

Abstract
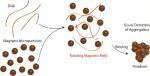
Combining DNA and superparamagnetic beads in a rotating magnetic field produces multiparticle aggregates that are visually striking, and enables label-free optical detection and quantification of DNA at levels in the picogram per microliter range. DNA in biological samples can be quantified directly by simple analysis of optical images of microfluidic wells placed on a magnetic stirrer without DNA purification. Aggregation results from DNA/bead interactions driven either by the presence of a chaotrope (a nonspecific trigger for aggregation) or by hybridization with oligonucleotides on functionalized beads (sequence-specific). This paper demonstrates quantification of DNA with sensitivity comparable to that of the best currently available fluorometric assays. The robustness and sensitivity of the method enable a wide range of applications, illustrated here by counting eukaryotic cells. Using widely available and inexpensive benchtop hardware, the approach provides a highly accessible low-tech microscale alternative to more expensive DNA detection and cell counting techniques.
INTRODUCTION
Determination of the nucleic acid content of biological samples is a keystone in genomic analysis techniques widely utilized in biomedicine and systems biology, medical diagnostics, and forensic science. Most techniques for the sequence-specific quantification of DNA/RNA rely on the polymerase chain reaction (PCR) to amplify target DNA sequences, which are subsequently probed via fluorescence, either directly or afterseparation by electrophoresis.1,2 Almost universally, DNA isolation from biosamples is required before quantification or sequence-specific interrogation. In some cases, the amount of DNA in the purified sample must be quantified prior to PCR to ensure the correct mass of template is available for amplification. Simple, sequence-independent quantification methods include spectrophotometry3 and fluorometry,2 while quantitative PCR (qPCR) allows quantification of specific sequences.4–7 Table 1 provides an overview of the most widely used techniques for DNA detection and quantification, with associated detection limits and implementation challenges. Here we report a new approach to detect, extract, and quantify the amount of DNA in biological samples, based on the visually detectable interaction of DNA with superparamagnetic beads. It is well-established that DNA binds to silica beads in an entropically driven process induced by high concentrations of a chaotrope (e.g., guanidine hydrochloride (GdnHCl)).8
Table 1.
Techniques for Detection and Quantification of DNA
| method | required equipment | limit of detection | sequence-specific | characteristics |
|---|---|---|---|---|
| UV absorbance | quartz optics, UV light source | 1000 pg/ μL2 | no | simple; poor sensitivity;2 susceptible to interferents4 |
| fluorescence | fluorometric optics, fluorescent dyes | 1 pg/ μLl | no | costly; sophisticated hardware |
| quantitative PCR | polymerases, fluorometric optics, thermocycling | 10 copies/50 μL (2 × 10−7 pg/ μL)5 | yes | expensive instrumentation; costly reagents |
Under these or similar conditions, commercial magnetic silica beads (a core of Fe3O4 surrounded by a layer of SiO2) interact with DNA, providing the basis for commercial DNA solid-phase extraction systems9 and the manipulation of beads in microfluidic environments through static magnetic fields.10 The magnetic interaction of paramagnetic beads is controlled by the strength of the external magnetic field. Under static conditions, the induced magnetic dipoles lead to the formation of linear chain assemblies of beads. It has long been recognized that these chains can be stabilized by the adsorption or grafting of polymers.11,12 This has been exploited to quantify the mechanical properties of polymer molecules by careful observation of the interparticle separation as a function of magnetic field strength.13,14 The stability of linear chain aggregates formed by polymer linking in the presence of a static magnetic field has been studied as a function of polymer grafting density, field strength, and salt concentration.15,16
In the presence of a rotating magnetic field (RMF), the induced dipole interaction is partially averaged. At high particle concentrations, this leads to phase transition phenomena into two-dimensional ordered structures, which have been studied closely in recent years.17–19 At lower bead concentrations, RMFs do not lead to aggregation of paramagnetic particles by themselves. However, as we show in the following, even very small concentrations (in the pg/L range) of adsorbing DNA can change the behavior drastically and lead to the formation of readily observable aggregates. This is the basis of the DNA quantification method presented here.
Two different chemical conditions for DNA-induced aggregation are described, both of which create unique, cost-effective pathways for DNA analysis. The sequence-independent approach (chaotrope-driven aggregation, CDA) facilitates a new, sensitive method to quantify DNA directly in crude samples or seamlessly via interface extraction, purification, and quantification in a single process. Hybridization-induced aggregation (HIA) represents a new label-free method for sequence-specific DNA detection and presents paradigm-shifting possibilities for low-cost, low-tech DNA analysis.
RESULTS
Overview of Two Approaches
With CDA, bead aggregation results from DNA binding to the silica surface of the paramagnetic beads in 6 M guanidine (GdnHCl);8,20 as will be demonstrated, the aggregation is sensitive to low concentrations of DNA and insensitive to the presence of proteins. Figure 1A shows 8-μm beads suspended in GdnHCl in a microfluidic well placed in a homogeneous RMF (i.e., the center of a laboratory magnetic stirrer) operating at 2000 rpm. The magnetic field generated by the stir plate was characterized using a Hall sensor. The peak-to-peak amplitude of the field component rotating in the plane of the well was ~30 mT, with a static normal component of 5 mT or less. Field variations over the sample volume are estimated at less than 10% (Figure S1). Without DNA, the beads are distributed homogeneously throughout the well when optimally positioned in a 3D magnetic field (Figure 1A.1); addition of 1 ng of lambda phage DNA (λ-DNA) forms large, stable aggregates within a few seconds (Figure 1A.2). The limit of detection (LOD) with λ-DNA is 62.5 pg of DNA, or a final concentration of ~3 pg/μL, which is still optically recognizable by the algorithm (discussed in a later section). The aggregates rotate visibly in the magnetic field (see Video S1), albeit not necessarily at the same frequency due to drag and other contributing physical factors. We refer to the aggregates as “pinwheels” since the condensed particles often show trailing arms as they rotate (Figure S2). It is critical to note that the type of pinwheel formation shown in Figure 1A.1 is unaffected by the presence of protein (e.g., 100 ng/μL human serum albumin), even when the DNA is presented in a solution containing a protein mass that exceeds that of the DNA by 5 orders of magnitude.
Figure 1.

Chaotrope-driven aggregation (CDA) forms “pinwheels” in a microfluidic chip well. (A) Magnetic silica beads (8 μm) in 20 μL of 6 M GdnHCl in a RMF. (A.1) In the absence of DNA, the beads are rapidly (20 s) distributed throughout the microwell. (A.2) Adding 1 ng of hgDNA and exposure to a RMF for 60 s causes the beads to aggregate into pinwheels that are easily detectable with the naked eye. (A.3) Schematic of a possible mechanism for CDA. (B) Superparamagnetic beads (1 μm) with adducted oligonucleotides specific to the 3′ and 5′ sequences of a DNA target under nonchaotropic conditions. (B.1) In the absence of target DNA, the beads are distributed throughout the microwell. (B.2) Response of beads to the addition of the DNA target in a RMF. (B.3) Schematic of a possible mechanism for hybridization-induced aggregation.
The second approach relies on sequence-specific hybridization rather than nonspecific chaotrope-driven binding. Beads functionalized with two oligonucleotides whose sequences are complementary to part of the sequence associated with a specific DNA target undergo HIA by the target in the presence of RMF. Obviously, in order for sequence complementarily to drive hybridization of the bead-appended oligonucleotides with the target, a non-denaturing, more physiological environment is required. As shown in Figure 1B, the detection modality is the same (aggregation), although the beads take on a slightly different appearance. In the absence of the DNA target, the oligonucleotide-appended beads remain distributed homogeneously throughout the well (Figure 1B.1); addition of a small mass of DNA (6 × 106 copies (0.79 pg/μL DNA) leads to the formation of large, stable aggregates within a few seconds at the requisite annealing temperature (25 °C) (Figure 1B.2).
The results given in Figure 1,B suggest that, with both the CDA and HIA scenarios, the target DNA functions as a “connector” to initiate bead aggregation (Figure S3), although in very different ways (Figure 1C). These involve extremely different chemical conditions, which likely plays a role in the slightly different appearance of the aggregates (Figure 1A2 vs B2). First and foremost, the bead size and shape differ significantly with the two approaches. CDA uses large, irregular-shaped 8-μm beads, while small, spherical 1-μm beads that are avidin-coated are used in HIA (Figure S4). Second, the highly denaturing environment of CDA, provided by a final concentration of 6 M GdnHCl, contrasts significantly the milder physiological conditions of HIA. As described by Melzak,8 the GdnHCl appears to denature most (but not all) of the DNA, and when crude samples are used, it denatures other cellular constituents (e.g., proteins) as well. The effect of this denaturing environment on the appearance of the aggregates may be clarified as more is understood about the mechanism.
It is important to note that diffusion and magnetic interactions alone are not effective at inducing aggregation, as neither DNA nor an RMF alone leads to pinwheel formation (Figure S4). Moreover, application of the RMF leads to localization of fluor-labeled DNA with the particle aggregates when observed via optical imaging (Figure S3), implicating DNA as directly involved in the aggregation. Remarkably, the aggregation effect is optically detectable at hgDNA concentrations less than 1 pg/μL. The interaction of DNA with silica particles is well known and forms the foundation for the vast majority of solid-phase purification methods used to recover DNA from complex samples.21 The distinction here is that the solid phase is not packed into a fixed space but rather manipulated in free suspension by the RMF. As one would expect from packed bed extraction methods, the chaotropic-driven pinwheel effect is reversible, in that the aggregates can be redispersed into solution upon removal of the underlying chaotropic driving force (Figure 2). This is evident in Figure 2 (step 1), where 800 nL of whole blood is lysed with 8 M GdnHCl and added to a poly(methyl methacrylate) (PMMA; PlexiGlass) chip-based microwell containing 12 μL of a 6 M GdnHCl bead solution (final DNA concentration 62.5 pg/μL to 2.5 ng/μL) and the RMF is applied. The RMF rapidly initiates a stable pinwheel formation within 90 s of the direct addition of the blood. This contrasts the addition of the same volume of plasma (lacking DNA but rich in protein (60–83 μg/μL)22), which fails to induce any appreciable aggregation, confirming the inertness of the pinwheel effect to non-DNA constituents in whole blood (discussed with Figure 4). Removal of the chaotrope (Gdn; Figure 2, step 2) followed by the immediate addition of 1× Tris-EDTA (TE; Figure 2, step 3) and reapplication of the RMF redisperses the particles on a time scale similar to that of the aggregation process. The supernatant can then be easily removed with the beads held in place by a magnetic field and used directly as input for PCR amplification (TE; Figure 2, step 4). A 10 μL aliquot of the eluant, which should be rich in DNA and mostly devoid of protein and other cellular components that could inhibit PCR, was added to a PCR master mix containing the appropriate primers for amplification of a 389-bp fragment of the β-globin gene. The result of electrophoretic analysis of that amplified product is given in the lower panel of Figure 2, which shows the dominant presence of a DNA fragment that is sized (relative to the internal standards) as 390 base pairs, well within the 2-bp standard deviation of this sizing method. This result was obtained with eluted DNA at 2.5 ng/μL, but PCR product from 62.5 pg/μL was detectable upon electrophoretic analysis. This demonstrates that the pinwheel-purified DNA from human whole blood is, indeed, free of blood-based PCR inhibitors (e.g., heme from hemoglobin,6 immunoglobulin G4) and the eluted DNA is amenable to effective PCR amplification.
Figure 2.
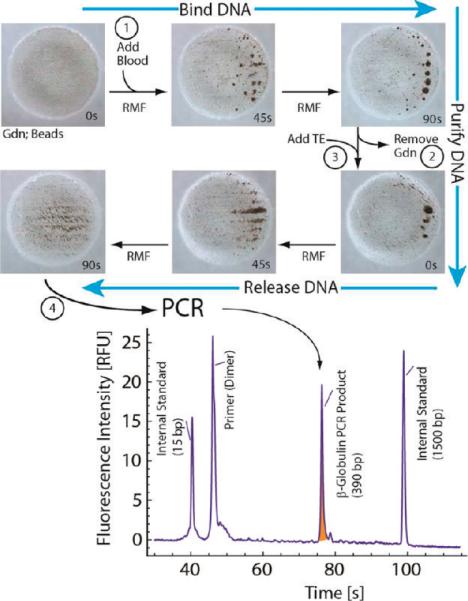
Schematic for pinwheel-based extraction of DNA from whole blood. (Upper) Whole blood (800 nL) added to 12 μL of GdnHCl containing ~6.4 × 105 8-μm magnetic beads and exposed to the RMF for 90 s. The formed pinwheels disaggregate after the chaotrope solution (GdnHCl) is replaced with Tris-EDTA buffer (TE), which leads to release of the DNA within 90 s. (Lower) Electropherogram of PCR-amplified products from an aliquot of elution buffer following bead dispersal. Product (shaded peak) is the 389-bp fragment of the β-globin gene.
Figure 4.

The pinwheel effect allows for quantification of DNA and white blood cell counting. (A) Bar graph showing that pinwheel activity is dominant for the plasma (buffy coat) fraction of whole blood, as expected since the white blood cells contain the DNA (fractions obtained from the same sample by centrifugation). (B) Comparison of cell counting via the Coulter counting method and the present bead-based approach: even when the cell count varies significantly, the accuracy of the method does not.
Quantification of DNA and Cell Counting Using Chaotrope-Driven Aggregation
The CDA of beads by genomic DNA goes beyond providing a simple qualitative method that signals the presence or absence of DNA in a complex sample (Figure 1), or the ability to do microscale purification of PCR-amplifiable DNA from whole blood. The visually distinct aggregates led us to question whether the degree of aggregation scaled with the concentration of DNA in the sample. Figure 3A shows a series of optical images captured by a digital camera following exposure of the same number of beads ((15.9 ± 0.6) × 104 beads) in multiple chip-based wells to a DNA mass ranging from 0 to 80 pg/μL. Images shown for the wells containing 0, 10, 20, and 80 pg of DNA/μL are clearly distinct in their aggregate formation, visually distinguishable, and, thus, easily quantified via optical image processing. The data points generated and plotted in Figure 3A used a high-resolution (12 megapixel) camera to capture images with the threshold gray level set so that dispersed beads and clusters are counted as “dark”, and areas in the image devoid of beads counted as “bright”. The number of dark pixels in the image is then used as a measure of aggregation, with homogeneous dispersion of the beads providing a “% Dark Area” value = 100% (no aggregation), while minimal % Dark Area values corresponded to nearly complete aggregation (a detailed description of the algorithm is given in the Supporting Information, Figures S5–7). The data points in Figure 3A are the average of triplicate image analysis of three separate wells containing the same mass of added DNA. The small standard deviation associated with those mean values demonstrates that a quantitative and precise measure of the DNA content can deciphered from image processing. Note the LOD is better than 10 pg/μL based on the width of the 95% confidence band. Even without optimization of the method, the dynamic range covers 2.5 orders of magnitude.
Figure 3.
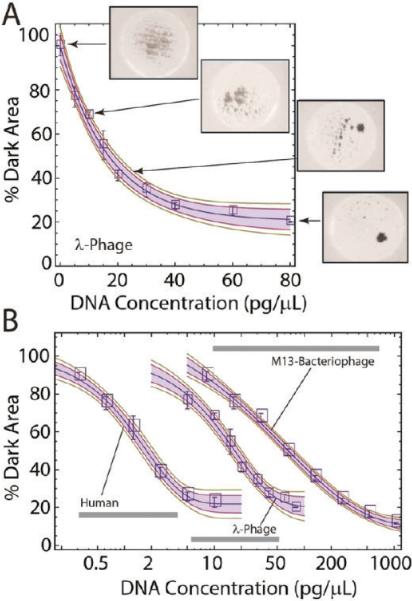
The pinwheel effect allows for quantification of DNA and cell counting. (A) Calibration curve illustrating the relationship between the mass of prepurified DNA and the extent of pinwheel formation (% Dark Area). The solid line represents a stretched exponential calibration function fitted to the data; the shaded area and green lines show the 95% and 90% confidence regions for this fit. (B) Pinwheel calibration curves for DNA from bacteriophage M13 (size 6.4 kbp), lambda phage DNA (48.5 kbp), and DNA in human whole blood (~100 kbp).
The logarithmic plot in Figure 3B compares optical calibration curves generated with DNA from three different sources. Bacteriophage M13, lambda phage, and human whole blood were chosen as DNA sources because their genomic DNA is inherently different in molecular mass (length), at 6.4, 48.5, and ~100 kbp, respectively. The plot suggests that neither the source nor length of DNA affects the basic shape of the aggregation response curve; however, the length has a profound effect and supports that the mass of DNA required for pinwheel formation increases as the fragment length decreases. This is revealing and provides some support for the mechanism illustrated in Figure 1C. Longer DNA strands more effectively aggregate the beads and, thus, do so at lower concentrations. Roughly order-of-magnitude increases in the mass of input DNA are required to achieve the same degree of aggregation as we decrease molecular mass from ~100 kbp to ~49 kbp and then to 6 kbp.
Nucleic acids are fundamental to all biological systems. As an analytical system that extracts and quantifies DNA for pennies of consumables (bead/reagents) using only trivial laboratory hardware, the pinwheel effect opens many avenues for rapid and inexpensive characterization of biological samples. Of paramount importance here is the ability to quantify DNA directly from crude samples using the pinwheel effect and subsequent image analysis. Given that DNA is confined to cells in most intact samples, we queried whether DNA quantification could exploited for enumerating cells in complex samples. Whole blood is considered the universal sample for clinical analysis and is not only a complex biosample but extremely heterogeneous in nature, in terms both of cell-based and non-cell-based constituents. Centrifugation separates whole blood into plasma (cell-free component), “buffy” coat (white cells), and red cell subfractions, with the vast majority of the sample DNA restricted to the buffy coat fraction (nucleated cells). Figure 4A shows the pinwheel effect upon addition of a 1-μL GdnHCl and beads. The buffy coat aliquot, rich in a diverse population of white blood cells, provides a remarkable pinwheel response. This is not surprising, as the vast majority of DNA in blood originates from white cells (25–62 ng/μL).23 Red blood cells are (mostly) anucleated and, therefore, as observed, should have minimal pinwheeling. There is a small mass of DNA in plasma (10–50 pg/μL), but it too shows minimal pinwheeling. This is not unexpected as the fragments are of low molecular mass (<1000 bp)24) and almost an order of magnitude smaller than the M13 DNA in Figure 3B.
Given the localization of the pinwheel effect to the white cell fraction, we evaluated whether DNA mass could represent samples from three patients underwent white cell enumeration using a Coulter Counter and the same samples used for DNA quantification via the pinwheel effect. Using an average mass of DNA per white cell of 6.25 pg,25 the number of white cells present in each sample was calculated for comparison with the Coulter Counter (standard deviation of ~2%). The pinwheel results obtained (triplicate analysis) yield results comparable to those obtained with the more sophisticated Coulter Counting method (Figure 4B). The second patient sample shows a poorer correlation with the Coulter Counter results, but these results from the sample fall outside the optimal range of the standard curve. These results show that the pinwheel assay can be applied to complex biological samples.
Detection with Specificity Using Hybridization-Driven Aggregation
The second method for driving bead aggregation involves DNA–DNA hybridization of target with specific oligonucleotides functionalized to the surface of the beads. Unlike CDA, this approach enables the visual detection of DNA in a sequence-specific manner. The schematic in Figure 5A illustrates this concept where 46- and 50-base oligonucleotides, complementary to the 5' and 3' ends of a 26-base target (connector), are adducted to separate beads. The oligonucleotide sequences were derived from a proximity ligation assay26 (hence, PL beads) and chosen because they effectively hybridize with the target at room temperature. The oligonuceotides were biotinylated and adducted to commercial 1-μm superparamagnetic beads derivatized with avidin; estimated probe coverage is 4.3 × 105 probes/bead. Analogous to what was shown generically in Figure 1B, the beads disperse homogeneously in the absence of the target (Figure 5B.i). However, addition of 6 × 106 copies of the 26-base target immediately leads to bead aggregation in the presence of a RMF (Figure 5B.ii)—this is roughly the detection limit using fluorescent DNA binding reagents.1 In fact, visually discernible pinwheeling is obvious with this target down to 6 × 104 copies (Figure 5B.iii), corresponding to a concentration of ~100 fM (0.0009 pg/μL). Figure 5B.iv shows the sequence-specificity of this phenomenon where little aggregation occurs when a 26-base connector sequence, having similar base composition but noncomplementary sequence, is exposed to the PL5' and PL3' beads, even at the extremely high concentration of 100 nM (6 × 1010 copies, or ~7900 pg/μL). The pinwheel response is observed over a range that spans 7 orders of magnitude (Figure S8).
Figure 5.

Hybridization-induced aggregation (HIA) of oligonucleotide-adducted beads in a RMF. (A) Principle of HIA with the 26-base PL target sequence, biotinylated to 1-μm superparamagnetic beads consisting of a ferromagnetic core and a polystyrene shell. (B) Collage of images illustrating specificity and sensitivity of PL beads to aggregate in a target-dependent manner to 6 × 106 and 6 × 104 copies of the 26-base target with minimal pinwheeling of 6 × 1010 copies of a 26-base nonspecific sequence (jumbled DNA sequence). (C,D) HIA induced by a 500-bp target DNA with images illustrating specificity of (C) PL beads and λ beads and (D) a combination of both to aggregate in a target- and temperature-dependent manner.
It is worthy of note that the PL sequences used in Figure 5B were specifically selected because hybridization was effective at 25 °C; however, typical oligonucleotide–target hybridization occurs at elevated annealing temperatures (55–70 °C). To demonstrate that hybridization-driven aggregation could be temperature-dependent, effective with longer targets, and agnostic with respect to single- or double-stranded targets, a 500-bp fragment (λDNA) of the lambda phage genome was used with oligonucleotide-adducted 1 μm beads (λ beads) designed to anneal with the λ target at 70 °C. As shown in Figure 5C, a positive result is indeed observed at annealing temperature, but not at 25 °C. For the PL beads in the presence of the PL target, the reverse is observed: aggregation occurs at 25 °C, but not at 70 °C. More extensive images involving other temperatures are given in Figure S9. When the two assays are combined in a single well, pinwheels form at both 25 and 70 °C (Figure 5D). From this result, it seems plausible that multiplexed pinwheel assays may be derived that can detect multiple targets in a single well by simply ramping through a select temperature range. The CDA approach is functional for quantification of sample DNA over a range from 1 to 80 pg/μL; for applications such as cell enumeration (shown earlier), reasonably accuracy is essential. While similar accuracy may be achievable with HIA with optimization, the detection response for many applications need only be “digital” or qualitative, i.e., signal the presence or absence of target sequences. Figure 6 shows this effect using the 26-base PL target, demonstrating a clear-cut pinwheeling with 104 copies. Using this and other targets, we have defined 80% Dark Area as the threshold for defining the presence of the target, although the number of copies providing that extent of pinwheeling is target-dependent.
Figure 6.
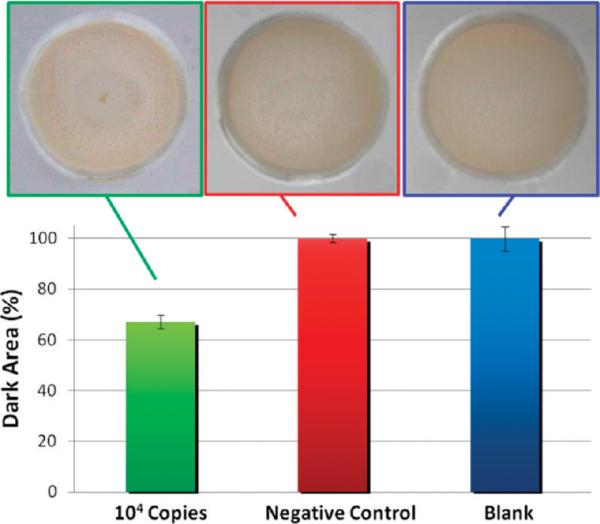
Digital response associated with HIA: semiquantitative HIA, demonstrating a clear digital distinction between the presence of the target sequence at a low concentration, with nonspecific sequences at high concentration, and with no DNA present. (A, left) 104 copies of the 26-base PL target. (B, middle) 1010 copies of the 26-base noncomplementary sequence yielding a % Dark Area similar to (C, right) no DNA present. A total of 4 μL of beads and a low aperture setting were required to generate digital response (hence, the darkness of the image). Each error bar represents the standard deviation associated with n = 4.
DISCUSSION
The sequence-specific detection of DNA through optical observation of magnetic bead aggregation is similar in principle to selective colorimetric detection described by Mirkin and co-workers27 using functionalized gold nanoparticles connected by a linker DNA strand. The aggregation of gold nanoparticles results in a color change due to a red shift in the surface plasmon resonance. The LOD for specific DNA detection has been reported at 0.5 pg/μL,27,28 and for nonspecific DNA detection at 66 pg/μL of MRSA total genomic DNA.29,30 It is noteworthy that the pinwheel system bears some resemblance to Scan-o-metric DNA detection, where a DNA-functionalized gold nanoparticle is linked to a surface with a target DNA linker through hybridization. The reported LOD for this method was ≥1 nM of target DNA (or 8 pg/μL), until the addition of a signal amplification step via silver deposition to decrease the LOD to 50 fM (or 0.0004 pg/μL).31 This illustrates that, with the only surface functionalization being the biotinylation of the beads with the appropriate oligonucleotides (for target specificity), the sensitivity that is comparable or better is obtained with pinwheeling.
The foregoing results clearly demonstrate the usefulness of DNA-induced aggregation of magnetic beads in a rotating magnetic field for DNA detection and quantification. They do not, however, provide quantitative insight into the details of the physics behind the phenomenon. The interaction of paramagnetic beads with adsorbing polymers in the presence of static magnetic fields has been studied quite closely, and this body of knowledge may provide important clues to understanding the pinwheel effect. The induced magnetic dipoles lead to attractive forces between superparamagnetic particles if they are aligned with the magnetic field, and to repulsion if their separation vector is perpendicular to the field. Therefore, the particles tend to form linear chains along the magnetic field lines, with the interparticle spacing controlled by the balance between the magnetic attraction and van der Waals/steric repulsion.32 Magnetic fields in the range of 10 mT are sufficient to produce attractive interactions several orders of magnitude above thermal energy.16 This is enough to keep the assemblies stable in static field, but not to overcome the hydrodynamic drag on the particles in the case of a field rotating at a frequency of several hertz. It is therefore not surprising that no aggregates are observed in a RMF in the absence of DNA. Li, Rogers, and Biswal have carefully studied the stability of colloidal chains of paramagnetic beads linked with DNA as a function of DNA grafting density, chain length, and magnetic field strength.16 In their experiments, however, the magnetic field was applied only for an incubation period, and the effects of permanent DNA linkages formed between the particles were observed after removal of the magnetic field. Nonetheless, some of the trends observed in their work are suggestive in the present context. For example, they found the stability of the particle assemblies to increase with the DNA chain length, which is in agreement with our observations. Clearly, more work needs to be done to provide a quantitative understanding of the pinwheel effect. The effective particle–particle interaction energy as a function of magnetic field strength, rotation frequency, and polymer grafting density needs to be studied systematically. Such experiments are currently underway in our laboratory and will be reported later.
In conclusion, we have shown that superparamagnetic silica beads aggregate in the presence of a rotating magnetic field and even small amounts of double-stranded DNA under chaotropic conditions. The resulting pattern of aggregation is related quantitatively to the concentration of DNA present, providing an inexpensive method for DNA quantification with a LOD that rivals or even surpasses those of conventional, fluorescence-based assays and provides about a 2.5 order of magnitude dynamic range. The effect is highly robust with respect to contamination with proteins and other biomolecules; it can be directly used to quantify the nucleic acid content of whole blood, ultimately representing white blood cell count. Our results also show that a similar aggregation phenomenon occurs under physiological (nonchaotropic) conditions if the beads are decorated with two different oligonucleotide sequences. In this case, the aggregation occurs only if DNA (single- or double-stranded) with a sequence complementary to both of the oligonucleotides attached to the beads is present, with obvious and important immediate applications in clinical settings.
METHODS
Reagents
Dynabeads MyOne Streptavidin C1 and ChargeSwitch superparamagnetic beads were purchased from Invitrogen (Carlsbad, CA). MagneSil superparamagnetic particles were purchased from Promega (Madison, WI). Biotinylated and unfunctionalized oligonucleotides were purchased from Eurofins MWG Operon (Huntsville, AL). Lambda phage genomic DNA (48.5 kb long) from E. coli infected with λCI857Sam7 in storage buffer [10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 1 mM EDTA] was purchased from USB (Cleveland, OH). M13 bacteriophage was obtained from New England Biolabs (Ipswich, MA). Whole blood samples were donated by consenting donors. EvaGreen dye was purchased from Biotium, Inc. (Hayward, CA). Guanidine hydrochloride (GdnHCl), 2-(4-morpholino)ethanesulfonic acid (MES, enzyme grade), hydrochloric acid, sodium hydroxide, acetone, ethanol, Taq DNA polymerase, 10× PCR buffer without MgCl2 (100 mM Tris-HCl, 500 mM KCl, pH 8.3), dNTPs, and MgCl2 were purchased from Fisher (Fair Lawn, NJ). 2-Amino-2-(hydrox-ymethyl)-1,3-propanediol (Trizma base, 99.9%) was purchased from Sigma (St. Louis, MO). All solutions were prepared in Nanopure water (Barnstead/Thermolyne, Dubuque, IA).
Microwell Fabrication
A VersaLASER system 3.50 from Universal Laser Systems (Scottsdale, AZ) was used to fabricate microwells in 1.5-mm-thick PMMA purchased from McMaster-Carr (Santa Fe Springs, CA). Each microwell device was prepared as a 4 × 4 matrix of 5-mm-diameter circular wells on a 4-cm square device. For MagneSil-based experiments, the device was designed using CorelDRAW, and wells were created in the solid 1.5-mm-thick PMMA by rastering the laser at 30% power three times to make wells of ~1 mm depth. For the remaining experiments (HIA), PMMA microwells were designed in AutoCAD, and the laser cut through the entire 1.5-mm-thick PMMA sheet. The layer with holes was thermally bonded to a blank PMMA piece of the same dimensions using established methods,33 creating wells with less surface roughness at the bottom. Microwells were sterilized with either 10% bleach or 2 M hydrochloric acid for 5–10 min and then rinsed with Nanopure water.
Reagent and Sample Preparation
Thirty microliters of stock Magnesil beads was washed once with deionized, distilled water (Nanopure) followed by one wash with GdnHCl solution (8 M, 1×TE, adjusted to pH 6.1 with 100 mM MES) and resuspended in 1 mL of GdnHCl solution to make the suspension, which was further diluted in the individual pinwheel assays. DNA samples were diluted serially with 1× TE buffer (10 mM Tris base, 1 mM EDTA, pH 8.0) to concentrations ranging from 320 to 5 pg/μL, and the DNA solutions were aliquoted and stored at −20 °C until use.
Assay Instrumentation
Images of the microwells were collected by using a T1i DSLR camera with MP-E 65 mm f/2.8 1–5× macro lens purchased from Canon U.S.A., Inc. (Lake Success, NY). A Thermix Stirrer model 120S magnetic stir plate was purchased from Fisher Scientific (Fair Lawn, NJ). A heated stir plate model 97042-642 from VWR (Batavia, IL) was used for experiments at elevated temperature (HIA). A Ledu compact desk magnifier lamp was purchased from Guy Brown Products (Brentwood, TN) and used without optics to provide lighting around the entire sample.
RMF Application
Three microliters of stock MagneSil suspension was added to each microfluidic well, followed by 12 μL of 8 M GdnHCl solution and 5 μL of sample. The mixture was exposed to a RMF generated by a magnetic stirrer rotating at 2000 rpm with a U-shaped bipolar magnet of 2800 G for 5 min. The RMF was turned off to image and restarted for 2 s before imaging again. The pixel resolution of each image was 4752 × 3168, and the field of view was 8.6 mm × 5.7 mm.
Image Processing
Images of magnetic beads with different amounts of lambda phage genomic DNA are shown in Figure S3. As the concentration of DNA increased, the beads started to aggregate, and correspondingly, the number of pixels representing the beads (i.e., % Dark Area) decreased. JPG image files (Figure S3) were processed into binary (Figure S4) with the gray level threshold set by an isodata algorithm34 written in Mathematica software (Supporting Information). For the HIA, the threshold was set in the saturation channel in HSB (hue-saturation-brightness) color space with the isodata algorithm. The number of dark pixels (dark area) was normalized to the negative control without DNA (Figure S5).
Fluorescent Pinwheel
ChargeSwitch superparamagnetic particles (Invitrogen, Inc.) were used to monitor the analogous electrostatic aggregation of DNA at low pH. One microliter of particle suspension was added to 9 μL of 100 mM MES buffer (pH 6.2) with 2 μL of lambda phage genomic DNA (10 ng/μL) and 2 μL of 20× DNA-binding fluorophor (EvaGreen, Biotium, Inc.) for fluorescence microscopy monitoring of pinwheel aggregation (Carl Zeiss, Axio Scope A1). Bright-field and fluorescent images were collected before and after application of the RMF.
Pinwheel-Based Extraction of DNA
Two microliters of whole blood was incubated with 18 μL of 8 M GdnHCl at room temperature for 60 min, during which DNA is expected to be released. The sample was then serially diluted and mixed with 12 μL of Magnesil beads. The final concentrations for pinwheel analysis were 62.5, 625, 1250, and 2500 pg/μL. The aggregates were washed with 20 μL of 6 M GdnHCl and 10 μL of 1× TE, and the DNA was eluted with 13 μL of 1× TE. Ten microliter portions of the eluted DNA at each starting concentration were amplified with PCR in a MyCycler thermal cycler (Bio-Rad Laboratories, Inc., CA), and the products were detected with a Bioanalyzer 2100 instrument (Agilent Technologies, Inc., CA).
Blood Cell Counting
Raw human blood samples were first incubated with GdnHCl solution in 1:10 volume ratio at room temperature for 30 min. The samples were then serially diluted in GdnHCl solution for pinwheel analysis. Dark area values were acquired and compared with the standard curve shown in Figure 3B.
Magnetic Particle Modification
The streptavidin-coated Dynabeads were coupled with the biotinylated oligonucleotides per the manufacturer's protocol. Briefly, 200 μL of vortexed particle suspension was washed three times with 1× binding/washing buffer [5 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, and 1 M NaCl] and incubated with 100 μL of the 1× binding/washing buffer with 0.2 μL of 30 mM biotinylated oligonucleotide solution on a rotisserie for 10 min. The beads were washed a further three times with the binding/washing buffer to remove any unbound oligonucleotides. Finally, the beads were resuspended in 500 μL of 1× PCR buffer [50 mM KCl, 10 mM Tris-HCl (pH 7.5)].
Hybridization-Induced Aggregation Assay
For the room-temperature experiments, 17 μL of 1× PCR buffer was added to the microfluidic well, followed by 1 μL of sample. A RMF (2000 rpm) was applied to the solution, and 1 μL portions of each of detection beads were added (1 μL of the 5′ modified and 1 μL of the 3′ modified). After 5 min, the RMF was removed, and a single picture was taken for each experiment. The temperature-dependent assays used a water bath to heat the 1× PCR buffer to the required temperature. The PMMA microfluidic well was heated on a heated stir plate to maintain the set temperature (45, 70, or 90 °C). The procedure is as above, except experiments were run for 3 min to maintain the desired temperature (within ±5 °C). Following 3 min, the heat began to fall by 3 °C min−1. Sequences of probes are provided in the Supporting Information.
Supplementary Material
Footnotes
The authors declare no competing financial interest.
Supporting Information Supplemental video demonstrating the pinwheel effect for visual detection of DNA with superparamagnetic micro-particles; experimental characterization of the magnetic field; further confirmation that DNA is critical to the formation of pinwheel aggregates; time course of pinwheel formation; steps of DNA quantitation with automated software analysis; photographs of sequence-specific DNA hybridization pinwheel assay and temperature dependence; isodata algorithm written in Mathematica for image thresholding and binarization. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Sassolas A, Leca-Bouvier BD, Blum L. J. Chem. Rev. 2007;108:109. doi: 10.1021/cr0684467. [DOI] [PubMed] [Google Scholar]
- (2).Voet D, Voet JG. Biochemistry. 3rd ed. J. Wiley; Sons: New York: 2004. [Google Scholar]
- (3).Gallagher SR. In Current Protocols in Molecular Biology. John Wiley & Sons, Inc.; New York: 2001. [Google Scholar]
- (4).Al-Soud WA, Jonsson LJ, Radstrom P. J. Clin. Microbiol. 2000;38:345. doi: 10.1128/jcm.38.1.345-350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Izreli S, Pfleiderer C, Lion T. Nucleic Acids Res. 1991;19:6051. doi: 10.1093/nar/19.21.6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K. J. Forensic Sci. 1994;39:362. [PubMed] [Google Scholar]
- (7).Burns M, Valdivia H. Eur. Food Res. Technol. 2008;226:1513. [Google Scholar]
- (8).Melzak KA, Sherwood CS, Turner RFB, Haynes CA. J. Colloid Interface Sci. 1996;181:635. [Google Scholar]
- (9).Otto P. J. Assoc. Lab. Autom. 2002;7:34. [Google Scholar]
- (10).Gijs MAM. Microfluid. Nanofluid. 2004;1:22. [Google Scholar]
- (11).Furst EM, Suzuki C, Fermigier M, Gast AP. Langmuir. 1998;14:7334. [Google Scholar]
- (12).Goubault C, Leal-Calderon F, Viovy J-L, Bibette J. Langmuir. 2005;21:3725. doi: 10.1021/la0471442. [DOI] [PubMed] [Google Scholar]
- (13).Goubault C, Jop P, Fermigier M, Baudry J, Bertrand E, Bibette J. Phys. Rev. Lett. 2003;91:260802. doi: 10.1103/PhysRevLett.91.260802. [DOI] [PubMed] [Google Scholar]
- (14).Koenig A, Hébraud P, Gosse C, Dreyfus R, Baudry J, Bertrand E, Bibette J. Phys. Rev. Lett. 2005;95:128301. doi: 10.1103/PhysRevLett.95.128301. [DOI] [PubMed] [Google Scholar]
- (15).Biswal SL, Gast AP. Phys. Rev. E. 2003;68:021402. doi: 10.1103/PhysRevE.68.021402. [DOI] [PubMed] [Google Scholar]
- (16).Li D, Rogers J, Biswal SL. Langmuir. 2009;25:8944. doi: 10.1021/la900370g. [DOI] [PubMed] [Google Scholar]
- (17).Ahniyaz A, Sakamoto Y, Bergström L. Proc. Natl. Acad. Sci. U.S.A. 2007;104:17570. doi: 10.1073/pnas.0704210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Osterman N, Poberaj I, Dobnikar J, Frenkel D, Ziherl P, Babić D. Phys. Rev. Lett. 2009;103:228301. doi: 10.1103/PhysRevLett.103.228301. [DOI] [PubMed] [Google Scholar]
- (19).Smallenburg F, Dijkstra M. J. Chem. Phys. 2010;132:8. doi: 10.1063/1.3425734. [DOI] [PubMed] [Google Scholar]
- (20).Breadmore MC, Wolfe KA, Arcibal IG, Leung WK, Dickson D, Giordano BC, Power ME, Ferrance JP, Feldman SH, Norris PM, Landers JP. Anal. Chem. 2003;75:1880. doi: 10.1021/ac0204855. [DOI] [PubMed] [Google Scholar]
- (21).Price CW, Leslie DC, Landers JP. Lab Chip. 2009;9:2484. doi: 10.1039/b907652m. [DOI] [PubMed] [Google Scholar]
- (22).Burtis CA, Ashwood ER, editors. Tietz Fundamentals of Clinical Chemistry. 5th ed. W. B. Saunders; Philadelphia: 2001. [Google Scholar]
- (23).Bain BJ. A beginner's guide to blood cells. 2nd ed. Blackwell; Malden, MA: 2004. [Google Scholar]
- (24).Swarup V, Rajeswari MR. FEBS Lett. 2007;581:795. doi: 10.1016/j.febslet.2007.01.051. [DOI] [PubMed] [Google Scholar]
- (25).Butler JM. Forensic DNA typing: Biology, Technology, and Genetics of STR Markers. 2nd ed. Elsevier Academic Press; Burlington, MA: 2005. [Google Scholar]
- (26).Leslie DC, Sohrabi A, Ikonomi P, McKee ML, Landers JP. Electrophoresis. 2010;31:1615. doi: 10.1002/elps.201000008. [DOI] [PubMed] [Google Scholar]
- (27).Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science. 1997;277:1078. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- (28).Reynolds RA, Mirkin CA, Letsinger RL. J. Am. Chem. Soc. 2000;122:3795. [Google Scholar]
- (29).Li H, Rothberg L. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14036. doi: 10.1073/pnas.0406115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Storhoff JJ, Lucas AD, Garimella V, Bao YP, Muller UR. Nat. Biotechnol. 2004;22:883. doi: 10.1038/nbt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Taton TA, Mirkin CA, Letsinger RL. Science. 2000;289:1757. doi: 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- (32).Fermigier M, Gast P, A. J. Magn. Magn. Mater. 1993;122:46. [Google Scholar]
- (33).Nayak N, Yue C, Lam Y, Tan Y. Microsystem Technol. 2010;16:487. [Google Scholar]
- (34).Ridler TW, Calvard S. Systems, Man Cybernetics, IEEE Trans. 1978;8:630. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.