

Abstract
The m.3243A>G variant in the mitochondrial tRNALeu (UUR) gene is a common mitochondrial DNA (mtDNA) mutation. Phenotypic manifestations depend mainly on the heteroplasmy, i.e. the ratio of mutant to normal mtDNA copies. A high percentage of mutant mtDNA is associated with a severe, life-threatening neurological syndrome known as MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes). MELAS is described as a neurovascular disorder primarily affecting the brain and blood vessels, but the pathophysiology of the disease is poorly understood.
We developed a series of cybrid cell lines at two different mutant loads: 70% and 100% in the nuclear background of a neuroblastoma cell line (SH-SY5Y). We investigated the impact of the mutation on the metabolism and mitochondrial respiratory chain activity of the cybrids. The m.3243A>G mitochondrial mutation induced a metabolic switch towards glycolysis in the neuronal cells and produced severe defects in respiratory chain assembly and activity. We used two strategies to compensate for the biochemical defects in the mutant cells: one consisted of lowering the glucose content in the culture medium, and the other involved the addition of L-arginine. The reduction of glucose significantly shifted the 100% mutant cells towards the wild-type, reaching a 90% mutant level and restoring respiratory chain complex assembly. The addition of L-arginine, a nitric oxide (NO) donor, improved complex I activity in the mutant cells in which the defective NO metabolism had led to a relative shortage of NO. Thus, metabolically induced heteroplasmy shifting and L-arginine therapy may constitute promising therapeutic strategies against MELAS.
Keywords: Mitochondria, mitochondrial disorders, mtDNA, MELAS, neuronal cells, L-arginine
1. INTRODUCTION
Mitochondrial oxidative phosphorylation carried out by respiratory chain complexes embedded in the mitochondrial inner membrane produces most of the energy required by eukaryotic cells [1]. Electrons produced by the oxidation of the substrate are transferred from complexes I and II to complex IV. The proton-motive force created by proton translocation through complexes I, III and IV is used by ATP synthase to phosphorylate ADP into ATP. The oxidation and phosphorylation processes are thus tightly coupled. However, in physiological and particularly in pathological conditions, electrons transported across the different respiratory chain complexes may accumulate and produce reactive oxygen species (ROS). Free radicals, generated mainly by complexes I and III, trigger cell-damaging oxidative stress.
The proteins of the respiratory chain complexes are encoded by a combination of mitochondrial and nuclear genes [2]. MtDNA is a 16,569 bp double-stranded circular DNA molecule encoding 13 polypeptides, 2 ribosomal RNAs and 22 transfer RNAs. Mutations in the mitochondrial or the nuclear genomes may thus lead to respiratory chain dysfunction, and ultimately to mitochondrial diseases. Since the initial description of the first human mtDNA mutation in 1988, more than 200 pathogenic mutations have been associated with a wide variety of human diseases [3]. One of the most frequently observed mtDNA mutations is the A-to-G transition at position 3243 of the tRNA(Leu (UUR)) gene, which has been shown to impair mitochondrial structure, methylation, amino-acylation and codon recognition of the tRNALeu [4]. This mutation, which gives rise to a wide range of phenotypes including diabetes, deafness, cardiomyopathy and intolerance to exercise, is predominant in about 80% of the patients with MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes; OMIM 540000) [5]. The variability of the phenotypes associated with the mutation may be partially explained by the level of heteroplasmy [6]. Indeed, mutant and wild type mtDNA copies coexist in cells in various proportions, resulting in different mutation loads in tissues or organs. The heteroplasmy level may thus shift during cell division due to random segregation of mutant or wild-type mtDNA. Hence, the ratio of mutant to normal mtDNA, and the specific tissue affected, may determine the severity of the disease [6]. The m.3243A>G mutation causes diabetes mellitus, with or without deafness [7]. However, with a higher proportion of mutants, the same mutation may lead to chronic progressive ophthalmoplegia, cardiomyopathy, or the complete MELAS syndrome [1]. Generally, a first episode of stroke in MELAS patients occurs between the ages of 5 to 15 years, although it may occur at any time from infancy to adulthood [5, 8]. In addition, maternal relatives may suffer from a variety of complex clinical problems involving the central nervous system, muscle, heart, kidneys and the endocrine system. Stroke-like episodes, which do not respect vascular territories, are usually reversible, allowing partial or almost complete recovery [8].
The specificity of MELAS with early-onset of stroke-like episodes is attributed to endothelial dysfunction since degenerative changes associated with several mitochondrial abnormalities, has been described in the endothelial and smooth muscle cells of cerebral small arteries [9–10]. Nitric oxide (NO), the main mediator of endothelial relaxation, is primarily generated by NO synthesis on L-arginine. Hence, L-arginine supplementation, which donates NO, alleviates endothelial dysfunction in MELAS patients suggesting that the NO metabolism plays an important role in the pathophysiology of MELAS [9, 11–12].
Several pathophysiological studies have reported a mitochondrial dysfunction associated with a severe reduction of the activities of complexes I and IV of the respiratory chain [13–14]. A reduction of aminoacylation of the tRNA(Leu(UUR) and the rate of mitochondrial protein synthesis has been reported in transmitochondrial mutant cell lines [15–16]. Mutant loads exceeding 90% have led to the decline of protein synthesis as well that of oxygen consumption of the cells [17]. These studies were performed on muscle tissue and fibroblasts from MELAS patients, and cybrid cells originating from 143B osteosarcoma cells; however, only a few authors have reported the mitochondrial effects of the m.3243A>G mutation on the organs most affected in the disease, i.e. the brain and blood vessels [8, 18].
Neuroblastoma cell lines such as SH-SY5Y cells have been used as a neuronal-like model to create transmitochondrial cybrids carrying deficient mitochondria from Parkinson’s or Alzheimer’s disease patients, showing that the mitochondrial defect was transferred to the cybrid cell lines [19–20].
To analyze the consequences of the m.3243A>G mutation on mitochondrial metabolism in a neuronal like model, we developed a series of SH-SY5Y transmitochondrial cybrid cells carrying 70% and 100% mutant loads, indicated below as 70% M and 100% M cybrids, respectively. Our study showed that the mutation modified the energetic metabolism of the cybrids, producing a metabolic switch towards glycolysis. Furthermore, the mutation severely altered the assembly and functions of the mitochondrial respiratory chain and induced an increase in oxidative stress.
No efficient treatment is currently available for mitochondrial disorders, including MELAS. A better understanding of the pathophysiological mechanisms triggered by the m.3243A>G mutation should help to define an effective therapeutic strategy for the disease [2]. Overexpression of the mitochondrial leucyl-tRNA synthetase in cybrid cells carrying the MELAS mutation resulted in the restoration of the respiratory chain function of the mutant cells [21]. Based on the hypothesis that stroke-like episodes are the consequence of a segmental impairment of vasodilation in small arteries, L-arginine has been proposed as a treatment for MELAS m.3243A>G syndrome patients during the acute stroke phase or long term use [9, 11–12, 22]. Thus, we used two experimental approaches to reduce mitochondrial dysfunction. The first approach involved cultivating the cybrid cells in a medium low in glucose so as to induce a heteroplasmic shift of the mutants to the wild type. The second approach consisted of treating the cybrid cells with L-arginine, known to be an NO donor, to investigate the role of L-arginine in the pathophysiology of MELAS.
2. MATERIAL AND METHODS
2.1 Cell culture and transmitochondrial cybrids
The SH-SY5Y human parental neuroblastoma cell line was obtained from the American Type Culture Collection (ATCC), and cultured in DMEM high-glucose medium (PAA) supplemented with 10% foetal bovine serum (FBS, PAA), 2 mM L-glutamine, and antibiotics (100 U/ml penicillin G, 2mM streptomycin and 0.25 μg/ml amphoptericin B, Institut de Biotechnologies Jacques Boy). Cells were grown in 75 cm2 flasks at 37°C in a humidified atmosphere (95% air and 5% CO2). Mitochondrial respiratory deficient fibroblasts were cultured in 2/3 DMEM F-12, 1/3 Amniomax (Gibco) medium supplemented with 10% SVF (PAA), 100μg/ml sodium pyruvate and 50μg/ml uridine (Sigma-Aldrich).
The generation of neuronal-like cybrid cell lines was realized as described elsewhere [23]. Briefly, SH-SY5Y cells were depleted of mtDNA with a rhodamine-6-G treatment (2.5 μg/ml) during 5 days. Fibroblasts from patients carrying the m.3243A>G mutation were enucleated by actinomycin D treatment (0.5 μg/ml for 14 hours). The cells were washed with complete DMEM and mixed SH-SY5Y/enuclated fibroblast cells were then fused with the addition of polyethyleneglycol (PEG 1500, Roche Applied Science). After 1 minute, the cells were washed and centrifuged twice (300g, 5 minutes) with DMEM high glucose, 10% FBS to eliminate the PEG, and the pellet was resuspended in the SH-SY5Y cell media. The medium was replaced by high-glucose DMEM, 10% FBS for three days to eliminate non-fused cells, and propagated to select independent clones.
2.2 Glucose, lactate and pyruvate measurements
Glucose and lactate concentrations in the culture media were determined by spectrophotometry, using appropriate enzymatic kits (Boerhinger) on a Hitachi-Roche apparatus (Roche Diagnostics). Pyruvate concentration was determined by measuring the NADH reduction in the presence of the lactate dehydrogenase enzyme using spectrophotometry at 340 nm.
2.3 MtDNA analysis and quantification of heteroplasmy
Total DNA was extracted from cells with High Pure PCR Template Preparation Kit (Roche Applied) in accordance with the manufacturer’s instructions. The PCR-amplified mtDNA fragments of control SH-SY5Y cells and 100% mutants were determined by full sequencing as described elsewhere [24]. The quantification of mutant loads in SH-SY5Y cybrids was determined by fluorescent RFLP (restriction fragment length polymorphism) and monitored on a regular basis during the experiments as shown before [25]. A 262 bp DNA fragment surrounding the m.3243A>G mutation was amplified by PCR using fluorescent hmtL3117 (5′-FAM-CCCTGTACGAAAGGACAAGAGAAATAACGCC-3′) and non fluorescent hmtH3381 (5′-CGTTCGGTAAGCATTAGGAATGCCATTGC-3′) primers. PCR products were digested with HaeIII restriction enzyme (New England Biolabs) which cleaves the mutant DNA into 3 fragments (a 123 bp fluorescent fragment, and 73 bp and 66 bp non-fluorescent fragments) and the wild-type DNA into 2 fragments (a 196 bp fluorescent fragment and a 66 bp non- fluorescent fragment). Fragments were separated on polyacrylamide gel capillaries of a fragment analyzer (3130xl Genetic Analyzer, Applied Biosystems) and fluorescent fragment size was determined by migration comparison with a fluorescent DNA size ladder (Genescan 600LIZ, Applied Biosystems). Data were analyzed with Peak Scanner software (Applied Biosystems) and the mutant load was calculated using mutant and wild type peak areas.
2.4 Enzymatic activity measurements of the respiratory chain complexes
Activities of complexes I, II and IV of the respiratory chain of SH-SY5Y parental and mutant cells were measured as described elsewhere [26]. Briefly, cell pellets were resuspended in a volume of 50 μl/million cells of cell buffer (saccharose 250 mM, TRIS 20 mM, EDTA 2 mM, BSA 1mg/mL, pH 7.4). After a freezing-thawing cycle, cells were centrifuged (16000 rpm, 1 min) and resuspended in the same volume of cell buffer. For complex I activity measurement, an additional step of sonication (6 cycles of 5 seconds with a 30 seconds stop) was required. One million cells were then incubated at 37°C in a reaction medium (KH2PO4 100 mM, pH 7.4, KCN 1mM, NaN3 2 mM, BSA 1mg/ml, ubiquinone-1 0.1 mM and DCPIP 0.075 mM). The reaction was started by adding 0.3 mM NADH and the disappearing rate of DCPIP was measured at 600 nm for 2 minutes. The unspecific activity was determined by adding rotenone 0.005 mM at the middle of the kinetic. For complex II activity, 0.5 million cells were added to a reaction buffer (KH2PO4 100 mM, pH 7.5, KCN 2mM, EDTA 0.1 mM, BSA 1mg/ml, rotenone 0.01 mM, antimycin 0.002 mg/ml, succinate 20 mM and DCPIP 0.08 mM) and the reaction was initiated by adding 0.005 mM of ubiquinone-1. The disappearing rate of DCPIP was measured at 600 nm and the background was subtracted by using thenoyltrifluoroacetone 200 μM. For complex IV, a volume equivalent to 0.1 million cells was added to the reaction medium (reduced cytochrome C 0.05 mM, BSA 1mg/ml, laurylmaltoside 0.250 mM) and the oxidation rate of reduced cytochrome C was monitored at 550 nm.
The enzymatic activities of the respiratory chain complexes were normalized with respect to citrate synthase activity, which is considered as a marker of mitochondrial mass. This activity was measured by adding 0.1 million cells to a prewarmed reaction mix (DTNB 0.15 mM, oxaloacetic acid 0.5 mM, acetyl coA 0.3 mM, triton X100 0.1%) and the appearing rate of coA-SH was measured at 412 nm.
2.5 Measurement of aconitase activity
Aconitase activity was measured on 1 million frozen SH-SY5Y cells resuspended in the cell buffer and incubated in the reaction buffer (Tris 50 mM, pH 7.4, MnCl2 0.5mM, sodium isocitrate 20 mM, triton 0.01%. The appearing rate of cis-aconitate was monitored at 240 nm. Activity was normalized with respect to citrate synthase activity as described above.
2.6 Oxygen consumption on permeabilized SH-SY5Y cells
Cells were trypsinized and the pellet was resuspended in the respiratory buffer (KH2PO4 10 mM, mannitol 300 mM, KCl 10 mM, MgCl2 5 mM, pH 7.4) containing 15 μg/million cells of digtonin. Cells were incubated at room temperature during 2.5 minutes and the digitonin action was stopped by adding five volumes of the respiration buffer supplemented with 1 mg/ml of BSA. Cells were centrifuged (800 rpm, 2.5 minutes), resuspended in the respiration buffer + BSA (50μl/million of cells) and placed in the oxygraph chamber (Oroboros). The oxygen consumption was measured in state II (5 mM malate + pyruvate), state III (5 mM malate + pyruvate + 1.5 mM ADP + 0.5 mM NAD or 5 mM succinate + 10 μM rotenone + 1.5 mM ADP + 0.5 mM NAD), state IV (8μg/ml oligomycin) and maximal cytochrome c oxidase capacity (4 mM ascorbate + 0.2 mM TMPD). Non-mitochondrial oxygen consumption was subtracted by adding antimycin (2.5μg/ml). After oxygraphic measurement, 400 μl of cell suspension was removed from the chamber and the protein concentration was measured using bicinchoninic acid.
2.7 ATP measurement
SH-SY5Y control and mutant cells were seeded in 24-well plates and at 80% of confluence, medium was removed and replaced by the extraction buffer (boric acid 0.2M, magnesium sulfate 10 mM, triton X100 0.1%, pH 9.2) for 10 minutes on ice. The supernatant was removed and centrifuged at 1000g for 5 minutes and immediately frozen at −80°C. ATP was then assayed with the luciferin-luciferase-based kit (ENLITEN ATP Assay System, Promega).
2.8 Deconvolution microscopy and mitochondrial network analysis
Mitochondria were labelled using Mitotracker® Green 100nM (Molecular Probes). Images were acquired with an inverted wide-field Leica microscope (DMI6000B, Microsystems) equipped with a Roper CoolSnap HQ2 camera (Roper Scientific) a high-sensitivity CCD camera for quantitative fluorescence microscopy. Metamorph® 7.7 software (Molecular Devices) was used for image acquisition and Huygens software (Scientific Volume Imaging) for deconvolution. Imaris 7.1.1® software (Bitplane) was used for 3D processing and morphometric analysis.
2.9 Western blotting
Frozen pellets of SH-SY5Y cells were resuspended in a 10μl/million of 1X protease inhibitor cocktail (Complete Protease Inhibitor Cocktail Tablets, Roche Applied Science). A volume corresponding to 40 μg protein was added to a sample buffer (Tris 0.1M, SDS 1.2%, glycerol 4.1M, bromophenol blue 2%, 2β mercaptoethanol 1.87M), boiled for 5 minutes and loaded on 12.5%. Proteins were separated by electrophoresis and transferred onto PVDF membranes (GE Healthcare). Membranes were blocked with TBS 1X (NaCl 0.8%, KCl 0.02%, Tris 0.3%, pH 7.4), Tween 0.01%, 5% non-fat milk for 2 hours and incubated with primary antibodies (MnSOD, Abcam, 1:2000; Total OXPHOS HumanWB Antibody Cocktail, Mitosciences, 1:1000) overnight at 4°C. Incubation with a secondary antibody (anti-mouse or anti-rabbit, GE Healthcare, 1:10000) was performed for two hours at room temperature. Proteins were detected by enhanced chemiluminescence (ECL plus, GE Healthcare) and quantified using Quantity One software (BioRad). Tubulin antibody (Calbiochem, 1:2000) was used as loading control.
2.10 Blue-Native PAGE of respiratory chain complexes
Frozen pellets of SH-SY5Y cells were resuspended in 1X PBS. Mitochondria were extracted by digitonin permeabilization (2mg/ml) and centrifugation (10,000g, 10 minutes). The mitochondria-enriched pellet was resuspended in the sample buffer (aminocaproic acid 1.5M, bis tris pH 7.0 75 mM, protease inhibitor cocktail 1X) and mixed with laurylmaltoside (2g/g protein). The suspension was incubated for 10 minutes on ice and centrifuged at 10,000g for 30 minutes. Isolated mitochondrial proteins were loaded onto a 4–15% bis-acrylamide gradient gel. The complexes were then transferred onto PVDF membranes. Membranes were saturated for two hours on TBS1X/Tween 0.01%/10% non-fat milk and incubated with primary antibody (for complex I: NDUFB6, 1:1000, for complex IV: COXI, 1:1000, and for complex V: V beta 1:1000, Mitosciences). Antibody against complex II 70 kDa, 1:1000 was used as loading control. Membranes were washed twice and incubated with secondary anti-mouse antibody (1:10000, GE Healthcare) and the complex assembly was visualized using enhanced chemilumiscence (ECL Plus, GE Healthcare).
2.11 Measurement of nitrates + nitrites
The concentration of nitrates + nitrites was measured on the culture supernatant of SH-SY5Y control and mutant cells using the colorimetric Nitric Oxide Synthase Assay Kit (Calbiochem), following the manufacturer’s instructions. Briefly, the reaction is based on the conversion of nitrites to an azo product by Griess reagents (sulfanilamide and N-(1-naphtyl) ethylenediamine). The azo product production was measured at 540 nm with a microplate spectrophotometer (SAFAS). Before the reaction, nitrates are converted to nitrites by nitrate reductase. A standard curve, performed with a known amount of nitrites was used to quantify the concentration of nitrates + nitrites.
2.12 Measurement of reduced glutathione, alanine and L-arginine
Glutathione (GSH), alanine and L-arginine concentrations were determined by mass spectrometry on whole lysates of SH-SY5Y cells. Briefly, cells were harvested by trypinisation with cold trypsin and were resuspended in a cold lysate medium (sucrose 250 mM, EGTA 0.5 mM, HEPES 10 mM, Triton 10%, antiproteinase 1X). Lysates were centrifuged (16,000 g, 5 minutes, 4°C) and the supernatant was immediately frozen in liquid nitrogen and kept at −80°C until measurement. Cell lysates were deproteinised and GSH, alanine and L-arginine concentrations were determined by mass spectrometry on an API 3000 triple quadripole apparatus (Applied Biosystems) using standard laboratory procedures.
3. RESULTS
3.1 The m.3243A>G mutation induces a metabolic switch in SH-SY5Y cybrid cells
The m.3243A>G mtDNA mutation is often associated with the MELAS syndrome, which is characterized by severe mitochondrial dysfunction associated with lactic acidosis and stroke- like episodes. The entire mtDNA sequencing of the SH-SY5Y cybrid cells (see supplemental Table 1) showed no other pathogenic mutations or variants that could modify mitochondrial expression, such as the 12300G>A, predicted to act as a mutant suppressor [27]. The sequence was deposited in the GenBank database under GenBank number JN989561. The quantification of mtDNA mutation load was carried out by fluorescent RFLP (restriction fragment length polymorphism) and monitored on a regular basis during the experiments as shown before [25].
To validate the cybrid cells as a MELAS model, and to determine whether the introduction of the m.3243A>G mutation in SH-SY5Y cells induces metabolic modifications; several metabolites including glucose, pyruvate and lactate were assayed in controls, heteroplasmic cybrids (70% M) and apparently homoplasmic cybrids (100% M). The glycolytic metabolism increased significantly in mutant cells (Fig. 1). After 3 days of culture, the glucose consumption was significantly higher in mutant cells relative to controls: 3.5 fold for 70% M cybrids, and nearly 6.5 fold for 100% M cybrids (Fig. 1A). Pyruvate production was not modified in 70% M cybrids but was slightly decreased in 100% M cybrids. Alanine concentration was significantly higher in 100% M cybrids. These findings underscore the major metabolic adaptation of mutant cells towards glycolysis, with a derivation of pyruvate from the Krebs cycle to the production of lactate and alanine, as commonly seen in MELAS patients. Lactate dehydrogenase (LDH) activity increased significantly in 70% M as well as 100% M cybrids, emphasizing the increase in the glycolytic metabolism (Fig. 1B).
Figure 1.

Cellular metabolism of the m.3243A>G SH-SY5Y cybrid cells. (A) Glucose consumption, pyruvate, lactate and alanine production in controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). (B) Enzymatic activity of lactate dehydrogenase in controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). The results are expressed as fold changes relative to mean control cell values. Data are presented as means ± sem of 4 independent experiments. Differences between groups were evaluated by a Mann-Whitney statistical test, the asterisk (*) indicates significant differences (p<0.05) compared to controls.
3.2 Mitochondrial impact of the m.3243A>G mutation on SH-SY5Y cells depends on the mutant load
To investigate the metabolic switch towards glycolysis induced by the introduction of the m.3243A>G mtDNA mutation in SH-SY5Y mutant cells, we studied the mitochondrial oxidative metabolism of the cybrids. In 100% M cybrids, respiratory rates supported by complexes I and II were 50% lower compared to control cells, whereas respiration with complex IV substrates (ascorbate + TMPD) was unaffected. However, in 70% M cybrids, respiratory rates driven by complexes I and IV increased significantly with the malate and pyruvate respiration being 2.5 times greater than in control cells (Fig. 2A). These results demonstrate that, in 100% M cybrids, the metabolism relies mainly on glycolysis because of mitochondrial respiratory chain dysfunction. The increased glycolytic metabolism in 70% M cybrids is correlated with an increase of respiratory chain activity. We hypothesize that in these cells, the oxidative phosphorylation of the remaining 30% wild type mitochondria compensates the energetic defect due to the mutant load. To investigate the consequences of the mitochondrial dysfunction on energy production, we measured the total ATP content of mutant cybrids and controls (Fig. 2B). ATP levels were not lower in the mutant cybrids compared to controls, underlining the crucial compensatory role of glycolysis in the energetic metabolism of mutant cells.
Figure 2.

Oxygen consumption assays on cybrid cells. (A) Mitochondrial oxygen consumption measured on digitonin-permeabilized controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). The results show complex I, complex II and complex IV-dependent respiratory rates. (B) Total ATP content of controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). The results are expressed as fold changes relative to mean control cell values. The data are presented as means ± sem of 4 independent experiments. Differences between groups were evaluated by a Mann-Whitney statistical test, the asterisk (*) indicates significant differences (p<0.05) compared to controls.
In addition, the analysis of the mitochondrial network showed that in 70% M and 100% M cybrids, the number of unconnected and fragmented mitochondria was 48% and 26% greater, respectively, than in controls (Fig. 3). This indicates that the mutation alters mitochondrial dynamics and fragmented mitochondria were much higher in 70% M compared to 100% M. The maximal enzymatic activities of respiratory chain complexes I, II and IV were significantly higher in 70% M cybrids than in controls (Fig. 4A). In 100% M cybrids, the activities of complexes I and IV were decreased, complex I being the most affected, with a residual activity of 25%. These results correlated with the western blot analysis of the quantity of respiratory chain complexes (Fig. 4B). Complex IV (COXII) and Complex I (20 kDa) subunits were increased in 70% M cybrids, but severely reduced in 100% M cybrids compared to controls. It has been suggested that residual COX activity and the presence of COX-positive fibres in muscle tissue from MELAS patients may be involved in the pathogenesis of this disorder [8]. To discriminate between the effects of the mutation on COX activity with respect to the mtDNA-encoded and the nuclear DNA-encoded subunits, we quantified the protein expression of COX II, which is mtDNA-encoded, and that of COXIV, which is nuclear DNA-encoded. The expression of the COX II subunit decreased in 100% M cybrids whereas COXIV appeared to be strongly expressed (Fig. 4B). This highlights the selective effect of the m.3243A>G mutation on mtDNA-encoded respiratory chain complex subunits, suggesting the existence of a regulatory mechanism of complex IV that compensates for the mitochondrial defect through a differential expression of nuclear DNA-encoded subunits.
Figure 3.

The m.3243A>G mutation affects the structure of the mitochondrial network. Control SHSY5Y, 70% M cybrids and 100% M cybrids were cultured on chamber-slides (Lab-Tek). Mitochondria were labelled with MitoTracker® Green 100 nM, 15min in DMEM-High Glucose, 10% FBS. Representative images are shown here. A colour code indicates the different types of mitochondria. Connected mitochondrial networks are shown in purple/blue whereas isolated and spherical mitochondria are shown in yellow/red. The histogram shows the percentage of connected or unconnected mitochondria per cell line. The results are the means of two independent experiments.
Figure 4.

Respiratory chain complex activities and assembly of the cybrid cells. (A) Maximal activity of complexes I, II and IV of controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). (B) Representative western blot analysis of subunits of Complex I (20 kDa), II (30 kDa), III (core 2), IV (COXII, mtDNA-encoded and COX IV, nuclear DNA-encoded) and V (F1α) and of a reference loading protein (α-tubulin). (C) Blue-Native PAGE analysis of the respiratory chain complex assembly of cybrid cells. Holoenzyme complexes and sub-complexes were visualized by enhanced chemiluminescence after primary antibody incubation (NDUFB6 for Complex I, COX I for Complex IV and II 70 kDa for Complex II used as loading reference).
Defects in the mtDNA-encoded subunits of respiratory chain complexes have been shown to interfere with the assembly of subunits of complexes I, IV and V [28]. We studied the assembly of respiratory chain complexes using the Blue-Native PAGE technique on controls, 70% M and 100% M cybrids. Complex I, the largest component of the respiratory chain, encompasses 45 structural subunits assembled in a structure of nearly 1000 kDa [29]. The assembly of nuclear DNA-encoded subunits requires fine coordination with mtDNA-encoded subunits in a sequential process involving several assembly intermediates of various molecular weights. In our study, complex I assembly was altered in mutant cells with the presence of low molecular weight assembly intermediates, most abundantly in 100% M cybrids (Fig. 4C). The Blue-Native PAGE results show that the respiratory chain complex assembly was severely altered in the mutant cells.
Modifications of the assembly and activity of the respiratory chain complexes may impair the respiratory function and lead to oxidative stress. We used four techniques to measure oxidative stress in the SH-SY5Y cybrid cells: the nitrite-nitrate assay, the reduction in GSH production, the protein expression of MnSOD, and the aconitase/fumarase ratio (Fig. 5). The results showed that the m.3243A>G mutation in SH-SY5Y cells triggered a significant decrease of the nitrite-nitrate concentration as a marker of reduced NO synthase activity (Fig. 5A). Moreover, GSH production was severely decreased in mutant cells compared to controls (Fig. 5B). However, the expression of MnSOD (a mitochondrial superoxide detoxifying enzyme) remained unchanged in the mutant cells (Fig. 5C). The decrease of both the GSH concentration and NO synthase activity strongly suggested that the mutant cells undergo a nitrosative stress as evidenced by the reduction of the aconitase/citrate synthase ratio in 70% M as well in 100% M cybrids (Fig. 5D). Indeed, aconitase, a Krebs cycle enzyme, is commonly used as an oxidative stress biomarker since it is highly sensitive to the presence of ROS. With the oxidative/nitrosative stress, aconitase undergoes reversible inactivation of its iron/sulphur clusters, and under conditions of prolonged oxidative/nitrosative stress, irreversible aconitase inactivation may occur [30]. Aconitase activity was normalized with respect to citrate synthase activity, another Krebs cycle enzyme that does not contain an Fe/S center and is thus insensitive to ROS. The decreased aconitase/citrate synthase ratio in cybrids compared to controls (Fig. 5D) is strongly indicative of the oxidative/nitrosative stress in the mutant cells.
Figure 5.

Oxidative stress measurements in cybrid cells. (A) The concentration of nitrates + nitrites concentration in the supernatant of controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). (B) GSH concentration in the supernatant of controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). (C) Western Blot analysis of antioxidant mitochondrial MnSOD. The beta subunit of complex V (Vbeta) was used as loading reference. (D) Ratio of aconitase/citrate synthase activity in controls (open bars), 70% M cybrids (grey bars) and 100% M cybrids (black bars). The results are expressed as fold changes relative to means of control cell values. The data are presented as means ± sem of 4 independent experiments. Differences between groups were evaluated by a Mann-Whitney statistical test; the asterisk (*) indicates significant differences (p<0.05) compared with controls.
3.3 L-arginine treatment improves mitochondrial enzyme activity and increases the formation of supercomplexes
Since NO is a potent regulator of the mitochondrial energetic metabolism, the significant decrease of NO synthase activity observed in the SH-SY5Y cybrids could lead to a respiratory chain defect. To compensate the reduced NO synthesis in the cybrids, the cells were incubated with 1 mM L-arginine (an NO-donor) for three days. The L-arginine levels were higher in treated cybrid cells and controls versus untreated cells, with an increase of L-arginine concentrations of 70%, 127% and 148% in controls, 70% M and 100% M cybrids, respectively (data not shown). L-arginine treatment led to a significant increase of complex I activity in controls and 100% M cybrids, with a 2- fold change in treated versus untreated cells, but did not affect the 70% M cybrids (Fig. 6A). Complex IV activity was higher in L-arginine-treated control cells but not in mutant cells (Fig. 6B). L-arginine did not modify the assembly of complex IV in controls or in cybrids (Fig. 6D). However, although L-arginine did not increase the activity of the respiratory complexes in 70% M cybrids, it stabilized the formation of supercomplex I:III (Fig. 6C).
Figure 6.

Mitochondrial respiratory chain enzyme assays and assembly of the arginine-treated cybrid cells. (A) The ratio of complex I/citrate synthase in controls (open bars), and in arginine-treated 70% M cybrids (grey bars) and 100% M cybrids (black bars). (B) The ratio of complex IV/citrate synthase in controls (open bars), and in arginine-treated 70% M cybrids (grey bars) and 100% M cybrids (black bars). The results are expressed as fold changes relative to means of control cell values. The data are presented as means ± sem of 4 independent experiments. Differences between groups were evaluated by a Mann-Whitney statistical test; the asterisk (*) indicates significant differences (p<0.05) compared with controls and the sharp sign (#) indicates significant differences between treated and untreated cells (p<0.05). (C) Blue-Native PAGE analysis of complex I assembly in arginine-treated cybrid cells. The holoenzyme complexes and sub-complexes were visualized by enhanced chemiluminescence after primary antibody incubation (NDUFB6). (D) Blue-Native PAGE analysis of complex IV assembly of arginine-treated cybrids (COX I antibody).
3.4 Heteroplasmy shifting can alleviate mitochondrial respiratory chain dysfunction
For mutant cells carrying the heteroplasmic mtDNA deletion, the ketogenic medium has been shown to shift the heteroplasmy level towards the wild type suggesting that a ketogenic diet may represent an interesting therapeutic approach in mtDNA-related diseases [31]. Since the mutant cells rely on glycolysis, we hypothesized that culturing mutant cells in a low-glucose medium forcing the cells to use oxidative phosphorylation would favour wild type cells and thereby reduce the mutant load. To test this hypothesis, the apparently homoplasmic 100% M cybrids were kept in low-glucose medium (0.5 g/l) for four weeks. After a high mortality phase, clones progressively appeared and finally recovered the same growth rate as the 70% M cybrids. The quantification of the mutant load revealed that the heteroplasmy level shifted to 90% M cybrids in these cells (Fig. 7A). Despite a longer four-week period of incubation in a low-glucose medium, the heteroplasmy level did not fall below 90% M. Table 1 shows that in 90% M cybrids, the enzyme activity of complex I was improved and complex IV was restored compared with 100% M cybrids. Moreover, in the 90% M cybrids, we observed an increase in the holoenzyme forms of complexes I and IV compared with 100% M cybrids (Fig. 7).
Figure 7.
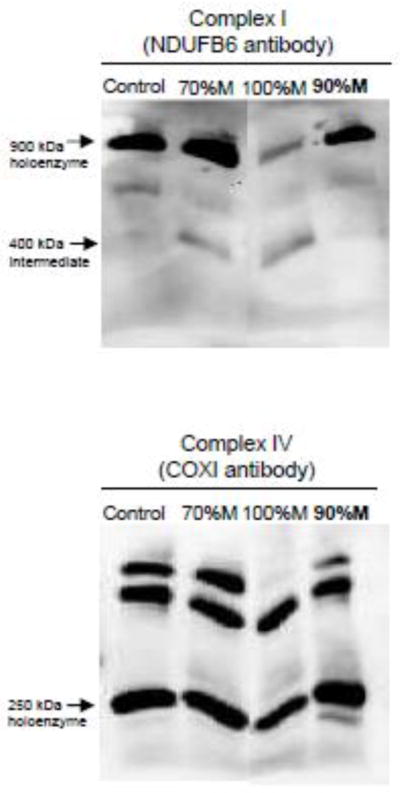
Blue-Native PAGE analysis of respiratory chain complex assembly of 90% M cybrids compared to 70% and 100% M cybrid cells and control cells. All cell lines were cultured in the same media. Holoenzyme complexes and sub-complexes were visualized by enhanced chemiluminescence after primary antibody incubation (NDUFB6 for complex I and COX I for complex IV).
TABLE 1.
Enzymatic respiratory chain complexes I and IV of the 90% M cybrid cells. Enzymatic activities of complexes I and IV normalized with citrate synthase activity of controls, 70% M cybrids, 90% M cybrids (in bold) and 100% M cybrids. The results are expressed as fold changes relative to means of control cell values. The data are presented as means ± sem of 4 independent experiments.
| Complex Activity/Citrate Synthase Ratio (relative to Control Cell)
|
||
|---|---|---|
| Complex I | Complex IV | |
| Control | 1.00 ± 0.16 | 1.00 ± 0.15 |
| 70% M | 1.67 ± 0.20* | 2.53 ± 0.16* |
| 100 % M | 0.19 ± 0.02* | 0.52 ± 0.04* |
| 90% M | 0.53 ± 0.04*# | 0.72 ± 0.12# |
Differences between groups were evaluated by a Mann–Whitney statistical test; the asterisk (*) indicates significant differences (p<0.05) of mutant cells compared with control cells; the sharp sign (#) indicates significant differences between 100% M cells cultured in high glucose medium and the 90% M (derived from 100% M cells) cultured in low glucose (p<0.05).
4. DISCUSSION
The consequences of the m.3243A>G mitochondrial mutation have been extensively studied on skin fibroblasts or osteosarcoma 143TK− derived cybrid cells. The mutation essentially inhibits mitochondrial protein synthesis with a deficiency of aminoacylation, leading to complex I deficiency [15–16]. However, the consequences of the mutation on neuronal cells close to the clinical MELAS phenotype have not yet received much attention. We have therefore created a neuronal-like model of the MELAS syndrome, which is known to be associated with recurrent focal neurological deficits and stroke-like episodes. The neuronal-like model consists of a series of transmitochondrial cybrid cell lines combining the nuclear background of a neuroblastoma cell line (SH-SY5Y) and enucleated fibroblast mitochondria carrying the m.3243A>G mutation at 70% and 100% mutant loads.
Our study shows as expected that the biochemical consequences of the m.3243A>G mutation in SH-SY5Y cells depend on the level of heteroplasmy, and that homoplasmic mutant cells are subject to severe reduction of respiratory chain complex activity, with complexes I and IV being the most affected. COXIV, a nuclear DNA-encoded complex IV subunit, is differentially expressed compared to COXII, an mtDNA-encoded subunit. This is in accordance with a previous study [32], suggesting that the m.3243A>G mutation accelerates mtDNA-encoded protein degradation and leads to a disequilibrium between nuclear DNA-encoded and mtDNA-encoded subunits, thereby impeding respiratory chain function. This hypothesis is reinforced by our Blue-Native PAGE findings that show that complex I is misassembled in 100% M cybrids. Surprisingly, Complex II driven respiration is decreased by 50 % in 100% M cybrids without reduction of enzymatic maximal activity emphasizing a functional effect on the respiratory chain when the mutation is apparently homoplasmic; a similar complex II reduction has been reported in a study on brain capillary endothelial cybrid cells carrying the m.3243A>G mutation [18].
In our model, the severe mitochondrial respiratory chain defects provoked by the m.3243A>G mutation were compensated by an increase in the glycolytic metabolism that ensured normal ATP levels in the cybrids. This increase was accompanied, particularly in 100% M cybrids, by high lactate and alanine concentrations as described in MELAS patients, in whom chronic lactic acidosis leads to cellular dysfunction [33].
Ketogenic diets reduce the percentage of deleted mtDNA molecules in the presence of large-scale heteroplasmic deletions [31]. Moreover, such diets slow down the progression of myopathy in the Deletor mouse model, a disease model for progressive, late-onset mitochondrial myopathy [34], and increase mitochondrial glutathione levels [35]. In our study, the respiratory defect caused by the m.3243A>G mutation in 100% M cybrids was compensated by increased glycolysis. We therefore hypothesized that metabolic interventions may effectively restore mitochondrial function in SH-SY5Y cybrid cells. A small decrease of the mutant load in cells may be enough to rescue the biochemical phenotype and then has the potential to modulate the clinical phenotype if you pass under the pathogenic threshold of the mutation. We first tested metabolic switching by maintaining the apparently homoplasmic mutant cybrids in a low-glucose medium (0.5 g/l instead of 4.5 g/l). After four weeks in this culture medium, the mutant load shifted from 100% M to 90% M, accompanied by the increased formation of complex I holoenzyme, improving complex I activity. However, keeping the cybrids for a longer time in the selective medium did not decrease the heteroplasmy level below 90% M.
The reason why we were not able to reduce below 90% of mutant load even after long term exposure is not well understood. Maybe more severe metabolic conditions ie no glucose or with the use of galactose should be a better way in reducing the mutant load. Further experiments are needed to better understand how heteroplasmy shifting might be useful. Indeed, the concept of reducing the mutant load and then increasing normal mtDNA as a means to correcting the biochemical defect has generated a lot of interest [31].
Interestingly, the mitochondrial network was analysed showing that in mutant cybrids, the number of fragmented and unconnected mitochondria was higher compared to control cells, as an indication of the alteration of mitochondrial dynamics due to the m.3243A>G mutation. Moreover, the percentage of fragmented mitochondria was much higher in the heteroplasmic mutant 70% M than the apparently homoplasmic 100% M. The aconitase/citrate synthase ratio was used as an index of ROS generation in cybrid cells. In parallel similar to the level of unconnected mitochondria in 70% M, the reduction of the aconitase/citrate synthase ratio was higher in 70% M compared to 100% M cybrids suggesting a direct link between ROS production and the modifications of mitochondrial fusion/fission machinery. This situation is similar to previous observation showing that a heteroplasmic mutation in ND5 gene carrying about 72% of mutant mtDNA appeared to be more deleterious producing more ROS and promoting tumorigenesis than the same mutation in a near homoplasmic state [36]. Increased oxidative stress and lipid peroxidation has been reported in cybrid cells harbouring 90% of mutated mtDNA copies [37]. Moreover, oxidative damages were significantly increased around stroke like brain lesions in MELAS patients [38]. In our model of neuronal-derived mutated cybrids, the mitochondrial respiratory chain impairment led to oxidative/nitrosative stress as shown by the reduction in the GSH level, in NOS activity and in the aconitase/citrate synthase activity ratio.
It has been hypothesized that oxidative stress may aggravate mitochondrial bioenergetic defects and inhibit the ubiquitin-proteasome pathway and protease activity in human cells harbouring mutant mtDNA, thereby leading to the accumulation of misfolded proteins, a condition that could trigger mitochondrial apoptosis [39]. Nitrosative stress may also be deleterious since S-nitrosylation has been shown to compromise the dynamics of the mitochondrial fission-fusion process, altering protein folding and leading to neurotoxicity [40]. However, nitric oxide is also an important second messenger especially in endothelial and neuronal cells. Indeed, increase in NO production through the stimulation of eNOS activity has been reported to protect against stroke through a wide range of cellular effects, including vasodilation, antithrombotic, anti-inflammatory and anti-proliferative activities [41]. One of the striking clinical symptoms in MELAS is the occurrence of stroke-like episodes, which is supposed to be linked to the NO metabolism [11]. Our study showed that NOS activity was significantly reduced in mutated cybrid cells, resulting in a decrease in the NO level. We therefore hypothesized that restoring the NO level in mutant cells would reduce mitochondrial dysfunction, and then as a consequence will be beneficial for ameliorating the neuronal and endothelial functions in MELAS patients. The action of L-arginine as an NO donor [11] makes it a good candidate for the treatment of the stroke-like episodes in MELAS patients. Indeed, the efficacy of L-arginine supplementation in MELAS patients, carrying either the m.13513G>A or the m.3243A>G mutations, has been shown to be beneficial in both short and long term treatments [11–12, 42]. L-arginine has been proposed as a treatment for MELAS m.3243A>G syndrome patients during the acute stroke phase or long term use [9, 12, 22]. L-arginine is administrated either by intravenous injections (0.5g/kg/day) or orally between crises at 0.15–0.3g/kg/day [22]. Since NO is the vasodilator, loss of NO would result in chronic vasoconstriction and transient ischemia. As the substrate of NOS, L-arginine should stimulate NOS, to generate more NO and alleviate the vasoconstriction. However, the consequences and biochemical rationale of L-arginine therapy on mitochondrial metabolism has not yet been studied. In our in vitro neuronal-like model, L-arginine treatment improved complex I enzymatic activity and stabilized the formation of supercomplexes I + III, which have recently been shown to be benefit cell metabolism while reducing the formation of ROS [43].
5. CONCLUSIONS
Our results emphasize the impact of the m.3243A>G mutation on neuronal mitochondrial metabolism and the usefulness of the MELAS neuronal-like model for the evaluation of therapeutic strategies against the disease. We tested two strategies likely to compensate for the biochemical defects produced by the mitochondrial mutation. The first strategy, involving the use of a low-glucose medium, shifted the heteroplasmy of the cybrids towards the wild type, significantly improving the mitochondrial function deteriorated by the m.3243A>G mutation. The second strategy, based on L-arginine treatment, improved the bioenergetic parameters of the cybrids, in which the mutation had led to defective NO metabolism, thus confirming the hypothesis that L-arginine, an NO donor, may constitute an effective treatment for the stroke-like episodes in the MELAS syndrome.
Supplementary Material
Research Highlights.
The m.3243A>G mutation induces a metabolic switch in SH-SY5Y cybrid cells
Oxidative stress contributes to the pathophysiology of MELAS
MELAS neuronal-like cells are a good model for the evaluation of therapeutic strategies
Heteroplasmy shifting can alleviate mitochondrial respiratory chain dysfunction
L-arginine a nitric oxide (NO) donor reduces complex I activity in MELAS neuronal-like model
Acknowledgments
The authors thank Stephanie Chupin for cell culture and Cédric Gadras for mass spectrometry analysis. We are grateful to Kanaya Malkani for critical reading of the manuscript. This work was supported by grants from the University Hospital of Angers; the Association contre les Maladies Mitochondriales (AMMi); and the Union Nationale des Aveugles et Déficients Visuels (UNADEV); and by National Institutes of Health grants [NS21328, AG24373, DK73691] awarded to DCW.
Abbreviations
- CNS
central nervous system
- COX
cytochrome C oxidase
- FBS
fetal bovine serum
- GSSG
glutathione disulfide
- GSH
glutathione
- LDH
lactate dehydrogenase
- MnSOD
manganese superoxide dismutase
- mtDNA
mitochondrial DNA
- MELAS
mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
- NO
nitric oxide
- RFLP
restriction fragment length polymorphism
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruiz-Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 5.Uusimaa J, Moilanen JS, Vainionpaa L, Tapanainen P, Lindholm P, Nuutinen M, Lopponen T, Maki-Torkko E, Rantala H, Majamaa K. Prevalence, segregation, and phenotype of the mitochondrial DNA 3243A>G mutation in children. Annals of neurology. 2007;62:278–287. doi: 10.1002/ana.21196. [DOI] [PubMed] [Google Scholar]
- 6.Shanske S, Pancrudo J, Kaufmann P, Engelstad K, Jhung S, Lu J, Naini A, DiMauro S, De Vivo DC. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A. 2004;130A:134–137. doi: 10.1002/ajmg.a.30220. [DOI] [PubMed] [Google Scholar]
- 7.Maassen JA, THLM, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, Raap AK, Janssen GM, Lemkes HH. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53(Suppl 1):S103–109. doi: 10.2337/diabetes.53.2007.s103. [DOI] [PubMed] [Google Scholar]
- 8.Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133–158. doi: 10.1196/annals.1444.011. [DOI] [PubMed] [Google Scholar]
- 9.Koga Y, Akita Y, Junko N, Yatsuga S, Povalko N, Fukiyama R, Ishii M, Matsuishi T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology. 2006;66:1766–1769. doi: 10.1212/01.wnl.0000220197.36849.1e. [DOI] [PubMed] [Google Scholar]
- 10.Sakuta R, Nonaka I. Vascular involvement in mitochondrial myopathy. Annals of neurology. 1989;25:594–601. doi: 10.1002/ana.410250611. [DOI] [PubMed] [Google Scholar]
- 11.Koga Y, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T. MELAS and L-arginine therapy: pathophysiology of stroke-like episodes. Ann N Y Acad Sci. 2010;1201:104–110. doi: 10.1111/j.1749-6632.2010.05624.x. [DOI] [PubMed] [Google Scholar]
- 12.Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K, Matsuishi T. MELAS and L-arginine therapy. Mitochondrion. 2007;7:133–139. doi: 10.1016/j.mito.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 13.King MP, Koga Y, Davidson M, Schon EA. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12:480–490. doi: 10.1128/mcb.12.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flierl A, Reichmann H, Seibel P. Pathophysiology of the MELAS 3243 transition mutation. J Biol Chem. 1997;272:27189–27196. doi: 10.1074/jbc.272.43.27189. [DOI] [PubMed] [Google Scholar]
- 15.Chomyn A, Enriquez JA, Micol V, Fernandez-Silva P, Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J Biol Chem. 2000;275:19198–19209. doi: 10.1074/jbc.M908734199. [DOI] [PubMed] [Google Scholar]
- 16.Park H, Davidson E, King MP. The pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR) decreases the efficiency of aminoacylation. Biochemistry. 2003;42:958–964. doi: 10.1021/bi026882r. [DOI] [PubMed] [Google Scholar]
- 17.Attardi G, Yoneda M, Chomyn A. Complementation and segregation behavior of disease-causing mitochondrial DNA mutations in cellular model systems. Biochimica et biophysica acta. 1995;1271:241–248. doi: 10.1016/0925-4439(95)00034-2. [DOI] [PubMed] [Google Scholar]
- 18.Davidson MM, Walker WF, Hernandez-Rosa E. The m.3243A>G mtDNA mutation is pathogenic in an in vitro model of the human blood brain barrier. Mitochondrion. 2009;9:463–470. doi: 10.1016/j.mito.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swerdlow RH. Does Mitochondrial DNA Play a Role in Parkinson’s Disease? A Review of Cybrid and Other Supportive Evidence. Antioxidants & redox signaling. doi: 10.1089/ars.2011.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD., Jr Origin and functional consequences of the complex I defect in Parkinson’s disease. Annals of neurology. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- 21.Li R, Guan MX. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol Cell Biol. 30:2147–2154. doi: 10.1128/MCB.01614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, Fujimoto S, Matsuishi T. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 23.Ishikawa K, Hayashi JI. Chapter 19 Generation of mtDNA-exchanged cybrids for determination of the effects of mtDNA mutations on tumor phenotypes. Methods in Enzymology. 2009;457:335–346. doi: 10.1016/S0076-6879(09)05019-8. [DOI] [PubMed] [Google Scholar]
- 24.Nochez Y, Arsene S, Gueguen N, Chevrollier A, Ferre M, Guillet V, Desquiret V, Toutain A, Bonneau D, Procaccio V, Amati-Bonneau P, Pisella PJ, Reynier P. Acute and late-onset optic atrophy due to a novel OPA1 mutation leading to a mitochondrial coupling defect. Mol Vis. 2009;15:598–608. [PMC free article] [PubMed] [Google Scholar]
- 25.Sarzi E, Brown MD, Lebon S, Chretien D, Munnich A, Rotig A, Procaccio V. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet A. 2007;143:33–41. doi: 10.1002/ajmg.a.31565. [DOI] [PubMed] [Google Scholar]
- 26.Desquiret V, Gueguen N, Malthiery Y, Ritz P, Simard G. Mitochondrial effects of dexamethasone imply both membrane and cytosolic-initiated pathways in HepG2 cells. Int J Biochem Cell Biol. 2008;40:1629–1641. doi: 10.1016/j.biocel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 27.El Meziane A, Lehtinen SK, Hance N, Nijtmans LG, Dunbar D, Holt IJ, Jacobs HT. A tRNA suppressor mutation in human mitochondria. Nat Genet. 1998;18:350–353. doi: 10.1038/ng0498-350. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochimica et biophysica acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 29.Lazarou M, Thorburn DR, Ryan MT, McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochimica et biophysica acta. 2009;1793:78–88. doi: 10.1016/j.bbamcr.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 31.Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Annals of neurology. 2004;56:662–669. doi: 10.1002/ana.20240. [DOI] [PubMed] [Google Scholar]
- 32.Janssen GM, Maassen JA, van Den Ouweland JM. The diabetes-associated 3243 mutation in the mitochondrial tRNA(Leu(UUR)) gene causes severe mitochondrial dysfunction without a strong decrease in protein synthesis rate. J Biol Chem. 1999;274:29744–29748. doi: 10.1074/jbc.274.42.29744. [DOI] [PubMed] [Google Scholar]
- 33.Okhuijsen-Kroes EJ, Trijbels JM, Sengers RC, Mariman E, van den Heuvel LP, Wendel U, Koch G, Smeitink JA. Infantile presentation of the mtDNA A3243G tRNA(Leu (UUR)) mutation. Neuropediatrics. 2001;32:183–190. doi: 10.1055/s-2001-17372. [DOI] [PubMed] [Google Scholar]
- 34.Ahola-Erkkila S, Carroll CJ, Peltola-Mjosund K, Tulkki V, Mattila I, Seppanen-Laakso T, Oresic M, Tyynismaa H, Suomalainen A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Human molecular genetics. 2010;19:1974–1984. doi: 10.1093/hmg/ddq076. [DOI] [PubMed] [Google Scholar]
- 35.Jarrett SG, Milder JB, Liang LP, Patel M. The ketogenic diet increases mitochondrial glutathione levels. Journal of Neurochemistry. 2008;106:1044–1051. doi: 10.1111/j.1471-4159.2008.05460.x. [DOI] [PubMed] [Google Scholar]
- 36.Park JS, Sharma LK, Li H, Xiang R, Holstein D, Wu J, Lechleiter J, Naylor SL, Deng JJ, Lu J, Bai Y. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Human molecular genetics. 2009;18:1578–1589. doi: 10.1093/hmg/ddp069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pang CY, Lee HC, Wei YH. Enhanced oxidative damage in human cells harboring A3243G mutation of mitochondrial DNA: implication of oxidative stress in the pathogenesis of mitochondrial diabetes. Diabetes Res Clin Pract. 2001;54(Suppl 2):S45–56. doi: 10.1016/s0168-8227(01)00335-7. [DOI] [PubMed] [Google Scholar]
- 38.Katayama Y, Maeda K, Iizuka T, Hayashi M, Hashizume Y, Sanada M, Kawai H, Kashiwagi A. Accumulation of oxidative stress around the stroke-like lesions of MELAS patients. Mitochondrion. 2009;9:306–313. doi: 10.1016/j.mito.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Wu YT, Wu SB, Lee WY, Wei YH. Mitochondrial respiratory dysfunction-elicited oxidative stress and posttranslational protein modification in mitochondrial diseases. Annals New York Academy of Sciences. 2010;1201:147–156. doi: 10.1111/j.1749-6632.2010.05631.x. [DOI] [PubMed] [Google Scholar]
- 40.Gu Z, Nakamura T, Lipton SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol Neurobiol. 41:55–72. doi: 10.1007/s12035-010-8113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawada N, Liao JK. Targeting eNOS and beyond: emerging heterogeneity of the role of endothelial Rho proteins in stroke protection. Expert Rev Neurother. 2009;9:1171–1186. doi: 10.1586/ern.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shigemi R, Fukuda M, Suzuki Y, Morimoto T, Ishii E. l-arginine is effective in stroke-like episodes of MELAS associated with the G13513A mutation. Brain Dev. 2010 doi: 10.1016/j.braindev.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 43.Lenaz G, Baracca A, Barbero G, Bergamini C, Dalmonte ME, Del Sole M, Faccioli M, Falasca A, Fato R, Genova ML, Sgarbi G, Solaini G. Mitochondrial respiratory chain super-complex I–III in physiology and pathology. Biochimica et biophysica acta. 2010;1797:633–640. doi: 10.1016/j.bbabio.2010.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.