

Abstract
Human CCRL1 belongs to the family of silent chemokine receptors. This transmembrane protein plays a role in blunting function of chemokines through binding to them. This will attenuate immune responses. Interaction between CCRL1 and CCL21 determines this immune extinction. Thus inhibiting the action of this atypical chemokine seems to stimulate immune responses especially in the case of suppressed and immune deficient conditions. In this study we predicted 3D structure of CCRL1 using comparative modeling and Hiddebn Markov Model algorithm. Final predicted model optimized by Modeller v9.8 and minimized regarding energy level using UCSF chimera candidate version1.5.3. ClasPro webserver was used to find interacting residues between CCRL1 and CCL21. Interacting residues were used as target for chemical inhibitors by simulated docking study. For finding potential inhibitors, library of KEGG compounds screened and 97 obtained chemicals docked against interacting residues between CCRL1- CCL21 and MolDock was used as docking scoring function. Results indicated that Hexadecanal is a potential inhibitor of CCRL1- CCL21 interaction. Inhibition of this interaction will increase intercellular level of CCl21 and interaction between CCL21 and CCR7 causes immune potentiaiton.
Keywords: CCRL1, CCL21, CCR7, Docking, MolDock, Hexadecanal
Background
Chemokines are small proteins fundamentally involved in cell migration, angiogenesis, proliferation and apoptosis [1]. Chemokines play multifaceted roles in numerous physiological and pathological states. Chemokines interact with major seven trans-membrane (7TM) receptors of immune system [2]. Interaction between CCL19 and CCL21 ligands and CCR7as their cognate functional receptor, play a fundamental role in lymph node metastasis in many solid tumors [3]. Human CCRL1 (Accession number 29169303) contains 350 amino acids and is expressed in various tissues as heart, thymus, liver, kidney, spleen, lymph node, placenta, brain, trachea, small intestine, colon, lung, astrocyte, T cell and dendritic cells [4]. CCRL1 belongs to the family of silent chemokine receptors like Duffy antigen receptor of chemokines (DARC). These receptors lack signaling elements which observed in traditional chemokines. This matter enabled them to have a broader spectrum of ligands. These chemokines activate no signaling cascade. Naturally, these proteins dose not induce a string calcium response. These receptors compete in binding to ligands and internalize and degrade ligands. It leads to chemokine depletion and inhibition of cell migration. Chemokine transcytosis which results in ligand transfer across certain barriers is also proposed [5]. It binds and clears the constitutively expressed chemokines considered as homeostatic chemokines compromising CC chemokine ligands CCL19 (ELC), CCL21 (SLC, 6Kine), CCL25 (TECK) and CXCL13 (BCA- 1, BLC). These haemostatic chemokines are involved in CCR7/CCL19 (CCL21), CCR9/CCL25 and CXCR5/CXCL13 axes which participate in the process of migration of T cells. Migration of T cells to the secondary lymphoid organs affords maturity to naive T cells and this is also associated with antigen presentation to these lymphatic organs which consequently arise defensive immune responses. An atypical chemokine receptor as CCRL1 disrupts this chemokine-chemokine receptor axis and consequently reduces defensive immune responses. Down-regulation of CCL19 and CCL21 in vitro and in vivo with consequently has been demonstrated using CCRL1. In the other word, CCRL1 is capable to blunt the actions of CCL19 and CCL21 through disrupting the interaction of these chemokines with their functional receptor. Indeed, xenographs transfected with this atypical chemokine binder indicated significant reduction in CCL25 and CXCL13 concentration with a high clearance capability for post-translational chemokine targeting. In this regard, natural decoy chemokine receptors were observed to potentially block attraction of these chemokines.
Chemokine sequestration using atypical receptors as decoy receptors [6] has a negative regulation on the development of proper immune responses. This chemokine scavenging might be a new therapeutic avenue for stimulation and potentiaiton of immune responses and treatment of immune-deficient diseases states. Therefore, the aim of this study is finding potential agonists for this protein.
Methodology
CCRL1 sequence obtained from NCBI database with Accession number 29169303 and CCL21 crystallographic structure retrieved from Protein Data Bank with PDB number 2L4N.
Prediction of 3-D structure:
In order to predict the 3D structure of CCRL1, three web servers were used. In the first approach we used automated mode of swissmodel. [7] This server predicts query 3D model using comparative modeling method based. This server used template of intracellular loop of the alpha 2A adrenergic receptor (1ho9) Chain: A for prediction of the query. As an alternative approach phyre2 server [8] was used. Any 20 identical amino acids between query and templates is a hit for this server. After finding similar structures, this server predicts query model based on identical folds. Phyre2 output model was predicted based on template of Family a G protein-coupled receptor-like (d1u19a) from family of Rhodopsin-like.The model constructed by this method showed high coverage (97%) and template alignment confidence (100%) values. Also a Hidden Markov Model based server used for modeling of CCRL1. SAM-T08 server [9] first makes HMM cluster from query and then aligns this cluster against a HMM database which is constructed from PDB structures and finally generates model based on HMM alignment. Therefore numbers of misaligned residues decrease by using this algorithm.
Model evaluation:
Q-mean score [10] was used for evaluation of predicted models. Q-mean is a parameter between 0-1. Scores reach near 1 are similar to crystallographic structure. But tranmembrane proteins even in crystallographic structures reach low score. The model Predicted by swissmodel reached score of 0.05 and score of phyre2 was 0.271 and SAM-T08 predicted model reached 0.440. Predicted models by Phyre2 and swissmodel did not cover all of query residues while in the SAM-T08 output all of 350 residues were modelled. Because of reaching highest score in Q-mean and complete query coverage, the SAM-T08 output was chosen for further study (Figure 1).
Figure 1.
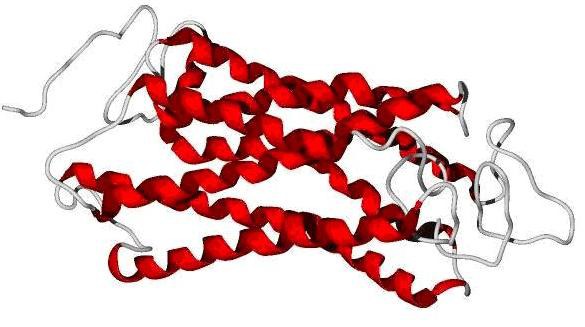
SAM-T08 predicted model of CCRL1
Model optimization:
Modeller v9.8 [11] was used for optimization of SAM-T08 predicted model. Optimization was performed in 10 steps. Also 100 steps of structure minimization were performed using UCSF chimera candidate version1.5.3. During this step the date interval was 10 and step size was set on 0.02.
Virtual screening:
Using FindSite server, the KEGG compounds library was screened for potentially binding ligands 97 poten ligands were obtained. These ligands were used for further simulated docking study in order to find best inhibitor structure.
Molecular docking study:
molegro virtual docker (MVD) 2011.4.3.0 used for computer simulated docking study. Before initiation of the docking operation, protein and ligand structures were prepared using MVD. For this purpose charges assigned to the model of protein and ligands structures and flexible torsions in ligands were detected by this software. This approach also identified probable missing explicit hydrogens and assigned possible missing bonds. Side chain minimization of the derived CCRL1 and CCL21 models was performed during which only torsion angles in the side chains of model were modified and other properties (bond lengths & backbone atom positions) were kept unchanged.
Discussion
Prediction of interacting residues between CCRL1 and CCL21:
Binding CCL21 to CCRL1 causes activation of CCRL1 inducing intracellular signal transduction and decreasingm the number of free CCL21 molecules. As the result formation of the CCL21- CCR7 complex which is an immune activator is decreased. This cause immune extinction in masked immune diseases. Thus, inhibition of the interaction between CCRL1-CCL21 causes formation of a CCL21-CCR7 complex and activation of the immune response. For preventing this interaction, interactive residues must be defined. To do this we used ClasPro [12], a rigid body based protein-protein docking webserver. In the output data, interaction of CCL21 with N-terminal of CCRL1 with least energy level (-979.9 in scale of Weighted Score) was selected and by using ligand scout software we detected that residues number 138, 142, 143, 144 and 226 are interacting residues of CCRL1.
Molecular docking study:
MolDock score [13] with a grid resolution of 0.30 Ȃ used as scoring function for docking. Internal electrostatic interaction and hydrogen bond between ligand and protein were permitted. MolDock SE was used as the docking algorithm and ten runs for ligands were carried out. After docking, energy minimization and optimization of hydrogen bonds performed. The energy threshold was 100.00 and similar poses were ignored. Docking radius was defined to cover amino acids positions 138, 142, 143, 144 and 226 which were predicted as interactive residues with CCL21. Using these docking radius results finding a chemical compound which can bind exactly in the interaction place and act as a potential inhibitor of interaction between CCRL1-CCL21. Docking results evaluated based on MolDock and Re-ranking score. Re-rank score is estimation for interaction. For the defined docking radius in Nterminal of CCRL1, the best pose derived from MolDock score was -142.991with Reranking score equal to -70.069. Table 1 (see supplementary material) describes energy level of five top pose of best inhibitor structure. Also 2D and 3D structure of the ligand is depicted in (Figure 2).
Figure 2.
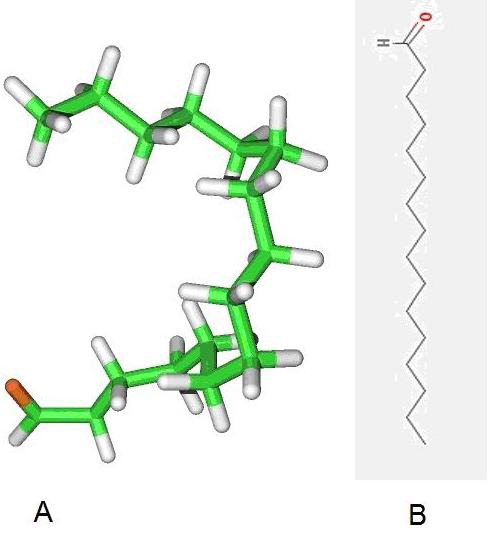
(A) 2D and (B) 3D structure of Hexadecanal
Identification of inhibitor structure:
PubChem database was used for identification of the inhibitor structure and Hexadecanal found as our query. Interaction between CCRL1 and Hexadecanal is depicted in (Figure 3). Hexadecanal (drug bank: DB03381) with chemical formula C16H32O is and aldehyde which drives from degradation of sphinganine. As a therapeutic application of hexadecanal, it has been reported that injection of this chemical can prevent growth of goiter in rat. [14] After increasing the intracellular level of hexadecanal, fatty acyl-CoA reductase oxidizes this chemical to hexadecanoyl-CoA. This product can follow fatty acids oxidation pathway in cell and detoxifies in this pathway. Inhibitory effect of hexadecanal on CCRL1-CCL21 interaction is due to its flexible carbon chain and its aldehyde functional group. It is probable that any similar and synthetic structure can have similar inhibitory effect.
Figure 3.

interaction between CCRL1 and Hexadecanal. This interaction is in the exact place of interacting residues of CCRL1 with CCL21
Conclusion
Based on obtained binding energy level between hexadecanal and CCRL1, it is mostly probable that binding this potential inhibitor to CCRL1 occupies interaction site of CCRL1 and CCL21. Occupation of this place prevents formation of CCRL1 and CCL21 complex. Thus, increased free CCL21 molecules result in its interaction with CCR7. After binding CCR7 to CCL21, this complex can potentiate immune responses through enhanced conveying of immune antigens to secondary lymphoid organs. Then, we suggest that any synthetic and drug-like compound similar to hexadecanal can be used as a potential inhibitor of CCRL1-CCL21 complex for further experimental analysis in order to stimulate immune responses. This might be valuable in the stimulating of attenuated immune system.
Supplementary material
Footnotes
Citation:Behjati et al, Bioinformation 8(7): 336-340 (2012)
References
- 1.A Mantovani, et al. Nat Rev Immunol. 2006;6:907. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- 2.S Thiele, et al. J Biol Chem. 2011;286:37543. doi: 10.1074/jbc.M111.243808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.HE Wiley, et al. J Natl Cancer Inst. 2001;93:1638. [Google Scholar]
- 4.S Krautwald, et al. Immunology. 2004;112:301. doi: 10.1111/j.1365-2567.2004.01859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.G Harlesden, A Rot. Eur J Immunol. 2006;36:1659. doi: 10.1002/eji.200636327. [DOI] [PubMed] [Google Scholar]
- 6.X Cheng, MC Hung. Clin Cancer Res. 2009;15:2951. doi: 10.1158/1078-0432.CCR-09-0141. [DOI] [PubMed] [Google Scholar]
- 7.F Kiefer, et al. Nucleic Acids Res. 2009;37:D387. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LA Kelley, MJ Sternberg. Nat Protoc. 2009;4:363. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 9.K Karplus. Nucleic Acids Res. 2009;37:W492. doi: 10.1093/nar/gkp403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.P Benkert, et al. Proteins. 2008;71:261. [Google Scholar]
- 11.N Eswaret, et al. Current Protocols in Bioinformatics. 2006;15:1. [Google Scholar]
- 12.D Kozakov, et al. Proteins. 2010;78:3124. doi: 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Azevedo WF Jr. Current Drug Targets. 2010;11:327. doi: 10.2174/138945010790711941. [DOI] [PubMed] [Google Scholar]
- 14.L Thomasz, et al. Mol Cell Endocrinal. 2010;317:141. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.