

Abstract
Oral ingestion of monosodium glutamate (MSG) to chronic alcoholic adult male mice at dose levels of 4 and 8 mg/g body weight for seven consecutive days caused a significant increase in lipid fractions, lipid peroxidation, xanthine oxidase, whereas the levels of superoxide dismutase, catalase, glutathione, and its metabolizing enzymes like glutathione peroxidase and glutathione reductase were significantly decreased in the arterial tissue. These observations suggested that ingestion of MSG to chronic alcoholic animals had no beneficial effect and thereby, could act as an additional factor for the initiation of atherosclerosis.
Keywords: Alcohol, lipid peroxidation, monosodium glutamate, oxidative stress and atherosclerosis
INTRODUCTION
Cardiovascular diseases (CVD) are the most alarming of the many health predictions for the new millennium Worldwide. According to WHO report - 2004,[1] 16.7 million people around the globe die because of CVD each year, that is, one-third of the total deaths. Current projections suggested that by the year 2020, India will have the largest CVD burden in the World. The underlining cause of this disease is atherosclerosis, which is a slowly progressive disease that begin in childhood, manifest middle age and later.
In the Modern World, the entrance of Chinese, Japanese, and ready to serve foods like 2-minute noodles, soups, sauces, etc., all containing monosodium glutamate (MSG), has tremendously increased especially in younger generation because of its palate pleasing favorite flavor. In the present era, MSG, an inducer of oxidative stress[2–8] and alcohol, a well-known factor of atherosclerosis,[9–13] is becoming a part of daily food, especially in younger generation. There are many reports in literature that the age for the establishment of atherosclerosis has become 25 to 30 years in the present, where it was about decade back. It is an alarming situation as number of premature deaths due to coronary heart disease is increasing tremendously. So, the present work was designed to observe the effect of MSG in chronic alcoholic adult male mice to ascertain that whether MSG in the presence of alcohol could act in synergism with alcohol for the initiation of atherosclerosis or not by studying its effect on oxidative stress markers like lipid peroxidation (LPO), antioxidant enzymes like xanthine oxidase (XOD), superoxide dismutase (SOD), catalase (CAT), glutathione (GSH) and its metabolizing enzymes such as glutathione peroxidase (GPx), glutathione reductase (GR) along with various fractions of lipid.
MATERIALS AND METHODS
Animals: Normal adult male mice (LAKA, US) weighing 25 to 30 g in body weight were procured from the animal house of Panjab University, Chandigarh, India. Animals were maintained on standard pellet diet (Hindustan Lever Ltd., Bombay) with free access to water.
Grouping: Animals were divided into four groups of six mice each and MSG at dose levels of 0, 4, and 8 mg/g body weight was given orally for seven consecutive days (that is from 31st day to 37th day of alcohol ingestion) to chronic alcoholic (30% ethanol/100 g body weight) adult male mice as follows:
Group-I: (Control): 0 mg MSG/g body weight.
Group-II: Alcohol for 37 days (30% ethanol/100 g body weight).
Group-III: Alcohol for 37 days + 4 mg MSG/g body weight (from 31st to 37th day orally).
Group-IV: Alcohol for 37 days + 8 mg MSG/g body weight (from 31st to 37th day orally).
This experimental design was approved by the Animal Experimental Ethics Committee of Panjab University, Chandigarh, and conducted according to Indian National Science Academy Guidelines for the use and care of experimental animals.
Sample preparation: After the dose period (38th day), animals were fasted overnight and sacrificed by decapitation. The arteries were removed, kept on ice, and washed with ice-cold saline. Ten percent homogenate was prepared in potassium phosphate buffer (100 mM, pH - 7.5) containing 0.15M KCl and was centrifuged at 1 000xg for 15 minutes in cold centrifuge (4°C). The supernatant stored was at 4°C and used for various biochemical assays.
Biochemical assays
Determination of Lipid fractions: The levels of total lipids, phospholipids, triglycerides, and free fatty acids were estimated by applying the methods of Frings et al., 1972,[14] Fiske and Suba Row, 1925,[15] and McGowan et al., 1983,[16] respectively. Total cholesterol levels were assayed by the methods of Zlatkis et al., 1953.[17]
Protein assay: The protein contents were estimated by Lowry et al.'s methods, 1951[18].
Lipid peroxidation: The LPO levels were assayed by measuring the pink color chromophore formed by the reaction of thiobarbituric acid (TBA) with malondialdehyde (MDA) according to the method of Beuge and Aust, 1978.[19]
Xanthine oxidase (EC 1.2.3.2): The activity of XOD was measured by the method of Fried et al. using nitro blue tetrazolium (NBT) which formed farmazan. The increase in the intensity of color with time was measured spectrophotometrically at 540 nm for 10 minutes.[20]
Superoxide dismutase (EC 1.5.1.1): The activity of SOD was assayed by applying the method of Kono.[21] The activity of SOD was measured by monitoring the rate of inhibition of NBT reduction. One unit is defined as the amount of enzyme which caused half-maximal inhibition of NBT reduction.
Catalase (EC 1.11.1.6): The CAT activity was estimated by the method of Luck, 1971,[22] in which decomposition of hydrogen peroxide (H2O2) catalyzed by this enzyme was measured by decrease in absorbance at 240 nm, taking 0.0394 mM-1cm-1 extinction coefficient and enzyme activity was expressed as nmol of H2O2 degraded/min/mg protein.
Reduced glutathione: GSH level was estimated by the method of Beutler et al., 1963,[23] using 5-5’ dithiobis2-notrobenzoic acid.
Glutathione peroxidase (EC 1.11.1.7): GPx activity was measured by using H2O2 as a substrate by applying the method of Rotruck et al., 1973.[24]
Glutathione reductase (EC 1.6.4.2): The activity of GR was estimated by measuring the change in absorbance at 340 nm due to NADPH utilization and GR activity was expressed as nmoles NADPH oxidized/min/mg proteins using an extinction coefficient of 6.22 mM-1cm-1 by the method of William and Arscott, 1971.[25]
Statistical analysis: Results of biochemical analyses are presented as mean value ± standard deviation (S.D.). The difference between control and test groups was analyzed by using Student “t” test (significant difference at P<0.05 confidence level). Correlation between the investigated groups was performed using test ONE-WAY ANOVA (one-way variance analysis).
RESULTS AND DISCUSSION
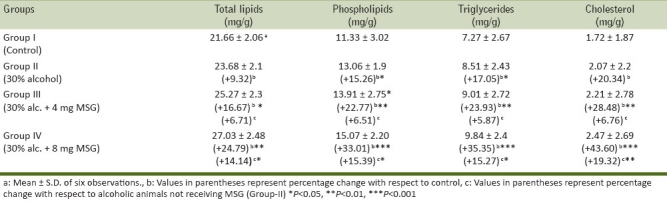
Ingestion of alcohol significantly increased various lipid fractions that are total lipids by 9.32%, phospholipids by 15.26%, triglycerides by 17.05%, and cholesterol by 20.34% as compared with control mice (Group-1). The maximal increase was observed in cholesterol level [Table 1]. It is well reported that alcohol is oxidized to acetaldehyde by alcohol dehydrogenase containing NAD+ and producing NADH + H+. NADH is used to synthesize glycerol and fatty acids and hence producing hyperlipidemia.[26,27] Furthermore, NADH by entering in electron transport chain produces ATP.[28] The high energetic state inhibits the normal oxidation of fat in fatty acid spiral and citric acid cycle and could be the reason for elevated level of various lipid fractions in arterial tissue in alcoholic animals. The oral ingestion of MSG at the dose level of 4 mg/g body weight and 8 mg/g body weight along with alcohol further increased the level of various lipid fractions (total lipids by 16.67% and 24.79%, phospholipids by 22.77% and 33.01%, triglycerides by 23.93% and 35.35%, and cholesterols by 28.48% and 43.60%) with respect to control animals in arterial tissue and a significant increase was also found in various lipid fractions in arterial tissue of MSG-ingested alcoholic animals as compared with alcoholic animals not receiving MSG [Table 1]. Previously, we have reported that MSG induced hyperlipidemia and hypertriglyceridemia in normal mice.[2,5,29] A significant increase in various lipid fraction levels suggested that MSG along with alcohol had no beneficial effect and therefore enhances hyperlipidemia.
Table 1.
Effect of oral ingestion of MSG at different dose levels (0, 4, and 8 mg/g body weight) for consecutive 7 days on various fractions of lipids in arterial tissue of chronic alcoholic adult male mice
The activity of XOD, a superoxide-initiating enzyme, was found to be significantly increased by 9.271% in chronic alcohol group (Group-II), 16.55% in 4 mg MSG/g body weight and 21.19% in 8 mg MSG/g body weight with respect to control animals and 6.66% and 18.90% (P<0.05) increase was observed in group-III and group-IV, respectively, with respect to chronic alcoholic group-II [Table 2]. XOD, a highly versatile enzyme that is widely distributed from bacteria to human, exists predominantly as NAD+-dependent xanthine dehydrogenase (XDH) that itself has no role in the start or potentiation of oxidative damage in cells. However, in many pathological conditions, XDH is converted into XOD.[30] XOD catalyses the oxidation of hypoxanthine/xanthine to uric acid and generates superoxide radical (O2.-). H2O2 formed from O2- could be converted into highly reactive hydroxyl radical (.OH) leading to oxidative stress as a result of oxidation of biological molecules. A significant increase in XOD activity in arterial tissue of 4 and 8 mg MSG/g body weight-ingested alcoholic animals could produce a burst of free radicals. Once O2 radical is produced, then H2O2 and OH are continuously produced by Haber-Weiss reaction and/or Fenton type reaction.[31] Oxygen radicals might cause the LPO of biomembrane through a chain reaction. The first step is the initiation reaction, which begins by taking out hydrogen atom from polyunsaturated fatty acid by oxygen radical. The second is the propagation and the final step is termination. The extent of LPO has often been determined by the TBA test, which has also been considered for the detection of MDA. A significant increase in LPO levels from 4.16 ± 0.19 to 5.89 ± 0.15 nmoles of MDA/min/mg protein in all the treated groups [Table 2] that was observed in the present study might lead to susceptibility of the biomembrane, which ultimately leads to tissue injury/damage.
Table 2.
Effect of oral ingestion of MSG at different dose levels (0, 4, and 8 mg/g body weight) for consecutive 7 days on LPO, XOD, CAT, in arterial tissue of chronic alcoholic adult male mice
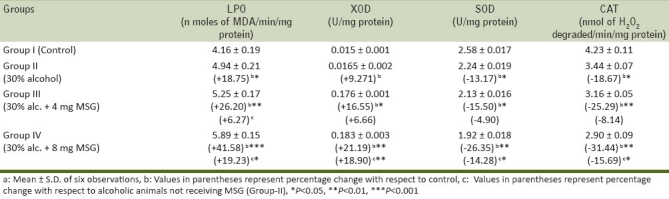
SOD, a superoxide radical scavenging enzyme level was decreased by 13.17% (P<0.05) in group-II with respect to Group-I and SOD activity significantly decreased by 15.50% (P<0.05) and 26.35% (P<0.01) in Group-III and Group-IV as compared with control animals, respectively [Table 2]. A similar trend was found in MSG-ingested alcoholic mice (Group-III and Group-IV) with respect to chronic alcoholic (Group-II) animals [Table 2]. SOD is considered the first line of defense against the deleterious effect of oxygen radicals in the cells and it scavenges reactive oxygen radical species by catalyzing the dismutation of O2- radical to H2O2 and O2. In mammals, three isozymes of SOD; CuZn-SOD, Mn-SOD, and extra cellular-SOD[32] CuZn-SOD are located primarily in the cytosol. CuZn-SOD consists of two protein subunits; each has an active site containing one Cu ion and one Zn ion. Cu ion serves as active redox site and Zn ion maintain the protein structure. Mn-SOD is located in mitochondrial matrix.[33] It has four subunits each with Mn ion. EC-SOD is present in plasma, bound to heparin sulfate ion, the surface of endothelial cells. EC-SOD is tetrameric glycoprotein, which contains Cu and Zn ion. The presence of SOD in various compartments of the our body enables it to dismutate O2.- radicals immediately and protects the cells from oxidative damage. A significant inhibition of SOD activity in arterial tissue of 4 mg MSG/g body weight-ingested alcoholic mice and above may result in an increased flux of O2- radical and hence reflects the tissue damage/injury.
The activity of CAT, another potent antioxidant enzyme, especially against the O2- radicals and singlet oxygen, was also significantly decreased in arterial tissue by 18.67% in Group-II, by 25.29% in Group-III, and by 31.44% in Group-IV, with respect to control animals [Table 2] and a similar trend in the activity of CAT was also observed in 4 and 8 mg MSG/g body weight-treated alcoholic mice (Group-III and Group-IV) as compared with chronic alcoholic (Group-II) animals [Table 2]. CAT protects cells from the accumulation of H2O2 by dismutating it to form H2O and O2 or by using it as an oxidant, in which it works as a peroxidase. Therefore, the decrease in the activity of CAT observed in present work could be due to less availability of NADH as MSG favors lipogenesis,[2] and hence MSG along with alcohol had no beneficial effect on CAT activity to reduce lipogenesis/oxidative stress.
GSH, a tripeptide, is maintained in reduced state by an efficient GPx/GR system. GSH, a potent endogenous antioxidant, helps to protect cells from a number of noxious stimuli including oxygen derived free radicals.[34,35] A significant decrease in GSH levels might be accompanied by a significant increase in LPO level. In the present work, the level of GSH is significantly decreased by 19.66% in chronic alcoholic mice (Group-II), 28.15% (P<0.01) in 4 mgMSG/g body weight, and 41.99% (P<0.001) in 8 mgMSG/g body weight-ingested alcoholic mice with respect to control mice and a significant decrease was also observed by 18.57% (P<0.05) and 27.79 (P<0.01) in 4 and 8 mgMSG/g body weight-ingested alcoholic mice (Group-III and Group-IV) as compared to alcoholic mice not receiving MSG [Table 3]. Reduced levels of GSH confirm an increased susceptibility to oxidative damage and this observation is an agreement with the reports that inverse relationship exists between LPO and GSH status. GSH depletion of 20 to 30% can impair the cell defense against the toxic action of xenobiotic and may lead to cell injury/death.[4,36]
Table 3.
Effect of oral ingestion of MSG at different dose levels (0, 4, and 8 mg/g body weight) for consecutive 7 days on GSH, GR, and GPx in arterial tissue of chronic alcoholic adult male mice
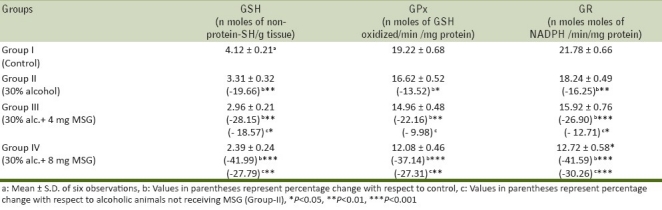
The activity of GPx, a selenium-containing enzyme, was found to be decreased by 13.52% (from 19.22 ± 0.68 to 16.62 ± 0.52 nmoles of GSH oxidized/min/mg protein, P<0.05), by 22.16% (from 19.22 ± 0.68 to 14.96 ± 0.48 nmoles of GSH oxidized/min/mg protein, P<0.01), and by 37.14% (from 19.22 ± 0.68 to 12.08 ± 0.46 nmoles of GSH oxidized/min/mg protein, P<0.001) in Group-II, Group-III, and Group-IV, respectively, with respect to control mice and a significant inhibition (P<0.01) in GPx activity was also found by 9.98% in Group-III and 27.31% (P<0.01) in Group-IV as compared with alcoholic group (Group-IV, Table 3). GPx catalyses the reduction of variety of H2O2 (ROOH and H2O2) using GSH as a substrate, thereby protecting mammalian cells against oxidative stress.[37] It is well reported that low activity of this enzyme may render the tissue more susceptible to LPO damage,. Accordingly, in the present work, we observed a significant decrease [Table 3] in GPx activity upon increase in LPO level [Table 2]. This observation is in accordance with the hypothesis that LPO and GPx might play a role in tissue damage.[38–40]
A significant decrease was observed in the levels of GR in different groups from 21.78 ± 0.66 to 12.72 ± 0.58 nmoles of NADPH /min/mg protein upon ingestion of 4 and 8 mg MSG/g body weight to alcoholic animals [Table 3]. The significant inhibition in the activity of GR in arterial tissue attributed to increased oxidation or decreased synthesis of GSH. The less availability of NADPH may also cause a decrease in GR activity.[41]
In conclusion, the aforementioned observations suggested that ingestion of MSG at dose levels of 4 mg/g body weight and above along with alcohol produced hyperlipidemia and increased the oxidative stress by altering the levels of oxidative stress markers like LPO, XOD, SOD, CAT, GSH, GPx, and GR in arterial tissue of adult male mice, thereby MSG along with alcohol had additive effect. Hence, MSG could act as an additional factor for the initiation of coronary artery disease/atherosclerosis.
ACKNOWLEDGMENT
The authors are grateful to the University Grants Commission, New Delhi, India, for providing financial assistance to conduct this work.
Footnotes
Source of Support: University Grants Commission, New Delhi, India.
Conflict of Interest: None declared.
REFERENCES
- 1.World Health Organization (WHO) Global Status Report on alcohol. 2004 [Google Scholar]
- 2.Malik VBT, Ahluwalia P. Studies on effect of MSG on various fractions of lipids and certain carbohydrate metabolic enzymes in liver and blood of adult male mice. ToxicolLett. 1994;74:69–77. doi: 10.1016/0378-4274(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 3.Ahluwalia P, Tewari K, Choudhary P. Studies on the effects of monosodium glutamate (MSG) on oxidative stress in erythrocytes of adult male mice. ToxicolLett. 1996;84:161–5. doi: 10.1016/0378-4274(95)03612-1. [DOI] [PubMed] [Google Scholar]
- 4.Choudhary P, Malik VBT, Puri S, Ahluwalia P. Studies on the effects of monosodium glutamate on hepatic microsomal lipid peroxidation, calcium, ascorbic acid and glutathione and its dependent enzymes in adult male mice. ToxicolLett. 1996;89:71–6. doi: 10.1016/s0378-4274(96)03786-1. [DOI] [PubMed] [Google Scholar]
- 5.Singh K, Ahluwalia P. Studies on the effect of MSG administration on some antioxidant enzymes in the arterial tissue of adult male mice. J NutrSciVitaminol (Tokyo) 2003;49:145–8. doi: 10.3177/jnsv.49.145. [DOI] [PubMed] [Google Scholar]
- 6.Singh K, Ahluwalia P. Alteration in some antioxidant enzymes in cardiac tissue upon MSG administration to adult male mice. Indian J Clin Biochem. 2005;20:43–6. doi: 10.1007/BF02893040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma J, Kuldip S, Ahluwalia P, Arvindpreet K. Alteration in serum homocysteine levels upon ingestion of monosodium glutamate to adult rats. J Life Sci. 2009;1:103–5. [Google Scholar]
- 8.Ahluwalia P, Jyoti S, Uma S, Kuldip S, Akhtar M. Effect of administration of monosodium glutamate on carbohydrate metabolizing enzymes in intestinal tissue of adult male mice. Inter Toxicol. 2005;12:93–6. [Google Scholar]
- 9.Pavlovic V, Pavlovic D, Kocic G, Sokolovic D, Jevtovic ST, Cekic S, et al. Effect of monosodium glutamate on oxidative stress and apoptosis in rat thymus. Mol Cell Biochem. 2007;303:161–6. doi: 10.1007/s11010-007-9469-7. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Cederbaum AI. Alcohol, oxidative stress and free radical damage. Alcohol Res Health. 2003;27:277–84. [PMC free article] [PubMed] [Google Scholar]
- 11.Husain K, Vazquez-Ortiz M, Lalla J. Down regulation of aortic nitric oxide and antioxidant systems in chronic alcohol-induced hypertension in rats. Hum ExpToxicol. 2007;26:427–34. doi: 10.1177/0960327106072993. [DOI] [PubMed] [Google Scholar]
- 12.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;341:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Oh SI, Kim CI, Chun HJ, Park SC. Chronic ethanol consumption affects glutathione status in rat liver. J Nutr. 1998;128:758–63. doi: 10.1093/jn/128.4.758. [DOI] [PubMed] [Google Scholar]
- 14.Frings CS, Fendley TW, Dunn RT, Queen CA. Improved determination of serum lipids by the sulfo-phospho-vanillin reaction. ClinChem. 1972;18:673–4. [PubMed] [Google Scholar]
- 15.Fiske CH, Subarow Y. The colorimetric determination of phosphorus. J BiolChem. 1925;66:375–400. [Google Scholar]
- 16.McGowan MW, Artiss JD, Strandbergh DR, Zak B. A peroxidase-coupled method for the colorimetric determination of serum triglycerides. ClinChem. 1983;29:538–42. [PubMed] [Google Scholar]
- 17.Zlatkis A, Zak B, Boyle AJ. A new method for the direct determination of serum cholesterol. J Lab Clin Med. 1953;41:486–92. [PubMed] [Google Scholar]
- 18.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folinphenol reagent. J BiolChem. 1951;193:265–75. [PubMed] [Google Scholar]
- 19.Beuge JA, Aust SD. Vol. 52. NY and London: Academic Press; 1978. Microsomal lipid peroxidation in methods in enzymology; pp. 302–10. [DOI] [PubMed] [Google Scholar]
- 20.Fried R, Fried LW. Xanthine oxidase (Xanthine dehydrogenase) Method of enzymatic analysis. In: Bergmeyers HU, editor. Vol. 2. New York, London: Academic Press; 1974. pp. 644–9. New York, London. [Google Scholar]
- 21.Kono Y. Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. ArchBiochemBiophys. 1978;186:189–95. doi: 10.1016/0003-9861(78)90479-4. [DOI] [PubMed] [Google Scholar]
- 22.Luck H. New York, London: Academic Press; 1971. Catalase, method of enzymatic analysis; pp. 855–93. [Google Scholar]
- 23.Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–8. [PubMed] [Google Scholar]
- 24.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–90. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 25.Williams CHJ, Arscott ID. Glutathione reductase. In: Tabor H, Tabor DW, editors. Methods in enzymology. 17B. New York (USA): Academic Press; 1971. pp. 503–9. [Google Scholar]
- 26.Zhang X, Li SY, Brown RA, Ren J. Ethanol and acetaldehyde in alcoholic cardiomyopathy: from bad to ugly en route to oxidative stress. Alcohol. 2004;32:175–86. doi: 10.1016/j.alcohol.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Vasdev S, Gill V, Singal PK. Beneficial effect of low ethanol intake on the cardiovascular system: possible biochemical mechanisms. Vasc Health Risk Manag. 2006;2:263–76. doi: 10.2147/vhrm.2006.2.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albano E. Alcohol, oxidative stress and free radical damage. ProcNutrSoc. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 29.Ceresosimo E, Williams P, Hoxworth B, Lacy W, Abumrad N. Glutamine blocks lipolysis and ketogenesis of fasting. Am J Physiol. 1986;250:E248–52. doi: 10.1152/ajpendo.1986.250.3.E248. [DOI] [PubMed] [Google Scholar]
- 30.Xu P, Huecksteadt TP, Harrison R, HoidalJR Molecular cloning, tissue expression of human xanthine dehydrogenase. Biochem Biophys Res Commun. 1994;199:998–1004. doi: 10.1006/bbrc.1994.1328. [DOI] [PubMed] [Google Scholar]
- 31.Gill P, Fernando F, Angela C. Encarnation and malondialdehyde: A possible marker of aging. Gerontol. 2002;48:209–14. [Google Scholar]
- 32.Marklund SL. Properties of extracellular superoxide dismutase from human lung. Biochem J. 1984;220:269–72. doi: 10.1042/bj2200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bast A, Haenen GR. Cytochrome P450 and glutathione: what is the Significance of their interrelationship in lipid peroxidation.Trends? BioSci. 1984;9:510–3. [Google Scholar]
- 34.Kim BJ, Hood BL, Aragon RA, Hardwick JP, Conrads TP, Veenstra TD, et al. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6:1250–60. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onyema OO, Farombi EO, Emerole GO, Ukoha AI, Onyeze GO. rats. Indian J Biochem Biophys. 2006;43:20–4. [PubMed] [Google Scholar]
- 36.Padmini E, Sundari BT. Erythrocyte glutathione depletion impairs resistance to haemolysis in women consuming alcohol. J ClinBiochemNutr. 2008;42:14–20. doi: 10.3164/jcbn.2008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakazawa H, Genka C, Fujishima M. Pathological aspects of active oxygens/ free radicals. JpnJ Physiol. 1996;46:15–32. doi: 10.2170/jjphysiol.46.15. [DOI] [PubMed] [Google Scholar]
- 38.Blum J, Fridovich I. Inactivation of glutathione peroxidase by superoxide radical. Arch BiochemBiophys. 1985;240:500–8. doi: 10.1016/0003-9861(85)90056-6. [DOI] [PubMed] [Google Scholar]
- 39.Raes M, Michiels C, Remacle J. Comparative study of the enzymatic defense systems against oxygen-derived free radicals: the key role of glutathione peroxidase. Free RadicBiol Med. 1987;3:3–7. doi: 10.1016/0891-5849(87)90032-3. [DOI] [PubMed] [Google Scholar]
- 40.Lapennna D, de Gioia S, Ciofani G, Mezzetti A, Ucchino S, Calafiore AM, et al. Glutathione related antioxidants defence in human atherosclerotic plaques. Circulation. 1998;97:1930–4. doi: 10.1161/01.cir.97.19.1930. [DOI] [PubMed] [Google Scholar]
- 41.Gul M, Kutay FZ, Temocin S, Hanninen O. Cellular and clinical implications of glutathione. Indian J ExpBiol. 2000;38:625–34. [PubMed] [Google Scholar]