

Abstract
Liquid crystals have a long history of use as materials that respond to external stimuli (e.g., electrical and optical fields). More recently, a series of investigations have reported the design of liquid crystalline materials that undergo ordering transitions in response to a range of biological interactions, including interactions involving proteins, nucleic acids, viruses, bacteria and mammalian cells. A central challenge underlying the design of liquid crystalline materials for such applications is the tailoring of the interface of the materials so as to couple targeted biological interactions to ordering transitions. This review describes recent progress toward design of interfaces of liquid crystalline materials that are suitable for biological applications. Approaches addressed in this review include the use of lipid assemblies, polymeric membranes containing oligopeptides, cationic surfactant-DNA complexes, peptide-amphiphiles, interfacial protein assemblies and multi-layer polymeric films.
Keywords: liquid crystals, biotic-abiotic interfaces, dynamic materials, lipid assemblies, polymeric multilayers, oligopeptides, bio-inspired materials
Introduction
Over the past several decades, a wide range of hard and soft materials have been explored for the design of interfaces that mediate desired interactions with biological systems. Examples of such materials include metal oxides (e.g., TiO21), self-assembled monolayers of organic molecules covalently attached to surfaces,2, 3 Langmuir-Blodgett films,4 grafted polymeric brushes,5, 6 hydrogels,7 and surfaces fabricated with a range of topographies by using micro- and nano-lithographic techniques.8 Surprisingly, although liquid crystalline phases are encountered in a variety of biological systems9-11 (e.g., concentrated DNA12, 13), only recently has the use of synthetic liquid crystals (LCs) as material interfaces to biological systems been explored.14-23 As summarized below, the dynamic and responsive properties of synthetic LCs appear potentially useful in a range of fundamental and applied biological contexts. The realization of this promise, however, requires advances in the interfacial design of liquid crystalline materials, the topic that is the subject of this review.
A wide range of molecules form liquid crystalline phases, including concentrated solutions of amphiphiles,10, 24-26 oligonucleotides,12, 13 rigid polymers dispersed in a solvent,27, 28 and small organic molecules that are typically rich in aromatic ring structures.29 While lyotropic LCs have been explored for their potential application in biological systems,30-32 in this review, we focus on the interfacial engineering of thermotropic liquid crystals such as those that form nematic phases that are largely immiscible with water (Figure 1A). This class of LCs can be used to define interfaces with aqueous phases at which biological interactions can be reported (see below). We begin this review by summarizing some of the key properties of thermotropic LCs that make them a promising class of materials for applications in biological systems:
LC ordering is responsive to interfacial interactions (Figure 1B).14-23 A remarkable property of low molecular weight LCs is their orientational ordering at interfaces. Many past studies of solid-LC interfaces have established that the orientational ordering of LCs is typically controlled by interfacial energetics on the scale of 10−3 - 1 mJ/m2.33-36 A change in the structure of a LC interface (e.g., as the consequence of a protein binding event) that perturbs the interfacial energetics of the LC on this scale can, therefore, potentially trigger an ordering transition in the LC material. Furthermore, because surface-induced ordering of a LC can extend a distance of up to 100 μm from an interface,35, 37, 38 biomolecular interactions at the interface of a LC can lead to ordering transitions in the bulk LC that are easily transduced by using optical or electrical methods.
LCs define interfaces at which molecules exhibit high mobilities (Figure 1C).16 A second key attribute of many LC materials that makes them promising for biological applications is their ability to define interfaces at which molecules exhibit high mobilities. In particular, as detailed below, at interfaces between LCs and aqueous phases, the mobilities exhibited by lipids are comparable to those found in biological membranes.16 The mobile nature of the interface of a LC thus permits both synthetic and biological molecules to reorganize in response to interactions.39 In some cases, the phenomena observed at LC interfaces mimic, in primitive ways, the dynamic properties and associated functionality encountered in biological membranes.
Synthetic LCs exhibit biologically relevant mechanical properties (Figure 1D).40, 41 Because of long-range orientational ordering of molecules, LCs are elastic media, with the introduction of curvature strain (twist, splay and bend) requiring that work be performed on the materials.38 Here we note several important consequences of the mechanical properties of LCs that are relevant to biological applications. First, the elastic energy stored in a strained LC material can be used to direct the interfacial organization of biomolecules,40 thus providing new routes to the design of biomolecular interfaces that can be actively controlled through engineering of strain within the LC. This approach to the design of LC interfaces can be viewed as bio-inspired, as strain within lipid bilayers of both bacterial and mammalian cells has been shown to lead to spatial targeting of lipids and proteins.42 Second, we note that the interaction of living cells with synthetic materials is strongly dependent on the mechanical properties of the materials.43, 44 As described below, it is possible to engineer LC composite materials to possess mechanical properties that influence the behaviors of cells. In the long term, it may be possible to actively control LC mechanical properties to dynamically direct cell behaviors.
Figure 1.

Illustrations of key attributes of liquid crystalline materials relevant to biological applications. (A) Long-range orientational ordering of mesogens in a nematic LC, 4′-pentyl-4-cyanobiphenyl (5CB); (B) Surface-sensitivity of orientational ordering of LCs; (C) LCs define dynamic and reconfigurable interfaces at which molecules possess high mobility; (D) Biologically relevant mechanical properties of LCs, including a responsiveness to forces generated by living mammalian cells.
Motivated by the potential utility of LC materials in biological applications (particularly, reporting biological interactions), below we address progress that has been made towards the design of interfaces of LC materials such that desired interactions are realized between the LC materials and biological systems. We focus this review on interfaces between aqueous phases and thermotropic LCs (Figure 1A) because the presence of the aqueous phase provides an environment that can preserve the activity of biological species (e.g., enzymes or cells). We refer the reader to a series of past studies in which LCs have been used to report the presence and organization of biomolecules arrayed on solid surfaces.34, 45-54 This latter topic lies beyond the scope of this review.
Approaches to Interfacial Design of LC Materials for Biological Applications
Lipid-Decorated LC Material Interfaces
The use of biological lipids to tailor the interfacial properties of synthetic LC materials for biological applications is addressed in this section.15-18, 23, 55-59 The approach is inspired by a prior literature demonstrating that solid surfaces, when decorated with lipids, can influence the ordering of supported films of thermotropic LCs in a manner that is dependent on lipid structure and organization on the solid surface.4, 45, 46 In the context of tailoring the interfacial properties of LC materials in contact with aqueous phases, two general approaches have been explored for delivery of lipid to the LC interface. The first approach involves the spontaneous adsorption of lipid from an aqueous dispersion onto the LC interface,15, 16, 18, 23, 59 and the second involves the transfer of a Langmuir monolayer of lipid prepared at the surface of water onto the LC-aqueous interface using the Langmuir-Schaefer technique.55, 60, 61 Below we review each of these methods for functionalization of LC interfaces, and then illustrate the utility of these tailored LC interfaces for reporting protein binding events.
Brake and coworkers reported that contact of an aqueous dispersion of lipid (in the form of vesicles) with the interface of a thermotropic LC (nematic phase of 5CB, as shown in Figure 1A) resulted in spontaneous formation of a monolayer of lipid at the LC-aqueous interface and an associated ordering transition in the LC.15 Figure 2 illustrates the essential results using the phospholipid l-dilaurylphosphatidylcholine (l-DLPC). Prior to contact of the LC with the aqueous dispersion of vesicles, the orientation of the LC was parallel to the interface (so-called planar anchoring), leading to a bright optical appearance of the LC hosted within a metallic grid (when viewed between crossed polars; Figure 2A and 2B). Upon contact of the LC with a dispersion of lipid, Brake et al. observed the LC to undergo an ordering transition from the planar orientation to the perpendicular orientation (Figure 2C and 2D).16 Quantitative measurements of epifluorescence intensity using fluorophore-labeled lipids (as described below) confirmed that the ordering transition in the LC was associated with formation of a monolayer of l-DLPC at the LC interface (Figure 2B and 2D).16 In addition, fluorescence-recovery after photobleaching (FRAP) experiments demonstrated that the phospholipids at the LC interface possessed diffusion coefficients (~6 ± 2 × 10−12 m2/s) that were comparable to those of lipid bilayers, including biological membranes.16 Overall, these results suggest that the lipid-decorated LC interface can be viewed as a primitive mimic of a biological membrane, where one leaf of the membrane is a lipid monolayer and the other “leaf” is comprised of the LC film with an ordering that is coupled to the first leaf.
Figure 2.

Optical images and schematic illustrations of the anchoring of 5CB at an aqueous interface immediately before and after injection into the aqueous phase of a dispersion of vesicles formed from l-DLPC. The scale bar is 300 μm. (A and B) Optical image and schematic illustration of the anchoring of 5CB before contact with the dispersion of l-DLPC. (C and D) Optical image and schematic illustration of the anchoring of 5CB after 2 h of contact with the aqueous dispersion of l-DLPC. Reproduced with permission from ref 15 (American Association for the Advancement of Science).
Several studies have addressed the origin of the molecular-level coupling between the LC and the lipid that gives rise to ordering transitions of the type shown in Figure 2.15, 29, 45, 46, 62, 63 In particular, at lipid-decorated interfaces of solids, non-linear optical measurements indicate that the mesogens of the LC penetrate into the lipids presented at the interface, leading to a change in the ordering of the tails of the lipid. The central role of the lipid tail-mesogen interaction in dictating the LC ordering is also supported by a series of additional experiments that have explored the influence of the tail structure (e.g., linear versus branched) as well as architecture (conventional amphiphile with single head group versus amphiphile with two head groups) of synthetic surfactants.64, 65 For example, whereas surfactants with linear (unbranched) tails typically cause perpendicular anchoring of LCs (SDS, LDS and L-DBS in Figure 3), the introduction of branching into the tail prevents the LC from assuming that orientation (BR-DBS in Figure 3). In addition, amphiphiles with “bolaform” architectures (FTMA in Figure 3) have been observed to also not cause the perpendicular orientation of LCs that is seen with conventional surfactants such as DTAB (Figure 3).65, 66
Figure 3.
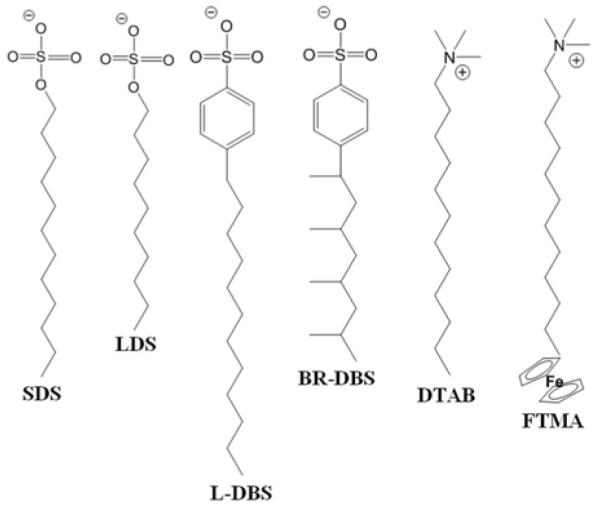
Chemical structures of surfactants; linear sodium dodecyl sulfate (SDS), linear dodecanesulfonate (LDS), linear dodecylbenzenesulfonate (L-DBS), and branched dodecylbenzenesulfonate (BR-DBS), octadecyltrimethylammonium bromide (DTAB), and 11- (undecylferrocenyl) trimethylammoniumbromide (FTMA). Reproduced with permission from ref 64 and 66 (American Chemical Society).
While the molecular structure of the lipid used to decorate the LC is a key factor that influences the ordering of the LC, a number of experiments also demonstrate that the organization of the lipid at the interface plays a central role in determining the ordering the LC. This is nicely illustrated by an experiment in which the interface of a LC was decorated with two lipids with identical tail structures by using the Langmuir-Schaefer technique, as mentioned above.67 As shown in Figure 4, whereas the monolayer of DPPC prepared with a density of 43 Å2/molecule leads to perpendicular ordering of the LC (left polarized light micrograph in Figure 4B; crossed polars), a monolayer containing a mixture of DPPC and DPPE-PEG2000, prepared with an identical density of tails as for DPPC, causes a tilted orientation of the LC (right polarized light micrograph in Figure 4B). This result indicates that the PEG2000 head-group of the DPPE-PEG2000 influences the organization of the lipids at the interface such that it triggers a change in the ordering of the LC relative to that of the DPPC.
Figure 4.

(A) Chemical structure of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DPPE-PEG2000). (B) DPPC induces homeotropic alignment of 5CB at the aqueous-LC interface (left polarized light micrograph), while the same areal density of DPPC/DPPE-PEG2000 induces planar LC alignment (right polarized light micrograph). Grid size is 283 μm. Reproduced with permission from ref 67 (Royal Society of Chemistry).
The key result mentioned above, namely that the organization of lipids at the LC interface can influence LC ordering, underlies one of the potential biological applications of lipid-decorated LC materials, that of reporting protein binding events. The exploration of lipid-decorated LC interfaces for reporting protein-lipid binding events was inspired, in part, by past studies of lipid monolayers at the surface of water in which protein binding from the aqueous sub-phase was demonstrated to trigger changes in the organization of the lipid. Specifically, by using grazing incidence x-ray diffraction, it was shown that specific binding of the protein phospholipase A2 (PLA2) to d-DPPC leads to reorganization of the lipid tails of the d-DPPC within a monolayer at the surface of an aqueous phase.68, 69 In contrast, addition of a protein that does not bind d-DPPC, such as BSA, did not trigger the ordering transition within the d-DPPC lipid monolayer. Recent studies have demonstrated that such protein-induced reorganization of lipid monolayers (caused by specific binding events) can trigger ordering transitions in LC films decorated with the lipids.18 As shown in Figure 5, the homeotropic ordering of nematic 5CB decorated with d-DPPC (center image in Figure 5) is perturbed by the addition of PLA2 to the aqueous phase in the presence of Ca2+, resulting in a bright optical appearance of the LC when viewed between crossed polars (right image in Figure 5). In contrast, when PLA2 was added in the absence of Ca2+ (5mM EDTA was added to scavenge any impurity divalent ions), the LC did not exhibit an ordering transition. The absence of the ordering transition is consistent with the known role of Ca2+ in mediating the specific binding of PLA2 to d-DPPC.71 Further support was obtained for the conclusion that specific binding of PLA2 to the d-DPPC mediated the ordering transition in the LC (in the presence of Ca2+) by adding proteins such as cytochrome-C and BSA, which do not specifically bind d-DPPC and do not trigger an ordering transition in the LC.
Figure 5.

Specific binding of PLA2 to a d-DPPC-decorated LC interface triggers an ordering transition in the LC. Prior to addition of PLA2, the d-DPPC-decorated LC interface exhibits homeotropic anchoring (center). Addition of PLA2 in the presence of Ca2+ triggers an ordering transition in the LC (right). A control experiment in which PLA2 was added in the absence of divalent cations (5 mM EDTA) did not trigger an ordering transition. The LC is nematic 5CB. Grid size is 283 μm. Reproduced with permission from ref 18 (American Chemical Society).
As mentioned in the Introduction to this review, a useful property of LC materials for biological applications is that they can be used to define LC interfaces at which adsorbed proteins and lipids possess lateral mobility (this contrasts to the surfaces of solids, at which adsorbed molecules typically exhibit limited molecular mobility).16, 17, 19, 40, 56, 57, 59-61 Here we note that in biological membranes, many functions of the membrane result from the ability of the proteins and lipids comprising the membrane to reorganize in a manner that is dependent on interactions of these species within the membrane.70, 71 In synthetic systems, it has been observed that interfaces at which lipids exhibit mobility can be used to form organized assemblies of proteins, including protein crystals.19 Because lipids hosted at LC interfaces also possess membrane-like mobility, it has also been observed that protein assemblies form at lipid-decorated LC interfaces, and that the assemblies possess morphologies consistent with those of two-dimensional crystals.16 An example is shown in Figure 6, involving the binding of streptavidin to a biotinylated lipid. Figure 6A shows a fluorescent micrograph of labeled streptavidin at the lipid-decorated interface of the LC. The branched nature of the protein assembly is consistent with prior reports of protein crystallization at interfaces.74 Figure 6B shows that formation of the protein assembly at the interface leads to patterned orientations of the LC. Figure 6C illustrates an important aspect of this phenomenon. For four LC interfaces that were decorated with different densities of biotinylated lipid (samples 1-4), the fluorescence intensity of the labeled streptavidin was recorded for regions of the LC interface that were homeotropic (i.e., unperturbed by the bound streptavidin) or tilted in orientation (perturbed). Inspection of this result reveals that the perturbed regions of the interface of the LC were all decorated by streptavidin at a similar density of protein. In contrast, the unperturbed regions of the LC interface exhibited fluorescent intensities that were either higher or lower than the perturbed regions of the interface. From this result, we make two conclusions. First, the uniform fluorescent intensity of the perturbed regions of the LC is consistent with formation of an organized protein assembly at this interface (e.g., a two-dimensional protein crystal). Second, the result obtained for the unperturbed region of the interface indicates that the LC is not simply reporting density of protein on the interface but rather the organization of the protein at the interface. Overall, these results highlight the opportunity presented by LC materials to report formation of lateral assemblies (including aggregated states) of proteins at interfaces.
Figure 6.
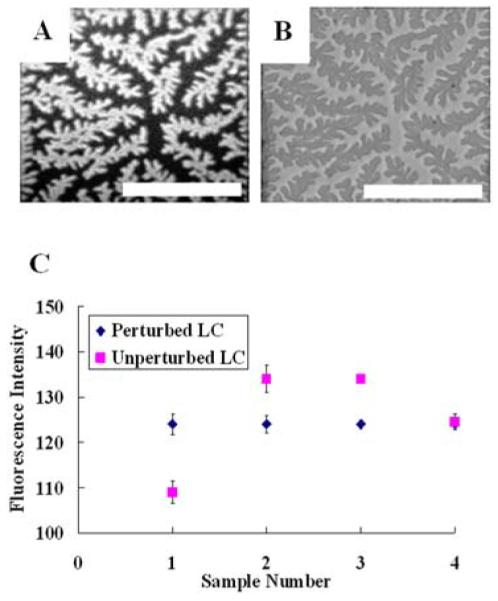
(A) Fluorescent micrograph of labeled streptavidin bound to a biotinylated lipid at an aqueous-LC interface with (B) the corresponding bright field micrograph of the LC texture. The scale bar represents 150 μm. (C) Fluorescent intensities of labeled streptavidin bound to biotinylated lipid presented at an LC interface decorated with differing lipid areal densities. The regions of the LC that displayed homeotropic alignment are indicated as unperturbed and regions that displayed a tilted LC alignment are indicated as perturbed. (A) and (B) reproduced with permission from ref 15 (American Association for the Advancement of Science).
We end this brief review of the use of lipids to tailor interfaces of LCs for biological applications by making two additional points. The first point is to comment that LC materials are not simply passive reporters of lipid organization at interfaces. Several investigations have revealed that the ordering of LCs can also play a role in directing lipid organization at interfaces.39, 67, 72 This point was revealed in a study by Gupta et al., where it was demonstrated that a bulk phase transition from an isotropic to nematic phase was accompanied by lateral phase separation of lipid that decorated the interface of the LC (Figure 7A-D).39 This observation led to the demonstration that the phase behavior of the lipid at the interface of the LC was driven by the elasticity of the LC. This mechanism of phase separation was also demonstrated with monolayers of water-soluble amphiphiles, and was shown to lead to the interesting situation where the geometry of the LC system (e.g., LC film thickness) dictates the interfacial phase behavior of the lipid (Figure 7E-I). Because the elastic energy within a film of LC can be manipulated in a variety of ways (geometry, as illustrated in Figure 7E-I, or, for example, application of an electric field), this result suggests that LC materials may permit the design of interfaces at which the interfacial phase behavior of amphiphiles and other molecular assemblies can be actively manipulated. The second additional point that we make is connected to the generality of the above-described approach for functionalization of interfaces of LC materials. In additional studies reported in the literature that we will not review in the interests of brevity, it has been demonstrated that a range of different types of lipids can be assembled at LC interfaces to control biological interactions, including glycolipids and PEGylated lipids.15, 16, 18, 39, 55, 56, 58-60, 67
Figure 7.
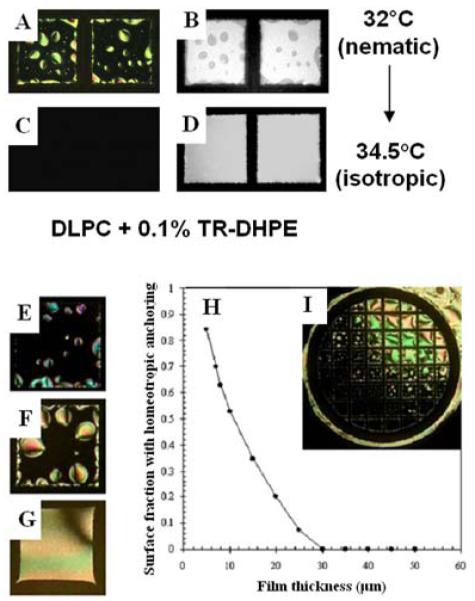
(A, C) Optical images (crossed polarizers) and (B, D) corresponding fluorescent images of a l-DLPC-laden aqueous-5CB interface prepared by the adsorption of lipid for 10 min from a 10 μM unilamellar vesicle solution of l-DLPC doped with 0.1% Texas Red-DPPE. (A, B) Nematic 5CB with a l-DLPC-laden interface. (C, D) Images of the samples in A and B heated to 34 °C (isotropic phase) and equilibrated at that temperature for 2 h. (E-G) Optical images of lipid-decorated films of nematic 5CB with thicknesses of (E) 5, (F) 20, and (G) 40 μm. (H) Prediction of fraction of LC area with homeotropic anchoring, and (I) corresponding LC alignment caused by l-DLPC at the aqueous-LC interface of a LC film with a continuous gradient of thicknesses (thinner at bottom left, thicker at top right). Grid size is 283 μm. Reproduced with permission from ref 39 (American Physical Society).
Interfacial Polymerization of Oligopeptides
The second approach to functionalization of LC materials for biological applications that we describe in this review involves the interfacial polymerization of oligopeptides. The approach was motivated by the observation that peptide-modified materials have potential utility in a range of biological applications, including the control of cellular behaviors, manipulation of protein-peptide interactions and monitoring enzymatic activities.73 One approach that has been demonstrated to permit formation of a peptide-containing film at the interface of a LC exploits the use of the LC as a solvent for a species that reacts with an oligopeptide.17 Specifically, as shown in Figure 8, this approach has been used to cross-link oligopeptides added to an aqueous phase with a reactive cross-linker (adipoyl chloride) added to the nematic phases of 5CB. The oligopeptide used in the study was SNKTRIDEANQRATK. In those studies, it was observed that a micrometer-thick membrane of cross-linked oligopeptide, with a mass fraction of water of ~0.63, was formed at the interface of the LC (Figure 8B and 8C). To illustrate the utility of the oligopeptide-containing polymer films formed at the LC interface for reporting biological interactions, the permeability of the membranes to lipid vesicles (diameters of 36 ± 2 nm) was investigated. The study built from the observations (as described above) that arrival of lipid at the interface of the LC will trigger ordering transitions in the LC.15, 16 Whereas a dispersion of DLPC vesicles triggered an ordering transition in the LC within 10 mins of contact with the LC, the presence of the oligopeptide membrane was observed to slow the initial appearance of the ordering transition to >1 hr. This result is an interesting one because the oligopeptide in the membrane is comprised of a sequence of amino acids that can be cleaved by the enzyme trypsin.
Figure 8.
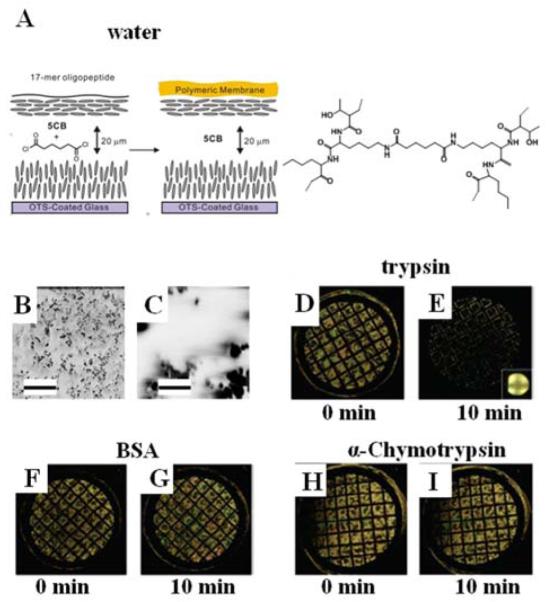
(A) Schematic illustration of the formation of an oligopeptide-containing membrane at the aqueous-LC interface through a cross-linking reaction of a 17-mer oligopeptide (see text for details) with adipoyl chloride. The LC is nematic 5CB. (B-C) SEM images of the oligopeptide membrane obtained at two different magnification levels. The scale bars in B and C are 10 μm and 1 μm respectively. (D-I) Polarized light micrographs of 5CB supporting a cross-linked oligopeptide membrane after incubation with the indicated protein solutions (21 μM, pH 8.0) for 2 h at room temperature, and subsequent contact with an aqueous dispersion of vesicles ofl-DLPC (0.10 mM, pH 8.0) for (D), (F), and (H) for 0 min and (E), (G), and (I) for 10 min at room temperature. Grid size is 283 μm. Reproduced with permission from ref 17 (American Chemical Society).
Trypsin is an enzyme that is known to cleave peptide bonds after (on the C-terminal side of) lysine (K) and argenine (R) if the next residue is not proline (P). It was hypothesized that the barrier properties of the membrane would, therefore, be changed by incubation of the membrane with trypsin. Indeed, it was observed that incubation of the membrane with trypsin resulted in accelerated transport of phospholipid to the interface of the LC, resulting in an ordering transition (Figure 8D and 8E). In contrast, incubation of the membrane with BSA (Figure 8F and 8G) or chymotrypsin (Figure 8H and 8I) did not accelerate the dynamics of the ordering transition upon contact with vesicles of DLPC. Overall, these results demonstrate that the ordering transition of 5CB observed with the trypsin arises from selective processing of the oligopeptide within the polymeric membrane formed at the interface of the LC. LC ordering transitions were observed with trypsin in the low nanomolar range. Overall, because oligopeptides can serve as selective substrates for a range of enzymes (e.g., metalloproteinases), oligopeptide-decorated interfaces of LCs offer the basis of general methods for reporting enzymatic interactions with oligopeptides that result in cleavage of the oligopeptide. We end this section by noting that additional studies involving oligopeptide-decorated LC materials have been reported.19, 50, 74 In particular, it has been demonstrated that enzymatic cleavage of oligopeptides can also be reported by conjugation of oligopeptides to carboxylic acid terminated monolayers of surfactants assembled on interfaces of LCs to form so-called peptide amphiphiles (Figure 9).19 In this case, cleavage of the oligopeptide leads to reorganization of the tails of the surfactant at the interface, which is reported as an ordering transition in the LC. Because the interface is decorated with a monolayer of oligopeptide, ordering transitions at these oligopepide-decorated interfaces were observed to be substantially more sensitive than those reported above using oligopeptide-based membranes at the LC interface.
Figure 9.

Immobilization of a 17-amino-acid oligopeptide at a lipid-laden aqueous-LC interface presenting carboxylic acid groups and tetra(ethylene glycol) groups through activation of the carboxylic acid by a water-soluble carbodiimide and N-hydroxysuccinimide. Reproduced with permission from ref 19 (Wiley-VCH).
Protein-Decorated LC Material Interfaces
A thermotropic LC such as 5CB can be viewed as a structured oil, and thus proteins adsorb to the hydrophobic (and negatively charged at neutral pH) interfaces of LCs, similar to that observed with a range of oil-water interfaces.77 While proteins alone, when adsorbed at LC interfaces, do not lead to pronounced changes in the orientations of LCs (relative to those observed when the LC is in contact with an aqueous phase), the presence of proteins can mediate a range of biological interactions with the interface of the LC that do lead to ordering transitions.
Here we focus on the interactions of living mammalian cells with the interfaces of LC materials that are decorated with proteins. The motivation for these studies is broadly based on the potential utility of LCs as responsive materials that can report interactions with cells (e.g., mechanical interactions or reorganization of protein matrices to which the cells attach) and also the use of LCs to create material environments that can be used to direct cell behaviors.
Prior to discussing experimental results that have demonstrated the feasibility of using protein-decorated LC materials to report cellular interactions, we wish to emphasize that an important enabling discovery in the past decade has been identification of LC materials that are not toxic to living mammalian cells.75 Indeed, early attempts to use LCs such as 5CB as materials on which cells are cultured were unsuccessful due to the toxicity of 5CB. However, as shown in Figure 10A, LCs with differing chemical functionality were found to exhibit substantially different toxicity towards mammalian cells in culture. A particularly interesting result was the finding that LCs containing fluorine-substituted aromatic groups are not measurably toxic to mammalian cells in culture (C Series and TL-205 in Figure 10). While a variety of factors may underlie the differences in the cytotoxicity of the LC materials shown in Figure 10A, a subsequent study of the interactions of 5CB and TL205 with liposomes did reveal that the two LCs differ substantially in their interactions with lipid bilayers (Figure 10B).76 Whereas 5CB was readily solubilized within bilayers of DPPC, leading to changes in the phase behavior of the lipids within the bilayers, TL205 was solubilized only sparingly and was observed to only slightly perturb the phase behavior of the lipid, as measured by DSC (Figure 10B). This result suggests that one factor that may underlie the relative toxicity of 5CB and TL205 is their interaction with cell membranes.
Figure 10.

(A) Relative intensities of fluorescence from CAL-AM added to 3T3 fibroblasts (black bars) and SV-40 HCECs (grey bars) after treatment of the cells with each LC indicated below the plot. The CAL fluorescence indicates the presence of living cells. (B) DSC endotherms of DPPC multilamellar vesicle suspensions following seven days of incubation at 37 °C in the absence or presence of either TL205 or 5CB. Reproduced with permission from ref 75 and ref 76 (Taylor & Francis Group).
Building from the results described above that identified TL205 as non-cytotoxic to cells in culture, protein-decorated interfaces of nematic phases of TL205 have been used to explore how LCs can report interactions with living cells.40 In this context, many past studies have established that extracellular matrix proteins (or fragments of these proteins) are generally required to be immobilized on the interfaces of synthetic materials in order to enable attachment of cells.78 To this end, in the studies described below, a protein mixture called Matrigel (Matrigel is an extract of bovine extracellular matrix proteins) was used to decorate the interface of nematic TL205 (Figure 11). Key issues addressed in the study were whether the protein-decorated interface of the LC would support attachment and proliferation of mammalian cells, and if so, whether interactions of cells with the protein-decorated interface of the LC would lead to ordering transitions within the LC. In initial studies, the cell type was a human embryonic stem cell. This cell type is known to attach and grow on soft substrates, and indeed it was observed that the cells grew and proliferated on the Matrigel-decorated interface of the LC (Figure 11).40 Interestingly, it was also observed that over the duration of a week of culture of the stem cells on the surface of the LC, an orientational ordering transition occurred within some regions of the LC (see polarized light micrographs in Figure 11). By using fluorescently labeled Matrigel, it was determined that the origin of the ordering transition was cell-driven reorganization of the Matrigel on the interface of the LC (see fluorescent micrographs in Figure 11). Specifically, the cells on the surface pulled the Matrigel layer across the surface of the LC. Upon reorganizing the Matrigel on the surface of the LC, adsorption of lipid from the cell culture media triggered an ordering transition in the LC in the areas of the interface of the LC that were not protected by the barrier protein layer. This result provides the first illustration of the potential utility of LCs as materials that can be used to report on the interactions of cells with surfaces.
Figure 11.

(A) Schematic illustration of human embryonic stem cells (hESCs) cultured at planar interfaces between aqueous phases and a thermotropic LC (TL205). (B and C) Polarized light images of TL205 following exposure to an Alexa Fluor-labeled Matrigel solution and subsequent seeding and culture of hESCs for 3 days (B) and 7 days (C). (D and E) Fluorescence images of TL205 following exposure to the Alexa Fluor-labeled Matrigel solution prior to seeding of hESCs (D) and after seeding and culture of hESCs for 3 days (E). Reproduced with permission from ref 40 (Wiley-VCH).
Whereas embryonic stem cells are known to attach to soft materials, many other cell lines (e.g., fibroblast cells) require that substrates possess mechanical moduli in the 1-10 kPa range in order for the cells to attach and grow at the interface of the material.43, 44, 80 For such cells lines, the mechanical properties of low molecular weight LCs (softness) have been reported to prevent attachment of the cells to the interface of the LC. Thus, in order to realize LC materials with potential for use with a range of cell types, LC composite materials that have tunable mechanical properties have been explored. A particularly promising class of LC composite materials is a so-called colloid-in-LC gel. These composite materials, which are comprised of a colloidal network of polymeric microparticles formed within a nematic LC,81-84 have been shown to possess bulk elastic moduli in the 1-10 kPa range, and as such, they exhibit mechanical properties that permit attachment and proliferation of the fibroblasts (Figure 12).41 Interestingly, the LC domains formed between the colloidal networks have been demonstrated to retain their responsiveness to interfacial interactions. Such materials blend the mechanical properties of soft solids with the responsiveness of low molecular weight LCs. In this respect, we note that LC gels formed from hydrogen-bonded molecular networks are also a promising class of responsive LC-based materials with mechanical properties that can be tuned for biological applications.85, 86
Figure 12.

Micrograph of fibroblast cells growing on a colloid-in-LC gel. The cells are stained with calcein-AM (scale bar = 20 μm) and the LC used to form the gel was E7; inset: corresponding polarized light image showing bright nematic LC domains confined by dark boundaries formed by colloids. Reproduced with permission from ref 41 (Wiley-VCH).
Functionalized Mesogens
All of the LCs described above were developed originally with an eye to optimization of dielectric and optical properties of LCs for use in displays. In contrast, only recently have the first designs of mesogens been reported with chemical functionality aimed at tailoring LC interfacial properties relevant to biological applications.79 Many past studies have reported that polyethyleneglycol (PEG)-decorated interfaces of materials exhibit low levels of non-specific adsorption of proteins, relative to those observed at charged or hydrophobic surfaces.3, 87, 88 This observation inspired the exploration of the interfacial properties of LCs that are “PEGylated”. Specifically, the mesogen shown in Figure 13A (EG4-LC) was investigated in mixtures with nematic 5CB.79 The investigation led to two key conclusions. First, the hydrophilic nature of the PEGylated mesogen led to the partitioning of the mesogen from the bulk of the LC to the aqueous interface. This modified the interfacial properties of the LC, specifically lowering the non-specific adsorption of proteins at the interface (Figure 13B). Second, the partitioning of the PEGylated mesogen to the LC resulted in an LC ordering transition. Whereas the LC free of PEGylated mesogen exhibited a parallel orientation at an aqueous interface, the PEGylated LC exhibited a perpendicular orientation. While this example is a relatively simple one, it does serve to illustrate the largely unexplored opportunity that exists to design mesogens with chemical functionality that is relevant to biological applications of LCs. For example, conjugation of biological binding groups to mesogens would likely provide a general and facile means to present the binding groups at the interfaces of LCs. Here we note that a synthetic lyotropic LC was recently reported in which a biological epitope was engineered into the mesogen.90
Figure 13.

(A) Molecular structure of the PEGylated mesogen EG4-LC. (B) Fluorescence intensities of fluorescently labeled BSA adsorbed at aqueous interfaces of either 5CB, octadecyltrichlorosilane, 1% EG4-LC in 5CB, 2% EG4-LC in 5CB, 5% EG4-LC in 5CB, 10% EG4-LC in 5CB, or PEG-treated glass. Each interface was contacted with 10 mM fluorescently labeled BSA (in phosphate buffer pH 7.4) for 2 h. Reproduced with permission from ref 79 (Wiley-VCH).
Interfacial Surfactant-DNA Assemblies
As discussed at the outset of this review, biological lipids provide a general and versatile way to design interfacial properties of LCs relevant to biological applications. Here we describe an example that illustrates how synthetic amphiphiles can also be used to tailor LC interfacial properties, particularly in the context of formation of interfacial complexes with oligonucleotides. As reported by Schwartz and coworkers, hybridization of oligonucleotides with complementary sequences at LC interfaces decorated with cationic amphiphiles can lead to LC ordering transitions.20 The experimental system explored by Schwartz et al. comprised a nematic LC into which octadecyltrimethylammonium bromide (OTAB) was dissolved. Upon contact of the LC/amphiphile mixture with an aqueous phase, perpendicular ordering of the LC was observed, consistent with adsorption of OTAB at the interface. The introduction of single-stranded DNA (ss-DNA) into the aqueous solution, however, resulted in association of the ss-DNA with the OTAB at the LC interface, and a concurrent ordering transition in the LC. Subsequent exposure of the ss-DNA/OTAB interfacial assembly to a complementary oligonucleotide sequence triggered the nucleation and growth of regions of the LC with a perpendicular orientation (Figure 14). The regions of the LC interface that underwent the LC ordering transition were con-firmed to coincide with locations at which oligonucleotides had undergone hybridization to form double-stranded DNA. These studies are particularly interesting in light of recent studies that have demonstrated that the nanostructure of complexes formed between ss-DNA differ from ds-DNA. For example, by using small angle x-ray scattering, it was demonstrated that CTAB complexes with ss-oligonucleotides possess a lamellar nanostructure whereas complexes of CTAB and ds-oligonucleotides possess a hexagonal nanostructure.91 The results reported by Schwartz et al. at the interface of a LC suggest that differences in the organization (nanostructure) of complexes formed by OTAB and ds-DNA or ss-DNA can give rise to distinct orientations of LCs. Finally, we note that it was also demonstrated that introduction of non-complementary oligonucleotides did not trigger an ordering transition in the LC, thus providing evidence that interfaces of LCs decorated with cationic amphiphiles can be used to probe the sequence-dependent interactions of DNA.
Figure 14.
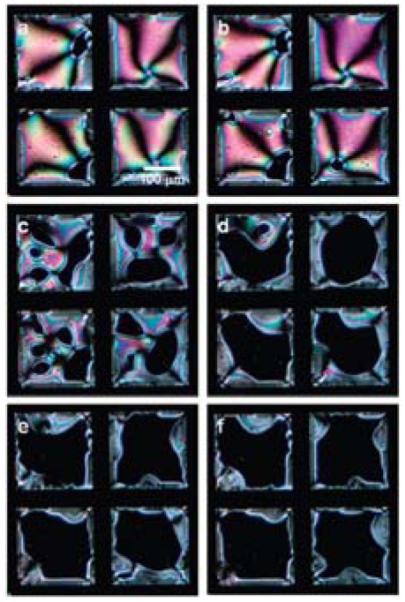
Polarized light micrographs of ordering transitions induced in a film of nematic E7 by hybridization of oligomers of DNA at aqueous interfaces of the LC. The LC contained a cationic surfactant and the LC was contacted with a 16 base-pair “probe” prior to incubation with the complimentary sequence (target DNA). The amounts of target DNA were (A) 0, (B) 100, (C) 200, (D) 300, (E) 400, and (F) 500 fmol. Grid size is 205 μm. Reproduced with permission from ref 20 (American Chemical Society).
Multilayer Polymer Films
The final approach to the design of LC interfaces for biological applications that we address in this review revolves around the use of polyelectrolyte multilayers (PEMs). This approach is a promising one because a large number of prior studies have demonstrated that PEMs formed on the surfaces of solids can be used to design properties relevant to a range of biological contexts.93 Recently, it has been demonstrated that PEMs can also be formed at the interfaces of LCs (Figure 15A).89 Because LC interfaces are more mobile and deformable relative to (rigid) solid surfaces, the first studies of LCs and PEMs focused on establishing the feasibility of fabricating PEMs on LCs, and determining if the LCs influenced the growth of the PEMs (relative to that observed on solids). Initial measurements of the growth of PEMs formed from poly(allylamine hydrochloride) (PAH; Figure 15B) and poly(styrenesulfonate) (PSS) at the aqueous-LC interface revealed growth characteristics similar to those measured at both hydrophobic and hydrophilic interfaces of solids. In contrast, however, the growth of PEMs from PAH and poly(acrylic acid) (PAA) at the aqueous-LC interface was found to differ substantially from the solids investigated. For example, in comparison to the growth of PEMs of PAH/PAA at the surface of glass (hydrophilic, charged surface), a higher rate of growth was observed at the aqueous-LC interface (Figure 15C). Subsequent imaging of the interface of the LC decorated with PEMs formed from PAH/PAA revealed an inhomogeneous and grainy appearance of the LC interface. This appearance was caused by a roughening of the LC interface. The observation suggests that the observed higher rates of growth of the PEMs on the LCs as compared to solid surfaces, is likely due to roughening of the deformable LC interface.
Figure 15.

(A) Schematic illustration of a PEM formed at the interface between an aqueous phase and a thermotropic LC. (B) Structures of the molecules used to form PEMs. (C) Fluorescence intensity during growth of PEMs from FITC-PAH/PAA either at the aqueous-5CB interface or glass-aqueous interface. Reproduced with permission from ref 89 (American Chemical Society). (D) Amphiphilic polyamine used in studies of polyelectrolyte formation with PSS.
A second significant conclusion of the study reported above was that pH-induced changes in the organization of PEMs formed from PAA and PAH at LC interfaces lead to changes in the ordering of the LCs.95 The coupling between formation of polyelectrolyte complexes and LC ordering was subsequently explored in a system comprised of an amphiphilic polyamine and PSS (Figure 15D).67 The amphiphilic design of the polycation achieved a strong coupling of the LC order to polyelectrolyte complexation. This result suggests that hydrophobically-modified polyelectrolytes represent a particularly useful class of polyelectrolytes for tailoring interfaces of LCs.
Whereas the studies described above involved micrometer-thick films of LCs, we note that several studies have recently demonstrated that polyelectrolytes can be used to manipulate the interfacial properties of LC droplets.92, 94-96 In one approach, dispersions of LC droplets were sequentially contacted with polyanions and polycations to encapsulate the LC droplets within PEMs.94 In a second approach, PEMs were fabricated on the surface of silica template particles (Figure 16A and 16B).92, 95 The silica template particles were subsequently dissolved by treatment with HF (Figure 16C). The resulting empty capsules formed by the PEMs were filled with LC (Figure 16D). In the interests of brevity, we refer the reader to other publications for details of experimental procedures.56, 92, 95, 96 Here we emphasize, however, that two key attributes of PEM-decorated LC droplets prepared by the second method above is that (i) the size of the LC droplets can be manipulated by the size of the silica template particle, and (ii) the interfacial properties of the LC droplets can be manipulated via changes in the composition of the PEM. These two attributes have provided an opportunity to understand how confinement of LCs within droplets can be used to tune the properties of LC systems for use in biological applications. Below we illustrate this opportunity by three examples. The first example addresses the effect of the droplet size on LC ordering.92 Thermodynamic arguments reported in the past for micrometer-sized LC droplets predicted that the orientation-dependent interfacial energy scales with the square of the droplet radius (~WR2, where W is the anchoring strength coefficient) whereas the bulk elastic energy of the LC droplet scales linearly with droplet radius (~KR, where K is the elastic constant of the LC).97-99 These thermodynamic considerations lead to the prediction that LC droplets having size R << K/W will avoid spatial variation of the orientation of the LC within the droplet (n(r) = constant, where n is so-called director of the LC) (Figure 16E and 16F). To test this prediction, LC droplets of different sizes, but with identical surface chemistry, were synthesized by using the above described procedure that employs a silica template. The results (Figure 16G to 16P) revealed that the ordering of LCs within droplets with constant interfacial chemistry changes with decreasing droplet size from bipolar (Figure 16I) to preradial (Figure 16N) and then to a radial ordering (Figure 16Q). The experimental observation of radial ordering in the smallest LC droplets was unexpected in light of the above-described prediction of a uniform LC orientation within small droplets. A refined analysis of the elastic energy of LC droplets that incorporated the energetic effects of saddle-splay (K24) was found to be consistent with the experimental observation of radial ordering of the LC. A key conclusion is that uniform ordering of LC within droplets is not observed in the limit of small droplet size. More broadly, by tuning the size of LC droplets at constant interfacial chemistry, these results demonstrate the subtle balance between bulk and surface energetics that controls the ordering of LC within droplets.
Figure 16.

(A-D) Preparation of LC droplets of predetermined sizes within polymeric multilayer shells. Polymeric shells were prepared by sequential deposition of PSS and PAH onto silica templates (B) and subsequent etching of the silica (C). The resulting polymeric shells were then filled with LCs (D). (E) Bipolar and (F) homogeneous director configurations. (G, J, L, O) Polarized and (H,K, M, P) bright-field optical micrographs of polymer-encapsulated 5CB droplets with (G,H) diameters of 8.0 ± 0.2 μm and bipolar LC ordering, (J, K) diameters of 1.0 ± 0.2 μm and preradial LC ordering (J and K show the end on views of the preradial ordering whereas L and M show side views), and (O, P) diameters of 0.70 ± 0.08 μm and radial LC ordering. Point defects in the LCs are indicated by white arrows. Cartoons in I, N, and Q show bipolar, preradial, and radial ordering of the LC droplets, respectively. The scale bars are 2 μm for G, H, and J-M and 1 μm for O and P. Reproduced with permission from ref 92 (Wiley-VCH).
The second example addresses the impact of the size of LC droplets on their response to interfacial adsorbates.56 To determine if manipulation of the size of LC droplets might provide the basis of a simple and general method to tune LC ordering transitions triggered by interfacial adsorbates, the bulk concentration of SDS needed to cause radial ordering of LC droplets as a function of droplet size was investigated. Significantly, the concentration of SDS that triggered radial ordering of the LC decreased continuously with a decrease in droplet size (Figure 17A). Overall, these results lead to the conclusion that control of LC droplet size in the micrometer range does allow the response of LCs to interfacial adsorbates to be tuned. This capability holds particular promise as a means to control the sensitivity and dynamic range of LC-based chemical and biological sensors. Such promise has recently been demonstrated in a study in which micrometer-size droplets of LCs (without PEMs) were shown to permit detection of bacterial lipids in the pg/ml concentration range (Figure 17B).57
Figure 17.

(A) Concentration of SDS needed to drive radial ordering of LC droplets of the indicated size. The LC was nematic 5CB. (B) Percentage of LC droplets (diameters of 4-8 μm) in aqueous solution exhibiting a radial configuration, plotted as a function of endotoxin (LPS) concentration. N indicates the number of independent experiments, and n indicates the total number of LC emulsion droplets that were analyzed. Reproduced with permission from ref 92 (Wiley-VCH) and ref 57 (American Association for the Advancement of Science).
Conclusion
This brief review has summarized a range of approaches that lead to LC materials with properties that are potentially suitable for biological applications. A central message of this review is that the responsive nature of LC materials provides new opportunities to design material interfaces that can report targeted biological interactions. A particularly intriguing aspect of LC-based approaches for reporting biological interactions is that the LC ordering transitions can provide information on the structure and organization of biological species at interfaces. This contrasts to many existing approaches for reporting biological species, where only the mass of species captured at an interface is reported (not their organization). LC materials, for example, hold promise for reporting the formation of protein and lipid assemblies at interfaces. To realize this promise, a number of key fundamental questions need to be addressed, particularly with respect to how the nano-scale structure of biomolecular interfaces impact the ordering of LCs. Whereas interactions involving the aliphatic tails of biological lipids play a clear role in orienting LCs at interfaces, there is increasing evidence that electrostatic phenomena (e.g., effects of electrical double layers) are also likely important at many LC interfaces decorated with biological molecules.
Several examples presented in this review demonstrate that LC materials also offer opportunities for the realization of material interfaces that can be used to direct and influence the behaviors of biological systems. At the molecular level, it is apparent that elastic stresses generated within thin films of LCs can influence the lateral organization of lipids at the aqueous-LC interface. In addition, there is emerging evidence that lipids can be organized locally within LCs via interactions of the lipids with topological defects within the LC. 57 This area of research defines many fundamental questions regarding how defects might be used to direct self-assembly processes in LCs. Finally, at the cellular level, several examples now exist where the behaviors of cells are influenced by the mechanical properties of LC composites (e.g., colloid-in-LC gels). In the long term, LCs thus offer the opportunity to design materials that can achieve two-way communication with biological systems, both reporting on and directing the behaviors of the biological systems.
ACKNOWLEDGMENT
This work was supported by the NSF under awards DMR-0520527 (MRSEC), DMR 0079983, the ARO (W911NF-10-1-0181), the DOE (DE - SC0004025) and by the National Institutes of Health (CA108467 and CA105730).
REFERENCES
- (1).Bozzini S, Petrini P, Tanzi MC, Zürcher S, Tosatti S. Langmuir. 2009;26:6529–6534. doi: 10.1021/la904066y. [DOI] [PubMed] [Google Scholar]
- (2).Prime KL, Whitesides GM. Science. 1991;252:1164–1167. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- (3).Li LY, Chen SF, Zheng J, Ratner BD, Jiang SY. J. Phys. Chem. B. 2005;109:2934–2941. doi: 10.1021/jp0473321. [DOI] [PubMed] [Google Scholar]
- (4).Kühnau U, Petrov AG, Klose G, Schmiedel H. Phys. Rev. E. 1999;59:578–585. [Google Scholar]
- (5).Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nat. Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- (6).Bhat RR, Chaney BN, Rowley J, Liebmann-Vinson A, Genzer J. Adv. Mater. 2005;17:2802–2807. [Google Scholar]
- (7).Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell JA. Biomacromolecules. 2002;3:710–723. doi: 10.1021/bm015629o. [DOI] [PubMed] [Google Scholar]
- (8).Curtis A, Wilkinson C. Biomaterials. 1997;18:1573–1583. doi: 10.1016/s0142-9612(97)00144-0. [DOI] [PubMed] [Google Scholar]
- (9).Vollrath F, Knight DP. Nature. 2001;410:541–548. doi: 10.1038/35069000. [DOI] [PubMed] [Google Scholar]
- (10).Chapman D. Ann. NY Acad.Sci. 1966;137:745–754. doi: 10.1111/j.1749-6632.1966.tb50196.x. [DOI] [PubMed] [Google Scholar]
- (11).Dogic Z, Fraden S. Langmuir. 2000;16:7820–7824. [Google Scholar]
- (12).Nakata M, Zanchetta G, Chapman BD, Jones CD, Cross JO, Pindak R, Bellini T, Clark NA. Science. 2007;318:1276–1279. doi: 10.1126/science.1143826. [DOI] [PubMed] [Google Scholar]
- (13).Zanchetta G, Nakata M, Buscaglia M, Bellini T, Clark NA. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1111–1117. doi: 10.1073/pnas.0711319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Brake JM, Abbott NL. Langmuir. 2002;18:6101–6109. [Google Scholar]
- (15).Brake JM, Daschner MK, Luk YY, Abbott NL. Science. 2003;302:2094–2097. doi: 10.1126/science.1091749. [DOI] [PubMed] [Google Scholar]
- (16).Brake JM, Daschner MK, Abbott NL. Langmuir. 2005;21:2218–2228. doi: 10.1021/la0482397. [DOI] [PubMed] [Google Scholar]
- (17).Park JS, Teren S, Tepp WH, Beebe DJ, Johnson EA, Abbott NL. Chem. Mater. 2006;18:6147–6151. [Google Scholar]
- (18).Brake JM, Abbott NL. Langmuir. 2007;23:8497–8507. doi: 10.1021/la0634286. [DOI] [PubMed] [Google Scholar]
- (19).Park JS, Abbott NL. Adv. Mater. 2008;20:1185–1190. [Google Scholar]
- (20).Price AD, Schwartz DK. J. Am. Chem. Soc. 2008;130:8188–8194. doi: 10.1021/ja0774055. [DOI] [PubMed] [Google Scholar]
- (21).Birchall LS, Ulijn RV, Webb SJ. ChemComm. 2008:2861–2863. doi: 10.1039/b805321a. [DOI] [PubMed] [Google Scholar]
- (22).Sivakumar S, Wark KL, Gupta JK, Abbott NL, Caruso F. Adv. Funct. Mater. 2009;19:2260–2265. [Google Scholar]
- (23).Hartono D, Qin WJ, Yang KL, Yung LYL. Biomaterials. 2009;30:843–849. doi: 10.1016/j.biomaterials.2008.10.037. [DOI] [PubMed] [Google Scholar]
- (24).Jeffrey GA, Wingert LM. Liq. Cryst. 1992;12:179–202. [Google Scholar]
- (25).Pomerantz WC, Cadwell KD, Hsu YJ, Gellman SH, Abbott NL. Chem. Mater. 2007;19:4436–4441. [Google Scholar]
- (26).Blackmore ES, Tiddy GJT. J. Chem. Soc. Farad. T. 2. 1988;84:1115–1127. [Google Scholar]
- (27).Hentschke R, Herzfeld J. Phys. Rev. A. 1991;44:1148–1155. doi: 10.1103/physreva.44.1148. [DOI] [PubMed] [Google Scholar]
- (28).Cheng SZD, Lee SK, Barley JS, Hsu SLC, Harris FW. Macromolecules. 1991;24:1883–1889. [Google Scholar]
- (29).Cognard J. Mol. Cryst. Liq. Cryst. 1982;1:1–77. [Google Scholar]
- (30).Shiyanovskii SV, Lavrentovich OD, Schneider T, Ishikawa T, Smalyukh II, Woolverton CJ, Niehaus GD, Doane KJ. Mol. Cryst. Liquid Cryst. 2005;434:587–598. doi: 10.1103/PhysRevE.71.020702. [DOI] [PubMed] [Google Scholar]
- (31).Luk YY, Jang CH, Cheng LL, Israel BA, Abbott NL. Chem. Mater. 2005;17:4774–4782. [Google Scholar]
- (32).Van Nelson JA, Kim SR, Abbott NL. Langmuir. 2002;18:5031–5035. [Google Scholar]
- (33).Cadwell KD, Alf ME, Abbott NL. J. Phys. Chem. B. 2006;110:26081–26088. doi: 10.1021/jp063211k. [DOI] [PubMed] [Google Scholar]
- (34).Clare BH, Guzman O, de Pablo JJ, Abbott NL. Langmuir. 2006;22:4654–4659. doi: 10.1021/la0535126. [DOI] [PubMed] [Google Scholar]
- (35).Jerome B. Rep. Prog. Phys. 1991;54:391–451. [Google Scholar]
- (36).Yaroshchuk O, Kravchuk R, Dobrovolskyy A, Qiu L, Lavrentovich OD. Liq. Cryst. 2004;31:859–869. [Google Scholar]
- (37).Collings PJ, Hird M. Introduction to Liquid Crystals. Taylor & Francis Ltd; 1997. [Google Scholar]
- (38).de Gennes PG, Prost J. The Physics of Liquid Crystals. Oxford University Press; London: 1994. [Google Scholar]
- (39).Gupta JK, Meli M-V, Teren S, Abbott NL. Phys. Rev. Lett. 2008;100:048301. doi: 10.1103/PhysRevLett.100.048301. [DOI] [PubMed] [Google Scholar]
- (40).Lockwood NA, Mohr JC, Ji L, Murphy CJ, Palecek SR, de Pablo JJ, Abbott NL. Adv. Funct. Mater. 2006;16:618–624. [Google Scholar]
- (41).Agarwal A, Huang E, Palecek S, Abbott NL. Adv. Mater. 2008;20:4804–4809. [Google Scholar]
- (42).Huang KC, Mukhopadhyay R, Wingreen NS. PLoS Comput. Biol. 2006;2:1357–1364. doi: 10.1371/journal.pcbi.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Georges P, Janmey P. J. Appl. Physiol. 2005;98:1547–1553. doi: 10.1152/japplphysiol.01121.2004. [DOI] [PubMed] [Google Scholar]
- (44).Discher DE, Janmey P, Wang YL. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- (45).Hiltrop K, Stegemeyer H. Ber. Bunsen-Ges. Phys. Chem. Chem. Phys. 1978;82:884–889. [Google Scholar]
- (46).Hiltrop K, Stegemeyer H. Ber. Bunsen-Ges. Phys. Chem. Chem. Phys. 1981;85:582–588. [Google Scholar]
- (47).Skaife JJ, Brake JM, Abbott NL. Langmuir. 2001;17:5448–5457. [Google Scholar]
- (48).Luk YY, Tingey ML, Dickson KA, Raines RT, Abbott NL. J. Am. Chem. Soc. 2004;126:9024–9032. doi: 10.1021/ja0398565. [DOI] [PubMed] [Google Scholar]
- (49).Clare BH, Abbott NL. Langmuir. 2005;21:6451–6461. doi: 10.1021/la050336s. [DOI] [PubMed] [Google Scholar]
- (50).Clare BH, Guzman O, de Pablo JJ, Abbott NL. Langmuir. 2006;22:7776–7782. doi: 10.1021/la0604578. [DOI] [PubMed] [Google Scholar]
- (51).Govindaraju T, Bertics PJ, Raines RT, Abbott NL. J. Am. Chem. Soc. 2007;129:11223–11231. doi: 10.1021/ja073203x. [DOI] [PubMed] [Google Scholar]
- (52).Lowe AM, Bertics PJ, Abbott NL. Anal. Chem. 2008;80:2637–2645. doi: 10.1021/ac800097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lowe AM, Ozer BH, Bai Y, Bertics PJ, Abbott NL. ACS Appl. Mater. Interfaces. 2010;2:722–731. doi: 10.1021/am900753v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chen CH, Yang KL. Langmuir. 2010;26:1427–1430. doi: 10.1021/la9033468. [DOI] [PubMed] [Google Scholar]
- (55).Meli MV, Lin IH, Abbott NL. J. Am. Chem. Soc. 2008;130:4326–4333. doi: 10.1021/ja077379a. [DOI] [PubMed] [Google Scholar]
- (56).Gupta JK, Zimmerman JS, de Pablo JJ, Caruso F, Abbott NL. Langmuir. 2009;25:9016–9024. doi: 10.1021/la900786b. [DOI] [PubMed] [Google Scholar]
- (57).Lin IH, Miller DS, Bertics PJ, Murphy CJ, de Pablo JJ, Abbott NL. Science. 2011;332:1297–1300. doi: 10.1126/science.1195639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).De Tercero MD, Abbott NL. Chem. Eng. Commun. 2009;196:234–251. doi: 10.1080/00986440802290060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lockwood NA, Abbott NL. Curr. Opin. Colloid In. 2005;10:111–120. [Google Scholar]
- (60).Lin IH, Meli MV, Abbott NL. J. Colloid Interface Sci. 2009;336:90–99. doi: 10.1016/j.jcis.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kinsinger MI, Buck ME, Meli MV, Abbott NL, Lynn DM. J. Colloid Interf. Sci. 2009;341:124–135. doi: 10.1016/j.jcis.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Huang JY, Superfine R, Shen YR. Phys. Rev. A. 1990;42:3660. doi: 10.1103/physreva.42.3660. [DOI] [PubMed] [Google Scholar]
- (63).Fletcher PDI, Kang NG, Paunov VN. ChemPhysChem. 2009;10:3046–3053. doi: 10.1002/cphc.200900405. [DOI] [PubMed] [Google Scholar]
- (64).Lockwood NA, de Pablo JJ, Abbott NL. Langmuir. 2005;21:6805–6814. doi: 10.1021/la050231p. [DOI] [PubMed] [Google Scholar]
- (65).Brake JM, Mezera AD, Abbott NL. Langmuir. 2003;19:6436–6442. [Google Scholar]
- (66).Brake JM, Mezera AD, Abbott NL. Langmuir. 2003;19:8629–8637. [Google Scholar]
- (67).Kinsinger MI, Lynn DM, Abbott NL. Soft Matter. 2010;6:4095–4104. [Google Scholar]
- (68).Dahmen-Levison U, Brezesinski G, Moehwald H. Thin Solid Films. 1998:327–329. 616–620. [Google Scholar]
- (69).Reichert A, Ringsdorf H, Wagenknecht A. Biochim. Biophys. Acta. 1992;1106:178–188. doi: 10.1016/0005-2736(92)90237-g. [DOI] [PubMed] [Google Scholar]
- (70).Song XD, Swanson BI. Anal. Chem. 1999;71:2097–2107. doi: 10.1021/ac981145f. [DOI] [PubMed] [Google Scholar]
- (71).Dietrich C, Bagatolli LA, Volovyk ZN, Thompson NL, Levi M, Jacobson K, Gratton E. Biophys. J. 2001;80:1417–1428. doi: 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Gupta JK, Abbott NL. Langmuir. 2009;25:2026–2033. doi: 10.1021/la803475c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Schuler M, Owen GR, Hamilton DW, De Wilde M, Textor M, Brunette DM, Tosatti SGP. Biomaterials. 2006;27:4003–4015. doi: 10.1016/j.biomaterials.2006.03.009. [DOI] [PubMed] [Google Scholar]
- (74).Bi XY, Yang KL. Biosens. Bioelectron. 2010;26:107–111. doi: 10.1016/j.bios.2010.05.018. [DOI] [PubMed] [Google Scholar]
- (75).Luk Y-Y, Campbell SF, Abbott NL, Murphy CJ. Liq. Cryst. 2004;31:611–621. [Google Scholar]
- (76).Lockwood NA, Meli MV, Surjosantoso A, Kim EB, De Pablo JJ, Abbott NL. Liq. Cryst. 2007;34:1387–1396. [Google Scholar]
- (77).Langmuir I, Waugh DF. J. Gen. Physiol. 1938;21:745–55. doi: 10.1085/jgp.21.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Hughes CS, Postovit LM, Lajoie GA. 2010;10:1886–1890. doi: 10.1002/pmic.200900758. [DOI] [PubMed] [Google Scholar]
- (79).Yang Z, Gupta JK, Kishimoto K, Shoji Y, Kato T, Abbott NL. Adv. Func. Mater. 2010;20:2098–2106. doi: 10.1002/adfm.201000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Engler AJ, Richert L, Wong JY, Picart C, Discher DE. Surf. Sci. 2004;570:142–154. [Google Scholar]
- (81).Anderson VJ, Terentjev EM, Meeker SP, Crain J, Poon WCK. Eur. Phys. J. E. 2001;4:11–20. [Google Scholar]
- (82).Petrov PG, Terentjev EM. Langmuir. 2001;17:2942–2949. [Google Scholar]
- (83).Vollmer D, Hinze G, Ullrich B, Poon WCK, Cates ME, Schofield AB. Langmuir. 2005;21:4921–4930. doi: 10.1021/la047090w. [DOI] [PubMed] [Google Scholar]
- (84).Hegmann T, Qi H, Marx VM. J. Inorg. Organomet. Polym. Mater. 2007;17:483–508. [Google Scholar]
- (85).Kato T, Kutsuna T, Hanabusa K, Ukon M. Adv. Mater. 1998;10:606–608. [Google Scholar]
- (86).Kato T. Science. 2002;295:2414–2418. doi: 10.1126/science.1070967. [DOI] [PubMed] [Google Scholar]
- (87).Herrwerth S, Eck W, Reinhardt S, Grunze M. J. Am. Chem. Soc. 2003;125:9359–9366. doi: 10.1021/ja034820y. [DOI] [PubMed] [Google Scholar]
- (88).Pale-Grosdemange C, Simon ES, Prime KL, Whitesides GM. J. Am. Chem. Soc. 1991;113:12–20. [Google Scholar]
- (89).Gupta JK, Tjipto E, Zelikin AN, Caruso F, Abbott NL. Langmuir. 2008;24:5534–5542. doi: 10.1021/la800013f. [DOI] [PubMed] [Google Scholar]
- (90).Pomerantz WC, Yuwono VM, Drake R, Hartgerink JD, Abbott NL, Gellman SH. J. Am. Chem. Soc. 2011;133:13604–13613. doi: 10.1021/ja204874h. [DOI] [PubMed] [Google Scholar]
- (91).Zhou SQ, Liang DH, Burger C, Yeh FJ, Chu B. Biomacromolecules. 2004;5:1256–1261. doi: 10.1021/bm034524d. [DOI] [PubMed] [Google Scholar]
- (92).Gupta JK, Sivakumar S, Caruso F, Abbott NL. Angew. Chem.-Int. Edit. 2009;48:1652–1655. doi: 10.1002/anie.200804500. [DOI] [PubMed] [Google Scholar]
- (93).Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv. Mater. 2006;18:3203–3224. [Google Scholar]
- (94).Tjipto E, Cadwell KD, Quinn JF, Johnston APR, Abbott NL, Caruso F. Nano Lett. 2006;6:2243–2248. doi: 10.1021/nl061604p. [DOI] [PubMed] [Google Scholar]
- (95).Sivakumar S, Gupta JK, Abbott NL, Caruso F. Chem. Mat. 2008;20:2063–2065. [Google Scholar]
- (96).Aliño VJ, Pang J, Yang K-L. Langmuir. 2011;27:11784–11789. doi: 10.1021/la2022215. [DOI] [PubMed] [Google Scholar]
- (97).Lavrentovich OD. Liq. Cryst. 1998;24:117–125. [Google Scholar]
- (98).Huang W, Tuthill GF. Phys. Rev. E. 1994;49:570–574. doi: 10.1103/physreve.49.570. [DOI] [PubMed] [Google Scholar]
- (99).Drzaic PS. Liquid Crystal Dispersions. World Scientific Publishing Company; Singapore: 1995. [Google Scholar]