(t) through the difference m(t) = m(t0) − M(t), where m(t0) = 10 µM in our experiments. The kinetic rate constants kon and koff represent processes of monomer addition and dissociation respectively, kb is for fibril breakage, and kn is for nucleation. Note that this model assumes that kb remains constant regardless of fibril length, and that fibril-fibril association is neglected. Finally, nc is the nucleation parameter, corresponding to the size and growth dimension of the nucleus.
We next determined the 5 kinetic constants by fitting experimental curves (Fig. 1A) with the model predictions. We used a modified Nelder-Mead optimization algorithm33,34 that imposes upper and lower bounds on parameter values (in-house code implemented on Matlab). In fitting, the average root mean square deviation between model predictions and experimental curves served as the optimization function. After multiple minimization runs we found that for all fitted curves nc ≈ 2. Because the nucleation number is often treated as an integer that represents the number of monomers in an aggregation nucleus,27 we fixed nc = 2 in all subsequent calculations. We find that the entire data set can be well fit by the model with the 4 parameters kon, koff, kb and kn, as seen for typical results in Figure 2B. In the following we examine how the aggregation process described by Equations 1 and 2 is modified due to cosolute addition. We do so by following changes in the fitting parameters with Ccs, the cosolute molal concentrations (Fig. 3). Model kinetic constants are presented in terms of products or ratios representing the various stages of the nucleation-dependent polymerization.
Figure 3.
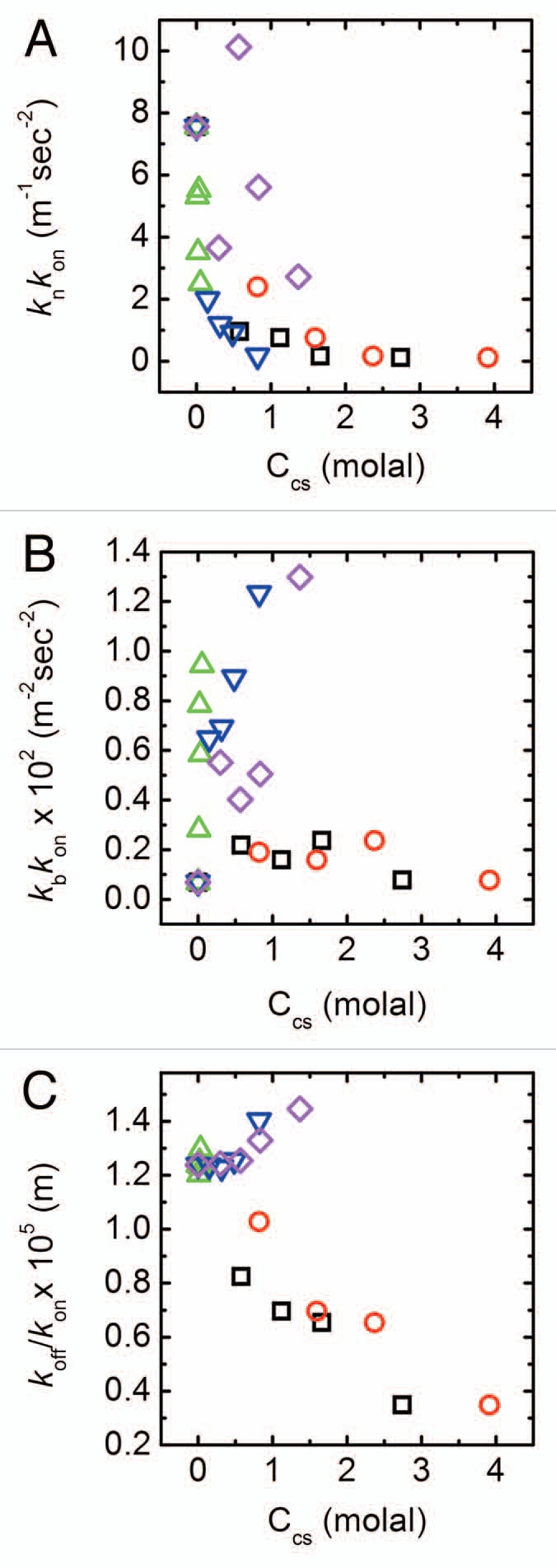
Changes in fibrillation kinetic constants with cosolute concentration. Results are shown in terms of best fitted values for (A) kn kon, which represents rates at the nucleation step, (B) kb kon that is related to the rate of filament population growth and (C) koff/kon representing the quasi-equilibrium constant for monomer-fibril dissociation. Symbols as in Figure 1D. Errors as determined by an average of 3 fits to each curve are <1%.
All Cosolutes Alter the Nucleation Phase
According to the kinetic scheme (Fig. 2A), the formation of new aggregating fibrils in solution may occur through either the spontaneous nucleation of monomers, or through the breaking of a single formed fibril into two shorter fibrils. These different processes can occur simultaneously. Overcoming the initial aggregation barrier at t0, however, is controlled entirely by nucleation. We find that the product knkon, representing the rate at the nucleation step, decreases with cosolute concentration irrespective of its chemical nature (Fig. 3A). At least for low cosolute concentrations, this can be consistent with an entropic or “crowding” model that describes the action of cosolutes primarily in terms of their available free volume in the presence of the nucleating monomers.8,9
Stabilizing cosolutes are generally excluded from the peptide interface, and as cosolute concentrations rise, the free volume available to peptide and cosolute molecules is depleted. By assuming a more compact conformation, the peptide effectively frees up some of the cosolute excluding volume, resulting in an entropic gain. Crowding by excluded cosolutes, therefore, thermodynamically stabilizes more compact peptide states. If nucleation requires an unfolded intermediate, as recent studies suggest,35 the cosolute induced compaction could result in longer nucleation times. Interestingly, we find that for all solutes, regardless of their chemical nature, the nucleation lag time scales with the nucleation constant with an exponent of ∼−0.25 (see Fig. 4A). This behavior highlights the importance of the nucleation rate to the initial stages of aggregation, and indicates that many excluded solutes may be acting through a common scaling law at this stage. This finding could explain the observed retardation of aggregation in the presence of polyols.
Figure 4.
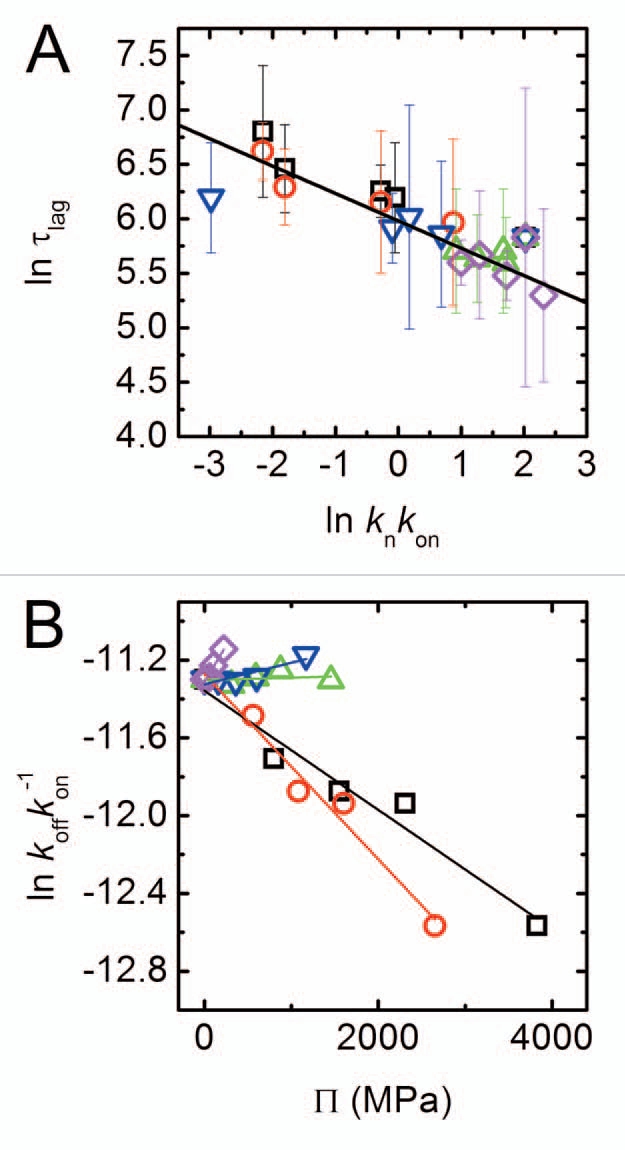
Scaling of kinetic constants. (A) Scaling of nucleation lag time τlag with nucleation rate knkon as determined from a fit to Equations 1 and 2. The scaling law is τlag ∼(knkon)−0.25. (B) Apparent monomer-fibril dissociation constant as a function of osmotic pressure. Lines are linear fits of the data; symbols as in Figure 1D.
Cosolutes may Differently Modulate Fibril Brittleness
The modification of knkon by cosolute concentration is insufficient to account for the fact that in the presence of PEGs the reaction proceeds with no significant change to nucleation rates. Changes in the product kbkon, related to the rate of filament population growth, with cosolute concentration may resolve this apparent conundrum. Figure 3B shows that while sorbitol and glycerol have a marginal effect on kbkon, added PEGs significantly increase the fibril breakage rate with concentration. This change also correlates with the length of the polymer: shorter PEG polymers show a more mild increase in kbkon compared with the longer PEG4000. This effectively higher rate of fibril breakage can act to shorten the nucleation lag time through additional fragmentation of already formed fibrils. Each break effectively creates two aggregating nuclei that can themselves break, as the process of fibril formation is further expedited. This effect may, therefore, counteract the decrease in the rate of the nucleation step.
Recent studies suggest that the morphology of the aggregating polymorph determines the physical properties of amyloid fibrils.30–32 It is not unreasonable to expect that in the presence of PEGs and polyols the solution conditions favor and dictate the formation of alternate peptide conformations that direct the formation of different fibril morphologies. These changes can then modulate fibril brittleness and differently modify the breakage rate, as seen for PEGs vs. polyols. It is tempting to speculate that changes in solution environments wrought by cosolutes may code for the mesoscopic material properties of the forming fibrils.
Monomer Dissociation Rate Decreases in Presence of Polyols
Comparing the effects of cosolutes on the dissociation rate of monomers from formed fibrils reveals additional differences between the two cosolute families (Fig. 3C). We find a significant, concentration dependant decrease in the koff/kon ratio for both sorbitol and glycerol. In contrast, for the smaller range of molal concentrations of the PEG molecules there appears to be a slight increase in this ratio. This correlates well with the fact that the maximal fluorescence Fmax increases with polyol concentrations, while remaining almost constant for PEGs.
Interestingly, koff/kon represents the effective monomers-fibrils equilibrium dissociation constant (Fig. 2A and second kinetic step) and can be related to the effective free energy associated with this process. The variation of the logarithm of this equilibrium constant with solution osmotic pressure, π, is shown in Figure 4B. The slope of the plot describes, through the Wymann linkage,13,17,36 the amount of cosolute excluding waters released in the association process of monomer to fibril. Both polyols show a negative slope, suggesting a smaller preferential hydration of the associated state than the dissociated. In contrast, for PEGs we find a smaller, positive slope that can possibly be interpreted in terms of larger preferential inclusion of PEGs with the dissociated monomer. Indeed, preferential inclusion has been shown to exist for PEGs with other biological molecules such as DNA.37
Implications of Cosolute Identity to Amyloid Fibril Formation
The analysis presented here may allow first hints into the complex effects that cellular solution conditions have on amyloid assembly. Recent studies show that compatible cosolutes do not always act as simple entropic crowders, and that their presence can result in dramatic changes to the way biological molecules associate and interact. For example, polyols have been shown to distort the hydrogen bond network of the aqueous environment.38 This, in turn, alters the preferred conformations solvated peptides adopt, and may also affect the physical properties of the aggregating species in amyloid formation processes. These effects become even more involved when considering possible kinetic mechanisms.
We have found significant disparities in the ways different cosolute families affect amyloid fibril formation. This disparity stresses that cosolutes can elaborately change the aqueous solvent properties. This change, in turn, should have ramifications to the conformational landscape of solvated peptides and proteins. Favoring and preferentially selecting specific states over others impacts the different stages in amyloid aggregation that should sensitively depend on the cosolute chemical identity. These effects should be considered, particularly when discussing peptide and fibril conformations obtained by crystallization in the presence of high cosolute concentrations,39 or under dehydration conditions that correspond to high osmotic pressure.40
By using a kinetic scheme to rationalize experimental results, we found first indications that the nucleation, monomer addition and fibril breakage steps could all serve as crucial points at which cosolutes act. These findings lead us to hypothesize that cosolutes may be altering not only rates of polymerization but also the material properties of the formed fibrils. It will be interesting to resolve the molecular mechanisms that determine how biological molecules react to different environmental conditions presented by various classes of cosolutes. This understanding may ultimately help guide new formulations directed at altering the process of amyloid formation in vivo.
Acknowledgments
We thank our colleagues Assaf Friedler, Dganit Danino, Regina Politi and Lior Ziserman, for their invaluable contributions to our original study on which this manuscript is based. We thank P. Lo Nostro for helpful discussions. DH acknowledges support from the Israel Science Foundation (ISF grants 1011/07, 1012/07). The Fritz Haber Center is supported by the Minerva Foundation, Munich, Germany.
Abbreviations
- PEG
polyethylene glycol
- TEG
triethylene glycol
- ThT
thioflavin T
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: The molecular basis for alzheimer's and parkinson's diseases. Mol Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Serpell LC. Alzheimer's amyloid fibrils: Structure and assembly. Biochim Biophys Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 5.Kodali R, Williams AD, Chemuru S, Wetzel R. Abeta(1–40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picotti P, De Franceschi G, Frare E, Spolaore B, Zambonin M, Chiti F, et al. Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: Insights into the effect of ph on protein fibrillogenesis. J Mol Biol. 2007;367:1237–1245. doi: 10.1016/j.jmb.2007.01.072. [DOI] [PubMed] [Google Scholar]
- 7.De Felice FG, Vieira MNN, Meirelles MNL, Morozova-Roche LA, Dobson CM, Ferreira ST. Formation of amyloid aggregates from human lysozyme and its disease-associated variants using hydrostatic pressure. FASEB J. 2004;18:1099–1101. doi: 10.1096/fj.03-1072fje. [DOI] [PubMed] [Google Scholar]
- 8.Asakura S, Oosawa F. On interaction between two bodies immersed in a solution of macromolecules. J Chem Phys. 1954;22:1255–1256. doi: 10.1063/1.1740347. [DOI] [Google Scholar]
- 9.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: Evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 11.Rösgen J, Pettitt BM, Bolen DW. Protein folding, stability and solvation structure in osmolyte solutions. Biophys J. 2005;89:2988–2997. doi: 10.1529/biophysj.105.067330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochachka PW, Somero GN. Biological adaptation. New York: Oxford University Press; 2002. [Google Scholar]
- 13.Harries D, Rösgen J. A practical guide on how osmolytes modulate macromolecular properties. Methods Cell Biol. 2008;84:679–735. doi: 10.1016/S0091-679X(07)84022-2. [DOI] [PubMed] [Google Scholar]
- 14.Politi R, Harries D. Enthalpically driven peptide stabilization by protective osmolytes. Chem Commun (Camb) 2010;46:6449–6451. doi: 10.1039/c0cc01763a. [DOI] [PubMed] [Google Scholar]
- 15.Cheung MS, Klimov D, Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc Natl Acad Sci USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders AJ, Davis-Searles PR, Allen DL, Pielak GJ, Erie DA. Osmolyte-induced changes in protein conformational equilibria. Biopolymers. 2000;53:293–307. doi: 10.1002/(SICI)1097-0282(20000405)53:4<293::AID-BIP2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 17.Parsegian VA, Rand RP, Rau DC. Osmotic stress, crowding, preferential hydration and binding: A comparison of perspectives. Proc Natl Acad Sci USA. 2000;97:3987–3992. doi: 10.1073/pnas.97.8.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Felitsky DJ, Record MT. Application of the local-bulk partitioning and competitive binding models to interpret preferential interactions of glycine betaine and urea with protein surface†. Biochemistry. 2004;43:9276–9288. doi: 10.1021/bi049862t. [DOI] [PubMed] [Google Scholar]
- 19.Timasheff SN. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc Natl Acad Sci USA. 2002;99:9721–9726. doi: 10.1073/pnas.122225399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukenik S, Politi R, Ziserman L, Danino D, Friedler A, Harries D. Crowding alone cannot account for cosolute effect on amyloid aggregation. PLoS ONE. 2011;6:15608. doi: 10.1371/journal.pone.0015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White DA, Buell AK, Knowles TPJ, Welland ME, Dobson CM. Protein aggregation in crowded environments. J Am Chem Soc. 2010;132:5170–5175. doi: 10.1021/ja909997e. [DOI] [PubMed] [Google Scholar]
- 22.Natalello A, Liu J, Ami D, Doglia SM, de Marco A. The osmolyte betaine promotes protein misfolding and disruption of protein aggregates. Proteins. Structure, Function and Bioinformatics. 2009;75:509–517. doi: 10.1002/prot.22266. [DOI] [PubMed] [Google Scholar]
- 23.Ignatov Z, Gierasch LM. Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc Natl Acad Sci USA. 2006;103:13357–13361. doi: 10.1073/pnas.0603772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, et al. Cyclohexanehexol inhibitors of abeta aggregation prevent and reverse alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 25.Hall D, Minton AP. Macromolecular crowding: Qualitative and semiquantitative successes, quantitative challenges. Biochim Biophys Acta. 2003;1649:127–139. doi: 10.1016/s1570-9639(03)00167-5. [DOI] [PubMed] [Google Scholar]
- 26.Hatters DM, Minton AP, Howlett GJ. Macromolecular crowding accelerates amyloid formation by human apolipoprotein c-ii. J Biol Chem. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 27.Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 28.Maynard AJ, Sharman GJ, Searle MS. Origin of β-hairpin stability in solution: Structural and thermodynamic analysis of the folding of a model peptide supports hydrophobic stabilization in water. J Am Chem Soc. 1998;120:1996–2007. doi: 10.1021/ja9726769. [DOI] [Google Scholar]
- 29.Morris AM, Watzky Ma, Finke RG. Protein aggregation kinetics, mechanism and curve-fitting: A review of the literature. Biochim Biophys Acta. 2009;1794:375–397. doi: 10.1016/j.bbapap.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Xue WF, Hellewell AL, Hewitt EW, Radford SE. Fibril fragmentation in amyloid assembly and cytotoxicity: When size matters. Prion. 2010;4:20. doi: 10.4161/pri.4.1.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue WF, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 33.Box M. A new method of constrained optimization and a comparison with other methods. Comput J. 1965;8:42. [Google Scholar]
- 34.Nelder JA, Mead R. A simplex method for function minimization. Comput J. 1965;7:308. [Google Scholar]
- 35.Calamai M, Chiti F, Dobson CM. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys J. 2005;89:4201–4210. doi: 10.1529/biophysj.105.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wyman J. Linked functions and reciprocal effects in hemoglobin: A second look. Adv Protein Chem. 1964;19:223–286. doi: 10.1016/S0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 37.Knowles DB, Lacroix AS, Deines NF, Shkel I, Record MT. Separation of preferential interaction and excluded volume effects on DNA duplex and hairpin stability. Proc Natl Acad Sci USA. 2011;108:12699–12704. doi: 10.1073/pnas.1103382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Politi R, Sapir L, Harries D. The impact of polyols on water structure in solution: A computational study. J Phys Chem A. 2009;113:7548–7555. doi: 10.1021/jp9010026. [DOI] [PubMed] [Google Scholar]
- 39.Streltsov VA, Varghese JN, Masters CL, Nuttall SD. Crystal structure of the amyloid-β p3 fragment provides a model for oligomer formation in alzheimer's disease. J Neurosci. 2011;31:1419–1426. doi: 10.1523/JNEUROSCI.4259-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]