

SUMMARY
Histone lysine acetylation is central to epigenetic control of gene transcription. Bromodomains of chromosomal proteins function as acetyl-lysine (Kac) binding domains. However, how bromodomains recognize site-specific histones remains unanswered. Here, we report three three-dimensional solution structures of the bromodomains of the human transcriptional coactivators CREB-binding protein (CBP) and p300/CBP-associated factor (PCAF) bound to peptides derived from histone acetylation sites at lysines 36 and 9 in H3, and lysine 20 in H4. From structural and biochemical binding analyses, we determine consensus histone recognition by the bromodomains of PCAF and CBP, which represent two different subgroups of the bromodomain family. Through bromodomain residues in the ZA and BC loops, PCAF prefers acetylation sites with a hydrophobic residue at (Kac+2) position and a positively charged or aromatic residue at (Kac+3), whereas CBP favors bulky hydrophobic residues at (Kac+1) and (Kac+2), a positively charged residue at (Kac−1), and an aromatic residue at (Kac−2).
INTRODUCTION
Chromatin, which packages all genomic DNA in eukaryotic cells, functions as a fundamental regulator that governs global dynamic changes of gene expression and silencing (Nightingale et al., 2006). Nucleosome functions as a building block of chromatin, consisting of DNA of 147 bp wrapped in two superhelical turns around a histone octamer that is formed by an H3–H4 tetramer and two H2A-H2B dimers. Nucleosome core particles are linked by short stretches of DNA bound to the “linker” histones H1 and H5 to form a nucleosomal filament that is folded into higher-order structure of chromatin fiber. Site-specific histone modifications, including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation largely in the N- and C-terminal residues, sets a dynamic platform for DNA-based processes within the nucleus (Jenuwein and Allis, 2001; Turner, 2002). The extremely dense, versatile nature of histone modifications argues that histone signaling is far more complex in information content than is cell-surface receptor signaling (Schreiber and Bernstein, 2002; Seet et al., 2006). Despite its fundamental importance to genomic stability and gene regulation, knowledge of the detailed underlying molecular mechanisms of histone signaling is lacking.
Histone lysine acetylation is highly dynamic and reversible and plays an important role in directing chromatin remodeling and gene transcription (Berger, 2002; Neely and Workman, 2002; Turner, 1998). The functional role of acetylation in histone-directed chromatin biology is highlighted by the discovery of bromodomains as acetyl-lysine binding domains (Dhalluin et al., 1999; Jacobson et al., 2000; Mujtaba et al., 2007) and is reinforced by the function of the “Royal” family proteins of chromodomains and Tudor domains, MBT (malignant brain tumor) domains, and more recently the PHD (plant homeo domain) fingers, as methyl-lysine-binding domains (Bannister et al., 2001; Lachner et al., 2001; Ruthenburg et al., 2007). Site- and state-specific histone lysine acetylation and methylation have now been recognized to serve as binding sites for effector proteins (Fischle et al., 2003; Yap and Zhou, 2006). Such molecular interactions have been postulated to constitute the readout of the so-called epigenetic histone code that dictates distinct functions in gene regulation in a sequential or combinatorial manner (Strahl and Allis, 2000; Turner, 2002).
The functional importance in regulating chromatin biology by bromodomain/acetyl-lysine interactions is underscored by a large number of bromodomain-containing chromatin-associated proteins and histone acetyltransferases (HATs; >170 proteins in humans; Jeanmougin et al., 1997). This fundamental mechanism modulating protein-protein interactions by bromodomain/acetyl-lysine recognition argues that bromodomains may contribute to site-specific histone acetylation, by tethering nuclear HATs to specific chromosomal sites (Manning et al., 2001; Travers, 1999), and to assembly of multiprotein remodeling complexes SAGA and NuA4 (Brown et al., 2001; Sterner et al., 1999) and SWI/SNF (Agalioti et al., 2002; Hassan et al., 2002). This mechanism helps elucidate the reported phenotypes linked to bromodomain mutation or deletion. For instance, the bromodomain module is indispensable for the function of GCN5 in yeast (Georgakopoulos et al., 1995; Syntichaki et al., 2000). Deletion of a bromodomain in HBRM in the human SWI/SNF complex causes decreased stability and loss of nuclear localization (Muchardt et al., 1998; Muchardt and Yaniv, 1999). Bromodomains of Saccharomyces cerevisiae Bdf1 are required for sporulation and normal mitotic growth (Chua and Roeder, 1995). Bromodomain deletion in members of the RSC remodeling complex causes a conditional lethal phenotype (Du et al., 1998) or a phenotypic inhibition on cell growth (Cairns et al., 1999). Moreover, transgenic mice with lymphoid-restricted over-expression of the double bromodomain-containing protein 2 (BRD2) develop splenic B cell lymphoma and leukemia (Greenwald et al., 2004).
Despite its importance, our understanding of sequence-specific histone recognition by the bromodomain is limited. Thus far, only a few structures of bromodomains bound to acetylated peptides derived from biological binding partners have been reported. These include the yeast GCN5 bromodomain/H4-K16ac complex (Owen et al., 2000), human PCAF bromodomain bound to the HIV Tat-K50ac peptide (Mujtaba et al., 2002), and human CBP bromodomain bound to the p53-K382ac peptide (Mujtaba et al., 2004). These complex structures, as well as the free-form structures of other single or tandem bromodomains from human TAFII250 (Jacobson et al., 2000), BPTF (Li et al., 2006), BRD2 (Nakamura et al., 2007), and BRG1 (Shen et al., 2007), and yeast Rsc4 (Vandemark et al., 2007), show that all bromodomains adopt a conserved structural fold of a left-handed, four-helix bundle, and two interhelical loops (i.e., the ZA and BC loops) at one end of the bundle form a hydrophobic pocket for acetyl-lysine binding. Although the bromodomain residues important for acetyl-lysine binding are largely conserved, sequence variations in the ZA and BC loops enable discrimination of different binding targets (Mujtaba et al., 2007). Sequence variations and amino acid deletions in the flexible ZA and BC loops suggest that these regions likely determine specificity of ligand binding by interacting with residues flanking the Kac in a target protein (Mujtaba et al., 2007). Thus, it is essential to study bromodomains in the context of their biologically relevant binding partners to understand their histone-binding selectivity.
In this study, we report three new, to our knowledge, three-dimensional solution structures of the human transcriptional coactivators PCAF and CBP bromodomains in complex with histone peptides derived from known acetylation sites in H3 at lysine 9 and lysine 36, and in H4 at lysine 20. Using combined NMR-binding and structural analyses, we have defined consensus histone recognition sequences for these two human bromodomains and their unifying features of histone recognition.
RESULTS AND DISCUSSION
Structures of the PCAF and CBP Bromodomains Bound to Histone Peptides
To understand the molecular basis of site-specific histone recognition by bromodomains, we sought to conduct structural analysis of the bromodomains from two prototypical coactivator histone acetyltransferases PCAF and CBP. Because of their major differences in the amino acid composition of the ZA loop that comprises bromodomain's ligand binding site, these two bromodomains represent two distinct subgroups of the large bromodomain family (Mujtaba et al., 2007). We chose lysine-acetylated peptides derived from specific and nonspecific bromodomain recognition sites in histones for the structural analysis—that is, histone H3-K36ac (ATGGV-Kac-KPHRYK, where Kac is an acetyl-lysine) and H4-K20ac (GGAKRHR-Kac-VLRDNIQ) as specific binding sites for the bromodomains of PCAF (Morris and Moazed, 2007) and CBP (Garcia et al., 2007), respectively, and H3-K9ac (ARTKQTAR-Kac-STGGKA) as a nonspecific binding site for PCAF. We solved the three-dimensional solution structures of the PCAF bromodomain in complex with a H3-K36ac peptide (Figures 1A, 1B, and 1C) and a H3-K9ac peptide (Figures 1D, 1E, and 1F), as well as the CBP bromodomain bound to a H4-K20ac peptide (Figures 1G, 1H, and 1I).
Figure 1. 3D Structures of Bromodomains Bound to Acetylated Histone Peptides.

The structures of the bromodomain/acetylated histone peptide complexes are shown in three different illustrations: stereoview of the backbone atoms (N, Cα, and C′) of 25 superimposed NMR structures of the complexes (left); ribbons representation of the average minimized NMR structure of the complexes in a similar orientation, prepared using Pymol (middle); and surface electrostatic potential representation of the protein with the peptide in a ball-and-stick depiction (right).
(A–C) The PCAF bromodomain/H3-K36ac complex.
(D–F) The PCAF bromodomain/H3-K9ac complex.
(G–I) The CBP bromodomain/H4-K20ac complex.
The conformations of the bromodomain proteins in all three complexes were well defined by the NMR data (Table 1). The structures of the PCAF and CBP bromodomains bound to the histone peptides adopt the conserved left-handed four-helix bundle (helices αZ, αA, αB, and αC; Figures 1B, 1E, and 1H), but the two proteins differ markedly in surface electrostatic potential (Figures 1C, 1F, and 1I). The overall structure in each case is similar to that of the free form (Dhalluin et al., 1999; Mujtaba et al., 2004), but conformational differences were observed in the ZA and BC loops, likely pertinent to accommodating peptide binding. This is supported by extensive NMR data, including changes of chemical shifts and NOE (nuclear Overhauser effect) patterns for the backbone amides, side-chain methyl groups, and aromatic rings of many protein residues. Because H3 and H4 residues flanking both sites of the acetyl-lysine are engaged in bromodomain recognition, the H3 or H4 peptides assume extended conformations when bound to PCAF or CBP bromodomain. However, because of the differences in the ways by which the two histone H3 peptides interact with the PCAF bromodomain (see below), the backbone orientations of the H3-K36ac and the H3-K9ac peptides are antiparallel to each other, lying across the hydrophobic binding site between the ZA and BC loops of the PCAF bromodomain (compare Figure 1B with Figure 1E).
Table 1.
Statistics of the NMR Structures of the Bromodomains Bound to Histone Peptides
| PCAF/H3-K36ac | PCAF/H3-K9ac | CBP/H4-K20ac | |
|---|---|---|---|
| NMR Distance and Dihedral Constraints | |||
| Distance constraints | 3541 | 3009 | 2885 |
| Intramolecular constraints | 3441 | 2958 | 2847 |
| Total NOE | 3394 | 2911 | 2797 |
| Intraresidue | 1078 | 1060 | 1097 |
| Interresidue | 2316 | 1851 | 1700 |
| Sequential (|i – j| = 1) | 662 | 586 | 568 |
| Medium range (|i – j| ≤ 4) | 774 | 630 | 662 |
| Long range (|i – j| > 4) | 880 | 635 | 470 |
| Hydrogen bonds | 47 | 47 | 50 |
| Intermolecular constraints | 100 | 51 | 38 |
| Total dihedral angle restraints | |||
| Phi | 70 | 70 | 72 |
| Psi | 70 | 70 | 72 |
| Ramachandra Analysis, %a,b | |||
| Most favorable region | 86.1 ± 1.6 | 85.0 ± 3.0 | 72.2 ± 2.1 |
| Additional allowed region | 12.3 ± 1.5 | 13.1 ± 2.6 | 22.2 ± 1.9 |
| Generously allowed region | 1.6 ± 0.6 | 1.2 ± 0.65 | 2.5 ± 1.7 |
| Disallowed region | 0.06 ± 0.24 | 0.7 ± 0.6 | 2.8 ± 1.1 |
| Structure Statisticsa,b,c,d | |||
| Violations (mean ± SD) | |||
| Distance constraints (Å) | 0.077 ± 0.0049 | 0.080 ± 0.011 | 0.12 ± 0.03 |
| Dihedral angle constraints (°) | 1.05 ± 0.11 | 0.97 ± 0.068 | 0.77 ± 0.22 |
| Max. distance constraint violation (Å) | 0.088 | 0.096 | 0.22 |
| Max. dihedral angle constraints (Å) | 1.33 | 1.09 | 1.21 |
| Deviations from idealized geometry | |||
| Bond lengths (Å) | 0.0093 ± 0.00014 | 0.0071 ± 0.00019 | 0.0064 ± 0.00026 |
| Bond angles (°) | 0.92 ± 0.016 | 0.78 ± 0.02 | 0.79 ± 0.027 |
| Impropers (°) | 0.77 ± 0.017 | 0.64 ± 0.019 | 0.71 ± 0.028 |
| Average pair-wise root mean square derivation (Å) | |||
| Heavy | 0.64 ± 0.066 | 0.83 ± 0.1 | 0.98 ± 0.13 |
| Backbone | 0.20 ± 0.035 | 0.34 ± 0.088 | 0.56 ± 0.15 |
Based on 25 lowest energy-minimized structures.
Protein include residues 725–827 for PCAF and residues 1087–1196 for CBP.
None of these final structures exhibits NOE-derived distance restraint violations greater than 0.5 Å or dihedral angle restraint violations greater than 5°.
The Lennard-Jones Potential was not used during any refinement stage.
Acetyl-Lysine Recognition
The side chain of the acetyl-lysine of all three histone peptides intercalates deep into the hydrophobic cavity of the bromodomains. For instance, in the PCAF bromodomain, the acetylated K36 in the H3-K36ac peptide interacts extensively with protein residues Ala757 and Tyr760 on one side and Tyr802 and Tyr809 on the other (Figure 2A), whereas the acetylated K9 in the H3-K9ac peptide interacts with Val752, Ala757, and Tyr802 (Figure 2C). For the CBP bromodomain, the acetylated K20 in the H4-K20ac peptide is immersed within a hydrophobic pocket lined with Val1115, Leu1120, Ile1122, Tyr1125, Tyr1167, and Val1174 (Figure 3A). In all three structures, the N-acetyl carbonyl oxygen of the acetyl-lysine forms a hydrogen bond with the side-chain amide hydrogen of a highly conserved asparagine (i.e., Asn803 in PCAF and Asn1168 in CBP) that serves as an anchoring point of contact that stabilizes the acetyl-lysine in a deep cleft of the protein. This explains that unacetylated histone peptides do not bind to the bromodomain of PCAF or CBP.
Figure 2. The Structural Basis of Histone Recognition by the Bromodomains of PCAF and GCN5.

(A) and (C) Recognition of the acetyl-lysine of H3-K36ac and H3-K9ac peptides by the human PCAF bromodomain, respectively.
(B) and (D) Recognition of amino acid residues flanking the acetyl-lysine of the H3-K36ac and H3-K9ac peptides by the human PCAF bromodomain, respectively.
(E) and (F) Recognition of the acetyl-lysine and its flanking residues of the H4-K16ac peptide by the yeast GCN5 bromodomain (PDB code: 1E6I), respectively.
(G) and (H) Recognition of the acetyl-lysine and its flanking residues of the HIV-1 Tat-K50ac peptide by the human PCAF bromodomain (PDB code: 1JM4), respectively.
In all four structures, side chains of protein or peptide residues are color-coded by atom types—that is, carbon (green for protein and yellow for peptide), oxygen (red), and nitrogen (blue).
Figure 3. The Structural Basis of Histone Recognition by the CBP Bromodomain.
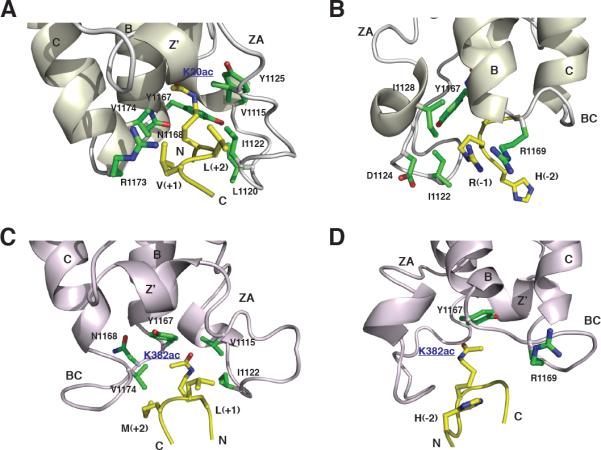
(A and B) Recognition of the acetyl-lysine and its flanking residues in the H4-K20ac peptide by the human CBP bromodomain, respectively.
(C and D) Recognition of the acetyl-lysine and its flanking residues of the p53-K382ac peptide by the human CBP bromodomain (PDB code: 1JSP), respectively.
Side chains of protein or peptide residues are color-coded by atom types in the same scheme as that in Figure 2.
Sequence-Specific Histone Recognition
Histone-binding selectivity is determined by bromodomain recognition of histone residues flanking the acetyl-lysine. The structures, which are supported by intermolecular NOEs in the NMR spectra, reveal that the PCAF bromodomain recognizes Pro(+2) and His(+3) at the H3-K36ac site (Figures 2A and 2B) but only Ala(−2) in H3-K9ac (Figure 2C), whereas the CBP bromodomain interacts with Val(+1), Leu(+2), Arg(−1), and His(−2) residues of the H4-K20ac peptide (Figures 3A and 3B). In the H3-K36ac peptide, Pro(+2) caps the opening of the hydrophobic pocket by interacting with side chains of Ala757 and Pro758 (Figure 2A), This network of intermolecular interactions stabilizes the bound peptide conformation and enables the acetyl-lysine side chain to form a hydrogen bond with Asn803 in the deep cleft. The H3-K36ac recognition by the PCAF BRD is further reinforced by the aromatic ring of His(+3) that lies in a shallow pocket outside of the BC loop and interacts with Tyr802 and Pro804 (Figure 2B).
For the H3-K9ac peptide, only the methyl group of Ala(−2) showed intermolecular NOEs to Ala757 and Pro758 of the protein (Figures 2C and 2D). Because PCAF bromodomain binding to H3-K36ac involves much more extensive intermolecular interactions than do those to H3-K9ac, the binding affinity of the former (dissociation constant [Kd] of 402 μM) is significantly higher than that of the latter (Kd of 1051 μM; Figure 4B), as determined using saturation transfer difference (STD) NMR spectroscopy (Meyer and Peters, 2003). The STD NMR is more suitable than the conventional NMR method, on the basis of ligand-induced chemical shift perturbations, for quantitative measurements of protein interactions with affinity in the μM-to-mM range, such as bromodomain/acetyl-lysine binding, because the former can be performed with protein concentrations much lower than the Kd. The STD-NMR results are consistent with the extent of chemical shift perturbations of the bromodomain residues upon the addition of the corresponding histone H3 peptide in 2D 15N-1H HSQC spectra (Figure 4C, red versus green signals). Moreover, because Pro(+2) in H3-K36ac and Ala(−2) in H3-K9ac are resided on the opposite sides of the acetyl-lysine in the histone peptides, their interactions with the same hydrophobic pocket of the bromodomain force two peptides to orient in anti-parallel directions. Moreover, the opposite location of a large positive amino acid with respect to the Kac—that is, Lys(+1) in H3-K36ac versus Arg(−1) in H3-K9ac—likely contributes to the reverse orientations of the two peptides when bound to the protein, owing to interactions with the negative electrostatic potential surface at the entrance of the Kac-binding pocket (compare Figure 1C with Figure 1F).
Figure 4. Unifying Features of Histone Recognition by the Bromodomains of PCAF and CBP.

(A) The unifying features of site-specific histone recognition by the PCAF and CBP bromodomains, as illustrated using the PCAF bromodomain/H3-K36ac complex. Three main ligand contact sites for the recognition of the acetyl-lysine, residues at (Kac+/−1) and/or (Kac+/−2) sites, and residues at (Kac+/−3) and beyond are color coded in red, blue, and green, respectively.
(B) Sequences of major histone lysine-acetylation sites that are preferably recognized by the PCAF or CBP bromodomain along with dissociation constants (Kd) of protein/peptide binding determined by STD NMR and fluorescent polarization binding assays as described in the Experimental Procedures. The amino acid sequences of the two N-terminal fluorescinated peptides of HIV-1 Tat-K50ac and p53-K382ac are Fl-GGGG-SYGR-Kac-KRRQRC and Fl-(β-Ala)-RHK-Kac-LMFK-NHC2H5, respectively.
(C) Comparison of the PCAF bromodomain binding to lysine-acetylated histone peptides as illustrated by two-dimensional 15N-1H HSQC spectra of the protein. The protein amide resonances in the NMR spectra are color-coded as the following: PCAF bromodomain in free (black), or in the presence of a H3-K9ac (green), H3-K14ac (red), and H3-K36ac (blue) peptide.
(D) NMR spectral comparison of the PCAF bromodomain in the free form (black), or in the presence of a H4-K16ac (blue), or H4-K20ac (red) peptide.
(E) NMR spectral comparison of the CBP bromodomain in the free form (black), or in the presence of an H3-K36ac (red) or H4-K20ac (blue) peptide.
(F) and (G) Fluorescent polarization titration of PCAF or CBP bromodomain binding to HIV-1 Tat-K50ac peptide (Fl-GGGG-SYGR-Kac-KRRQRC) or p53-K382ac peptide (Fl-GGGG-SYGR-Kac-KRRQRC), respectively. The concentration of the corresponding fluorescein-labeled peptide used in each study was 10 nM, as described in the Experimental Procedures.
The H4-K20ac peptide binding to the CBP bromodomain causes line-broadening of protein residues located at the peptide-binding site, which is not the case for PCAF/H3 peptide binding. This finding is consistent with the observation that the binding affinity of the former (Kd of 218 μM, Figure 4B) is in an intermediate exchange regime on the NMR time scale and higher than that of the latter. As such, fewer intermolecular NOEs were observed and assigned unambiguously for the CBP bromodomain/H4-K20ac complex (Table 1). Nevertheless, an extended conformation of the H4-K20ac peptide, when bound to the CBP bromodomain, was determined by intermolecular NOEs assigned to histone residues flanking both sides of the acetyl-lysine. Specifically, backbone atoms of Val(+1) and Leu(+2) in the H4 peptide form pair-wise interactions with those of the protein residues Leu1120 and Leu1121, with the main chains aligned in an antiparallel fashion (Figure 3A). This intermolecular interface is secured by interactions with the side chains of Val(+1) and Leu(+2), which point to opposite directions. These two residues in the peptide further interact with Arg1173 in αC of the protein. H4-K20ac recognition is reinforced by Arg(−1) binding to Ile1122 and Tyr1167 and forms electrostatic interactions with Asp1124 (Figure 3B). Additionally, His(−2) contributes to CBP bromodomain binding by interacting with Tyr1167 and Arg1169 in the BC loop (Figure 3B). The latter residues are not conserved in the bromodomain family, thus underscoring the selective nature of H4-K20ac recognition by the CBP bromodomain.
Differences of Histone Binding Selectivity by Bromodomains
To extend our understanding of bromodomain/histone binding selectivity, we compared our structures to the available complex structures of the PCAF bromodomain bound to HIV-1 Tat-K50ac (SYGR-Kac-KRRQR; Mujtaba et al., 2002), the GCN5 bromodomain to H4-K16ac (A-Kac-RHRKILRNSIQGI) (Owen et al., 2000), and the CBP bromodomain to p53-K382ac (SHLKSKKGQSTSRHK-Kac-LMFK; Mujtaba et al., 2004). To facilitate this structural analysis, we systematically measured the binding affinities of the PCAF and CBP bromodomains to various lysine-acetylated peptides by STD-NMR (Figure 4B). Notably, the affinity of individual bromodomain binding to acetyl-lysine in histones, as shown in this study as well as those reported for the bromodomains of Rsc4 (Vandemark et al., 2007) and BRG1 (Shen et al., 2007), is generally much weaker than that of chromodomain (Bernstein et al., 2006) or PHD finger (Li et al., 2006) binding to methyl-lysine (Kd of 1–100 μM). These differences are likely not just due to the use of different biophysical methods, because we confirmed that the affinities of PCAF or CBP bromodomain binding to respective HIV-1 Tat-K50ac or p53-K382ac peptide measured by STD-NMR are comparable to those obtained in a fluorescence polarization assay (Figure 4B). The latter is commonly used for measuring methyl-lysine binding by the chromatin binding domains.
Our structural comparison confirms that the mode of acetyllysine recognition is conserved in all three bromodomains. Consistent with their high sequence identity of about 95%, the PCAF and GCN5 bromodomains share a great deal of similarity in his-tone binding. However, PCAF and CBP bromodomains exhibit distinctive features in molecular recognition. As shown in the PCAF bromodomain/H3 peptide complexes, the GCN5 bromodomain has a limited number of contacts with two flanking residues at (Kac+2) and (Kac+3) in the H4-K16ac peptide. Binding of His(+2) by aromatic rings of Phe367 and Tyr406 of GCN5 (Figure 2E) is reminiscent of PCAF recognition of Pro(+2) in H3-K36ac (Figure 2A), or Ala(−2) in H3-K9ac (Figure 2C). Thus, it appears that recognition of flanking residues at (Kac+2) site and, to a lesser extent, at (Kac+3) in the target histone sequence anchors the conformation of the bound peptide, thereby reinforcing the acetyl-lysine binding in the hydrophobic pocket. Unlike PCAF or GCN5, however, the CBP bromodomain recognizes the hydrophobic residue at (Kac+1) and (Kac+2) sites in the target sequence. Consistent with the structure of the CBP/H4-K20ac complex, in the CBP bromodomain/p53-K382ac complex, Leu(+1) of the p53 peptide forms hydrophobic interactions with Val1115, Ile1122, and Val1174 of the protein (Figures 3C and 3D). Notably, although this major recognition site for one or two residues flanking the acetyl-lysine is located similarly in the region between the ZA and BC loops in PCAF and CBP bromodomains, the two amino-acid insertions (including Leu1120 and Ile1121) in the ZA loop in the CBP bromodomain distinguishes itself in its histone-binding selectivity.
The second major binding site for the acetyl-lysine flanking residues is located at the backside of the BC loop. As shown in the GCN5 bromodomain/H4-K16ac complex structure, the guanidinium group of Arg(+3) forms two hydrogen bonds to the backbone carbonyl groups of Glu409 and Tyr414 (Figure 2F). Similarly, the imidazole ring of His(+3) in H3-K36ac sits in the pocket located similarly around the BC loop in the PCAF bromodomain and forms hydrogen bonds with backbone carbonyls of Pro758, Tyr802, and Asn803 (Figure 2B). Moreover, similar to His(−2) in the H4-K20ac peptide, in the CBP bromodomain/p53-K382ac complex, His(−2) of p53-K382ac locates at the back of the BC loop and interacts with Arg1169 and Tyr1167 of the CBP bromodomain (Mujtaba et al., 2004; Figure 3D). Thus, His(−2) in H4-K20ac or p53-K382ac peptide plays a functional role in bromodomain recognition that is similar to that of Arg(+3) in H4-K16ac or His(+3) in H3-K36ac. The residue of (Kac−2) or (Kac+3) in the second interaction site likely works together with the acetyl-lysine in the conserved hydrophobic pocket like a clamp that holds around the BC loop. Collectively, these sequence-specific electrostatic and hydrophobic interactions confer the target binding selectivity and affinity of a particular bromodomain.
The highly specific association of the PCAF bromodomain and HIV-1 Tat at K50ac site is dependent on interactions with residues located at different positions flanking the acetyl-lysine. For instance, Tyr(−3) at the Tat-K50ac site shows intermolecular NOEs with the PCAF residues Val763 and Glu801 that are resided in the backside of the ZA and BC loops, which is analogous to the recognition of His(+3) by the PCAF bromodomain, or Arg(+3) by the CBP bromodomain (Figure 2H). Additionally, both Arg(+3) and Gln(+4) in the Tat sequence form hydrogen bonds with Glu756 of the PCAF bromodomain instead of carbonyls of the BC loop (Figure 2G). Without these hydrogen-bonding interactions, the PCAF bromodomain/Tat-K50ac binding is completely abolished (Mujtaba et al., 2002). Note that Glu756 is not conserved and is present in only a small subset of bromodomains, including GCN5. Because of a lack of a structurally analogous Glu756, the CBP bromodomain does not bind to HIV Tat-K50ac.
Conclusion
As the acetyl-lysine binding domain, the bromodomain, which is present in a large number of chromatin-associated proteins and histone acetyltransferases, plays a fundamental role in regulation of chromatin remodeling and gene transcription (Mujtaba et al., 2007). The solution structures of the PCAF and CBP bromodomains, in complex with lysine-acetylated histone H3 and H4 peptides reported here, provide new insights into the molecular basis of how these two prototypical bromodomains recognize sequence-specific acetylyation sites in histones—a question that remained unanswered until this study (Figure 4A). Our study shows that for PCAF or PCAF-like bromodomains, the preferred acetyl-lysine-binding site in histones or cellular proteins should have a hydrophobic residue at (Kac+2) and an aromatic or positively charged residue at (Kac+3), whereas for the CBP or CBP-like bromodomains, the consensus target sequence should contain hydrophobic residues at (Kac+1) and (Kac+2) and a positively charged residue at (Kac−1), as well as an aromatic residue at (Kac−2) (Figure 4B).
The new knowledge of histone-binding selectivity helps identify preferred histone recognition sites by bromodomain proteins. For instance, we predicted that histone H3-K14ac (ARKSTGG-Kac-APRKQLA) is likely a preferred site for the PCAF bromodomain, because it has a hydrophobic proline at (Kac+2) and a positively charged arginine at (Kac+3) (Figure 4B). In addition, a small hydrophobic Ala(+1), and guanidinium group of Arg(+3) at this site could enhance bromodomain binding by forming hydrophobic and electrostatic interactions, respectively. Indeed, the H3-K14ac peptide binds to the PCAF bromodomain with an affinity better than that of H3-K36ac (Kd of 128 μM versus 402 μM), as determined by STD-NMR (Figure 4B). This result is also supported by the chemical shift perturbations of the bromodomain resonances in two-dimensional15N-HSQC spectra induced by the corresponding H3 peptides binding (Figure 4C). Moreover, H4-K16ac (GLGKGGA-Kac-RHRKVLR) and H4-K20ac (GGAKR HR-Kac-VLRDNIQ) sites both have an Arg(+3). Because the former has a histidine at (Kac+2) and the latter has a leucine, we predicted and confirmed by STD-NMR titration that bromodomain binding to the H4-K16ac site is weaker than binding to the H4-K20ac site (Kd of 365 μM versus 247 μM; Figure 4D). Similarly, because the H3-K36ac site contains a nonoptimal sequence for the CBP bromodomain, it binds much more weakly than does the H4-K20ac to the CBP bromodomain (Kd of 218 μM versus 122 μM; Figure 4E).
The unifying features of site-specific histone recognition by the bromodomains of PCAF and CBP emerging from the structures can be defined as three major contact points in the conserved bromodomain fold, as illustrated in the structure of the PCAF bromodomain/H3-K36ac peptide complex (Figure 4A). First, acetyl-lysine is recognized by most, if not all, of the bromodomains in a hydrophobic pocket embedded between the ZA and BC loops at the bottom of the left-handed four-helix bundle (Figure 4A, red region). The bromodomain residues engaged in acetyl-lysine recognition are among the most highly conserved amino acid residues in the large bromodomain family, which includes Tyr760, Tyr802, and Asn803 in PCAF. The latter forms a hydrogen bond with the amide nitrogen of the acetyl-lysine. Second, ZA and/or BC loop residues resided at the entrance of the acetyl-lysine binding pocket interact, with one or two amino acid residues at (Kac+/−1) or (Kac+/−2) adjacent to the acetyl-lysine in the target protein (Figure 4A, blue region). These interactions reinforce the binding of the acetyl-lysine of the target sequence. Third, the additional ZA and BC loop residues that face the backside of the bromodomain (i.e., opposite the αZ helix) form hydrophobic and/or electrostatic interactions with target sequence residues at (Kac+/−3) or even further away from the Kac (Figure 4A, green region). In such a way, the (Kac+3) residue recognition by the bromodomain clamps on the BC loop together with the acetyl-lysine side chain that is bound inside the hydrophobic pocket of the bromodomain.
Although many bromodomains use similar regions of the conserved structural fold to interact with target proteins, sequence variations in the highly flexible ZA and BC loops, with change, addition, and deletion of amino acids, contribute to their distinct histone-binding selectivity. As illustrated in our structural analyses of the PCAF and CBP bromodomains, functional diversity of bromodomains is likely achieved by evolutionary changes of the structural features at the ligand-binding sites outside the conserved scaffold. Because of low protein sequence similarity in the ligand-binding sites, the unifying features of PCAF and CBP bromodomains in histone recognition as defined here are useful for identifying new biological ligands and assigning their functions. Given the high degree of sequence variations and structural dynamics of the ZA and BC loops, we expect that additional modes of molecular recognition by bromodomains likely exist, and their understanding requires new structural analysis of these bromodomains in complex with their biological ligands that may go beyond histones.
EXPERIMENTAL PROCEDURES
Sample Preparation
Expression and purification of the recombinant PCAF and CBP bromodomains in poly-his tag form has been described elsewhere (Mujtaba et al., 2002, 2004). The lysine-acetylated peptides were prepared on a MilliGen 9050 peptide synthesizer (Perkin Elmer) using Fmoc/HBTU chemistry. Acetyl-lysine was incorporated using Fmoc-Kac with HBTU/DIPEA activation. N-terminal fluorescein isocyanate-tagged peptides were prepared on solid-phase 4-formyl-3-methoxy-phenyloxymethyl polystyrene resin (200–400 mesh) (Bachem) with Py-BOP/HOBT as coupling reagents. Peptides were purified by reverse-phase HPLC on a C18 column and were characterized by ESI-TOF mass spectrometry.
Protein Structure Determination by NMR Spectroscopy
NMR samples contained a protein/peptide complex of ~0.5 mM in 100 mM phosphate buffer (pH 6.5) that contains 5 mM perdeuterated DTT and 0.5 mM EDTA in H2O/2H2O (9/1) or 2H2O. All NMR spectra were collected at 30°C on NMR spectrometers of 800, 600, or 500 MHz. The 1H, 13C, and 15N resonances of a protein of the complex were assigned by triple-resonance NMR spectra collected with a 13C/15N-labeled and 75% deuterated protein bound to an unlabeled peptide (Clore and Gronenborn, 1994). The distance restraints were obtained in three-dimensional 13C- or 15N-NOESY spectra. Slowly exchanging amides, identified in two-dimensional 15N-HSQC spectra recorded after a H2O buffer was changed to a 2H2O buffer, were used with structures calculated with only NOE distance restraints to generate hydrogen-bond restraints for final structure calculations. The intermolecular NOEs were detected in 13C-edited (F1), 13C/15N-filtered (F3), three-dimensional NOESY spectrum.
Structure Calculations
Structures of the PCAF or CBP bromodomain/histone peptide complexes were calculated with a distance geometry-simulated annealing protocol using the X-PLOR program (Brunger et al., 1998), as described elsewhere (Mujtaba et al., 2004). Manually assigned NOE-derived distance restraints were used to calculate initial structures. ARIA (Nilges and O'Donoghue, 1998) assigned distance restraints agree with structures calculated using only the manually determined NOE distance restraints. Ramachandran plot analysis of the final structures was performed with Procheck-NMR program (Laskowski et al., 1996).
Protein/Peptide Binding by Saturation Transfer Difference NMR
Affinity of bromodomain binding to lysine-acetylated peptides was measured using STD-NMR spectroscopy (Meyer and Peters, 2003). The NMR pulse program uses the WATERGATE scheme for water suppression and a presaturation by alternating on and off resonance frequencies after each scan to eliminate in-homogeneity artifacts in signal difference accumulations. On resonance irradiation was set on frequency at −400 Hz (−0.8 ppm) for PCAF signal of Pro747 HG2 and −822 Hz (−1.6 ppm) for CBP signal of Val1157 HG21, whereas off resonance irradiation was tuned at 22,253 Hz (44 ppm) where no protein or peptide signals were present. The NMR resonance of the acetyl methyl group (~1.90 ppm) was used to derive binding disassociation (Kd) of all peptides. Selective saturation of the protein was achieved by a train of Gauss-shaped pulses of 49 ms length, truncated at 1%, and separated by a 1 ms delay. Sixty selective pulses were applied with a total saturation of 3 s and a recovery delay of 2 s. A total number of scans was 256–10,000. Protein concentrations (15 μM) were determined by UV absorbance, whereas peptide concentrations were calibrated in 1D-NMR of DMSO (0.2 mM) in the same NMR buffer. The dissociation constants were determined by using nonlinear regression analysis with SigmaPlot (Version 10.01).
Fluorescent Polarization
Fluorescent polarization was performed for PCAF and CBP bromodomains' binding to two fluorescinated peptides of HIV-1 Tat-K50ac (Fl-GGGG-SYGRKac-KRRQRC) and p53-K382ac (Fl-(β-Ala)-RHK-Kac-LMFK-NHC2H5), respectively. Protein concentrations were determined by absorbance spectroscopy using calculated molar extinction coefficients (ε280 = 25,440 M−1 cm−1 and 17,420 M−1 cm−1 for CBP and PCAF, respectively). Peptide concentrations were determined using absorbance spectroscopy (ε494 = 68,000 M−1 cm−1 for the fluorescinated peptides). Fluorescent polarization assay was performed in polypropylene 96-well plates (Costar) with 10 nM fluorescein-labeled peptide and varying concentrations of a protein in 25 mM Tris-buffer (pH 7.4), 100 mM NaCl, and 1 mM Imidazol in 100 μl. Measurements were obtained after 10 min incubation of the peptide and the protein at 25°C using a fluorescence polarization reader (Tecan). Dissociation constants were determined as a one-site model (Roehrl et al., 2004) by fitting the binding data with SigmaPlot to the following equation: Fb = {(Kd + P + R) − [(Kd + P + R)2 − (4 × P × R)]1/2}/(2 × P), where Fb is fraction of bound labeled ligand, Kd = dissociation constant, R = total protein concentration, and P = total fluorescent-peptide concentration.
ACCESSION NUMBERS
Coordinates for the three-dimensional solution structures of the PCAF bromodomain/H3-K9ac and H3/K36ac peptide complexes, as well as the CBP bromodomain/H4-K20ac peptide complex, are being deposited in the Protein Data Bank, with accession codes 2RNW, 2RNX, and 2RNY.
ACKNOWLEDGMENTS
We thank S. Yan for technical support of the initial protein preparation. M.-M.Z. is supported by funds from the Dr. Golden and Harold Lamport Chair, the National Institutional Institutes, and National Science Foundation, and acknowledges generous access to the New York Structural Biology Center's NMR facilities.
REFERENCES
- Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Berger SL. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Duncan E, Masui O, Gil J, Heard E, Allis C. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 2006;26:2560–2569. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, Workman JL. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;292:2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Schlichter A, Erdjument-Bromage H, Tempst P, Kornberg RD, Winston F. Two functionally distinct forms of the RSC nucleosome-remodeling complex, containing essential AT hook, BAH, and bromodomains. Mol. Cell. 1999;4:715–723. doi: 10.1016/s1097-2765(00)80382-2. [DOI] [PubMed] [Google Scholar]
- Chua P, Roeder G. Bdf1, a yeast chromosomal protein required for sporulation. Mol. Cell. Biol. 1995;15:3685–3696. doi: 10.1128/mcb.15.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods Enzymol. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Carlson J, Zeng L, He C, Aggarwal K, Zhou M. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Du J, Nasir I, Benton BK, Kladde MP, Laurent BC. Sth1p, a Saccharomyces cerevisiae Snf2p/Swi2p homolog, is an essential ATPase in RSC and differs from Snf/Swi in its interactions with histones and chromatin-associated proteins. Genetics. 1998;150:987–1005. doi: 10.1093/genetics/150.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis C. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CC, et al. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos T, Gounalaki N, Thireos G. Genetic evidence for the interaction of the yeast transcriptional co-activator proteins GCN5 and ADA2. Mol. Gen. Genet. 1995;246:723–728. doi: 10.1007/BF00290718. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, Faller DV, Denis GV. E mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood. 2004;103:1475–1484. doi: 10.1182/blood-2003-06-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- Jacobson RR, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- Jeanmougin F, Wurtz JM, Le Douarin B, Chambon P, Losson R. The bromodomain revisited. Trends Biochem. Sci. 1997;22:151–153. doi: 10.1016/s0968-0004(97)01042-6. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis C. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan E, Wysocka J, Allis C, Patel D. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning ET, Ikehara T, Ito T, Kadonaga JT, Kraus WL. p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Mol. Cell. Biol. 2001;21:3876–3887. doi: 10.1128/MCB.21.12.3876-3887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Morris CA, Moazed D. Centromere assembly and propagation. Cell. 2007;128:647–650. doi: 10.1016/j.cell.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J. Mol. Biol. 1999;293:187–198. doi: 10.1006/jmbi.1999.2999. [DOI] [PubMed] [Google Scholar]
- Muchardt C, Bourachot B, Reyes JC, Yaniv M. ras transformation is associated with decreased expression of the brm/SNF2α ATPase from the mammalian SWI-SNF complex. EMBO J. 1998;17:223–231. doi: 10.1093/emboj/17.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Farooq A, Carlson J, Ott M, Verdin E, Zhou M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell. 2002;9:575–586. doi: 10.1016/s1097-2765(02)00483-5. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, Sanchez R, Zeleznik-Le N, Ronai Z, Zhou M. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell. 2004;13:251–263. doi: 10.1016/s1097-2765(03)00528-8. [DOI] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Umehara T, Nakano K, Jang M, Shirouzu M, Morita S, Uda-Tochio H, Hamana H, Terada T, Adachi N, et al. Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J. Biol. Chem. 2007;282:4193–4201. doi: 10.1074/jbc.M605971200. [DOI] [PubMed] [Google Scholar]
- Neely KE, Workman JL. Histone acetylation and chromatin remodeling: which comes first? Mol. Genet. Metab. 2002;76:1–5. doi: 10.1016/s1096-7192(02)00014-8. [DOI] [PubMed] [Google Scholar]
- Nightingale KP, O'Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 2006;16:125–136. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Nilges M, O'Donoghue S. Ambiguous NOEs and automated NOE assignment. Prog. Nucl. Magn. Reson. Spectrosc. 1998;32:107–139. [Google Scholar]
- Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, Travers AA. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000;19:6141–6149. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrl MH, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- Shen W, Xu C, Huang W, Zhang J, Carlson J, Tu X, Wu J, Shi Y. Solution structure of human Brg1 bromodomain and its specific binding to acetylated histone tails. Biochemistry. 2007;46:2100–2110. doi: 10.1021/bi0611208. [DOI] [PubMed] [Google Scholar]
- Sterner DE, Grant PA, Roberts SM, Duggan LJ, Belotserkovskaya R, Pacella LA, Winston F, Workman JL, Berger SL. Functional organization of the yeast SAGA complex: distinct components involved in structural integrity, nucleosome acetylation, and TATA-binding protein interaction. Mol. Cell. Biol. 1999;19:86–98. doi: 10.1128/mcb.19.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Topalidou I, Thireos G. The Gcn5 bromodomain co-ordinates nucleosome remodelling. Nature. 2000;404:414–417. doi: 10.1038/35006136. [DOI] [PubMed] [Google Scholar]
- Travers A. Chromatin modification: how to put a HAT on the histones. Curr. Biol. 1999;9:R23–R25. doi: 10.1016/s0960-9822(99)80037-2. [DOI] [PubMed] [Google Scholar]
- Turner BM. Histone acetylation as an epigenetic determinant of long-term transcriptional competence. Cell. Mol. Life Sci. 1998;54:21–31. doi: 10.1007/s000180050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Vandemark AP, Kasten MM, Ferris E, Heroux A, Hill CP, Cairns BR. Autoregulation of the rsc4 tandem bromodomain by gcn5 acetylation. Mol. Cell. 2007;27:817–828. doi: 10.1016/j.molcel.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Zhou MM. Structure and function of protein modules in chromatin biology. Results Probl. Cell Differ. 2006;41:1–23. [PubMed] [Google Scholar]