

Abstract
Background
Several bone marrow-derived cell populations may possess angiogenic activity, including cells termed endothelial progenitor cells. Decreased numbers of circulating angiogenic cell populations have been associated with increased cardiovascular risk. However, few data exist from large, unselected samples, and the genetic determinants of these traits are unclear.
Methods and Results
We examined the clinical and genetic correlates of early outgrowth colony forming units (CFUs) in 1,799 participants of the Framingham Heart Study (mean age, 66 years; 54% women). Among individuals without cardiovascular disease (n=1612), CFU number was inversely related to advanced age (P=0.004), female sex (P=0.04), and triglycerides (P=0.008), and positively related to hormone replacement (P=0.008) and statin therapy (P=0.027) in stepwise multivariable analyses. Overall, CFU number was inversely related to the Framingham risk score (p=0.01) but not with prevalent cardiovascular disease. In genome-wide association analyses in the entire sample, polymorphisms were associated with CFUs at the MOSC1 locus (P=3.3×10−7) and at the SLC22A3-LPAL2-LPA locus (P=4.9×10−7), a previously replicated susceptibility locus for myocardial infarction. Furthermore, alleles at the SLC22A3-LPAL2-LPA locus that were associated with decreased CFUs were also related to increased risk of myocardial infarction (P=1.1×10−4).
Conclusions
In a community-based sample, early outgrowth CFUs are inversely associated with select cardiovascular risk factors. Furthermore, genetic variants at the SLC22A3-LPAL2-LPA locus are associated with both decreased CFUs and an increased risk of myocardial infarction. These findings are consistent with the hypothesis that decreased circulating angiogenic cell populations promote susceptibility to myocardial infarction.
Keywords: Genetics, epidemiology, coronary disease, cardiovascular disease, cells
Background
Endothelial injury is recognized as a central event in the development and progression of atherosclerosis.1 Since mature endothelial cells possess limited ability to regenerate,2 intense efforts have been made to identify factors that might otherwise aid in restoring and maintaining the endothelium. Accordingly, much attention has focused on cells termed endothelial progenitor cells (EPCs), a heterogeneous population of bone-marrow derived cells in the peripheral circulation that are believed to have endothelial reparative properties through a variety of mechanisms.3 Experimental studies suggest that EPCs, variably defined, may be capable of promoting neovascularization in the setting of arterial ischemia4–6 and re-endothelialization following mechanical arterial injury,7 though more recent data are conflicting on this point.8, 9
Prior studies have reported an inverse association of EPC-related phenotypes with cardiovascular risk factors and aggregate cardiovascular risk,10, 11 although these associations are not consistent across all studies.12, 13 EPC phenotypes have also been inversely correlated with subclinical markers of endothelial dysfunction,11, 14–16 and incident cardiovascular events.17, 18 However, most prior investigations have been small case-control studies or case series that have varied widely with respect to patient selection and illness acuity.19, 20 Furthermore, in the absence of EPC-specific markers,21 many different methods have been used in attempts to quantitate EPCs, including in vitro colony assays, fluorescent microscopy, and flow cytometry for endothelial and hematopoeitic cell surface markers.22 These methods have limited ability to differentiate true endothelial precursor cells from other cell types with pro-angiogenic and vasculogenic properties.3, 23 In fact, recent evidence suggests that phenotypes previously defined as EPCs likely include several different cell populations of varying lineage and with distinct functional roles with respect to postnatal vasculogenesis.21, 24–26
A widely used EPC-related phenotype is early outgrowth colony forming units (CFUs), which arise from culturing peripheral blood mononuclear cells as originally described by Hill et al.11 Although CFUs are not direct endothelial precursors, prior experimental studies suggest that CFUs are highly angiogenic and their formation appears dependent on cell populations with known vascular promoting activities, including monocytes and T cells.3, 24, 25, 27 Furthermore, CFUs have been inversely related to cardiovascular risk factors,11 vascular dysfunction,11 and adverse cardiovascular outcomes17 in selected patient samples. As such, CFUs may represent an important cell-based marker of circulating angiogenic potential and risk for cardiovascular disease (CVD). Therefore, we sought to investigate the clinical correlates of CFUs in a large, community-based ambulatory cohort. In addition, we applied a genome-wide analysis to identify potential genetic determinants of CFU variation. We also examined whether genetic variants associated with lower CFU number might, in turn, be related to higher cardiovascular risk, which would support the hypothesis that decreased number of circulating angiogenic cells and CVD can arise from shared genetic mechanisms.
Methods
Because of the heterogeneity in phenotypes referred to as EPCs,8 we use CFU to describe our early outgrowth colony measurement in the interest of clarity. We use terms such as EPC phenotypes or EPC-related traits when referring to findings in the broader literature that draw upon diverse definitions of EPCs.
Study Sample
The Framingham Heart Study was established in 1948, when 5,209 residents of Framingham, MA were enrolled in a longitudinal cohort study designed to identify risk factors for CVD.28 In 1971, an additional 5,124 participants (offspring of the original cohort subjects, and their spouses) were enrolled in the Framingham Offspring Study.29 All Offspring study participants receive routine examinations approximately every 4 years. Of the 3,021 participants who attended the eighth Offspring examination cycle (2005 through 2008), 1,799 had assessment of CFUs. Individuals with and without CFU assessment had similar clinical characteristics and cardiovascular risk factor profiles (Supplemental Table 1). Of the total sample with CFUs assessed, the majority (n=1,612) of participants did not have a documented history of CVD (history of myocardial infarction [MI], coronary insufficiency, stroke, and heart failure) (Table 1). Participants underwent a standardized medical examination and laboratory assessment of cardiovascular risk factors (Supplemental Methods). All participants were also followed longitudinally through 2007, during which data regarding incident cardiovascular events were collected and adjudicated (Supplemental Methods). All participants gave informed consent. The institutionalreview board of the Boston University School of Medicine approved allstudy protocols.
Table 1.
Sample Characteristics
| Characteristic | Total Sample N=1,799 |
Non-CVD Sample N=1,612 |
|---|---|---|
| Age, y | 66 ± 9 | 66 ± 9 |
| Female, % | 54 | 57 |
| Systolic blood pressure, mm Hg | 129 ± 17 | 128 ± 17 |
| Diastolic blood pressure, mm Hg | 74 ± 10 | 74 ± 10 |
| Body mass index, kg/m2 | 28 ± 5 | 28 ± 5 |
| Fasting glucose, mg/dL | 106 ± 23 | 106 ± 23 |
| Total cholesterol, mg/dL | 186 ± 37 | 189 ± 36 |
| LDL cholesterol, mg/dL | 105 ± 31 | 108 ± 31 |
| HDL cholesterol, mg/dL | 57 ± 18 | 58 ± 18 |
| Triglycerides, mg/dL | 119 ± 69 | 117 ± 68 |
| Hypertension, % | 63 | 60 |
| Diabetes mellitus, % | 13 | 11 |
| Cigarette smoking, % | 7 | 7 |
| Medications, % | ||
| ACE inhibitors | 24 | 22 |
| Angiotensin receptor blockers | 8 | 7 |
| Beta-blockers | 28 | 23 |
| Calcium channel blockers | 14 | 12 |
| Diuretics | 23 | 21 |
| Hormone replacement | 4 | 8 |
| Statins | 41 | 37 |
| Framingham risk score | 9 ± 4 | 9 ± 4 |
| Prevalent cardiovascular disease, % | 10 | — |
CVD, cardiovascular disease. Values shown are means ± standard deviation or percents.
Assessment of Early Outgrowth CFUs
All blood specimens for the CFU assay were collected from fasting participants in the morning between 8 and 9 A.M. Each blood sample underwent initial centrifugation and the resulting buffy coat was further processed for CFU characterization within 4 hours of specimen collection according to an established protocol11 with modifications (Supplemental Methods).
Genotyping
Genotyping was performed in FHS by Affymetrix (Santa Clara, CA), using the Affymetrix 500K GeneChip array and a custom-designed gene-centric 50K molecular inversion probe, according to an established methodology (Supplemental Methods).30
Statistical Analyses
Because it had a skewed distribution that included zero values, square-root transformation of CFU yielded the best normally distributed data (skewness=1.12), and was performed prior to analyses. We also tested logarithmic transformation (after adding 1 to account for zero values), but this resulted in over-correction of right skewness leading to a left skewed distribution (skewness= −0.59). We analyzed clinical correlates of CFU based upon prior reports12, 17 in 1,612 participants free of CVD at the time of the assay. Age- and sex-adjusted linear regression models were used to examine the relation of CFU with the following covariates: age, sex, systolic blood pressure (SBP), diastolic blood pressure (DBP), body mass index (BMI), fasting glucose, total cholesterol, HDL cholesterol, LDL cholesterol, log-triglycerides, hypertension (SBP ≥ 140 mm Hg or DBP ≥ 90 mm Hg or taking anti-hypertensive medications), diabetes, smoking status, and selected medications (angiotensin converting enzyme inhibitors, angiotensin receptor blockers, beta-blockers, calcium channel blockers, diuretics, hormone replacement therapy (HRT), and statins). Stepwise regression models were used to examine multivariable associations of CFU, with variables having P values <0.10 in the age- and sex-adjusted analyses. We also performed analyses relating CFU to the Framingham risk score31 in the sample free of CVD, and then in men and women separately (of the same sample). In the total sample, we examined the relations of CFU with prevalent CVD. Our sample size, with N=1,799 observations and a fixed significance level α=0.05, afforded 80% power to detect a significant result for a variable accounting for 0.44% of the variance in CFU. A P value of <0.10 was the criterion for covariates to enter and remain in stepwise regression models, and a two-tailed P value of <0.05 was considered significant.
In secondary analyses, we repeated the following analyses using generalized estimating equations32 to account for correlations among related individuals (siblings) in the study sample (SAS PROC GENMOD): 1) age- and sex-adjusted analyses of the relation between CFU and the clinical covariates listed above (and in Supplemental Table 2), and 2) a single multivariable model adjusting for the clinical covariates that were associated with CFU in age- and sex-adjusted analyses with a P value threshold of at least <0.10. All analyses were performed using SAS statistical software, version 9.1.3.
In order to estimate heritability, the residuals of the age- and sex-specific model were were rank normalized, and the heritability was computed using the Sequential Oligogenic Linkage Analysis Routines (SOLAR, version 2.1.4).33
Genetic Association Analyses
To maximize statistical power, we performed the genetic association analyses in the total study sample, including participants with previous CVD. To account for potential confounding by population stratification, we used principal components analysis to define principal components of ancestry;34 because no principal components were associated with CFU, adjustment for these components was not included in regression modeling. We created age- and sex-adjusted residuals of square-root transformed CFU using models that did not account for family structure, because age- and sex-adjusted residuals from models that either did or did not account for family structure were highly correlated (r=0.99996, P<0.0001). These residuals were tested for association with each imputed (~2.5M) SNP under an additive genetic model using the linear mixed effects model in the R- kinship package to account for relatedness.35 We restricted our analyses to additive genetic models in order to limit the number of statistical tests. The use of analyses that assume an additive model is considered relatively robust to misspecification of the true model for a quantitative trait such as CFU.36 We selected an a priori genome-wide statistical significance threshold of 5×10−7, which is the threshold used by the Wellcome Trust Case-Control Consortium and accounts for ~1 million independent tests given the known high correlation among the majority of identifiable SNPs.37 For 2.5 million tests, this threshold provides an expectation of <1.25 false-positive results across the genome. A threshold for moderate genome-wide statistical significance (5×10−6)37 was considered to identify all potential loci of interest. For the top chromosomal loci, we verified that each locus had a directly-genotyped SNP with a similar association to that observed with the most significantly associated imputed SNPs. Regional plots for top hits showed high LD between the directly-genotyped SNP (red diamond) and the imputed SNPs with the lowest p-values (squares with darker red coloring denote high LD) (Supplemental Figure 1). Given the uniqueness of measuring CFUs in large samples, a replication sample was not available for the present study. Therefore, in the absence of a replication sample, we sought to further determine if any genetic determinants of CFU identified in the study sample were, in turn, related to MI. Thus, SNPs with associations with CFU were tested in survival analyses, using an empirical robust (sandwich) standard error, for their association with adjudicated recognized MI in both Original and Offspring Cohorts (N=4,449) occurring over 36 years of follow up between 1971 and 2007 (Supplemental Figure 2), encompassing the entire duration of the Framingham Offspring study.
Results
Clinical characteristics of the total sample and the subset without prevalent CVD are shown in Table 1. The mean (± standard deviation) age was 66 ± 9 years, and 54% were women. The distribution of absolute CFU counts in the total sample is shown in the Supplemental Figure 3, and there was no significant difference in CFU distribution between individuals with and without prevalent CVD as well as individuals with and without a prior history of MI. In the total sample, the median (interquartile range) CFU number was 38.9 (45.3) per well.
Clinical Correlates
In age- and sex-adjusted analyses performed in individuals without prevalent CVD, CFU was inversely associated with older age, female sex, higher triglycerides, and hormone replacement therapy (Table 2). Although women overall had lower CFU than men, CFU number in women taking HRT was not significantly different from that of men (P=0.154). The age- and sex-adjusted associations remained significant in multivariable-adjusted analyses, where CFU was also higher among individuals on statin therapy. Overall, however, clinical covariates accounted for a small proportion of the total variation in CFU (R2 = 0.02).
Table 2.
Clinical Correlates of Colony Forming Units
| Covariate | Age- and Sex-Adjusted | Multivariable-Adjusted | ||
|---|---|---|---|---|
| Regression Coefficient (standard error) | P value | Regression Coefficient (standard error) | P value | |
| Age | −0.069 (0.025) | 0.005 | −0.074 (0.023) | 0.004 |
| Female Sex | −0.099 (0.050) | 0.046 | −0.101 (0.050) | 0.044 |
| Body mass index | −0.009 (0.024) | 0.71 | — | |
| Height | −0.045 (0.038) | 0.24 | — | |
| Weight | −0.025 (0.028) | 0.37 | — | |
| Systolic blood pressure | −0.009 (0.026) | 0.73 | — | |
| Diastolic blood pressure | −0.003 (0.026) | 0.90 | — | |
| Fasting glucose | −0.016 (0.026) | 0.54 | — | |
| Total cholesterol | −0.009 (0.026) | 0.73 | — | |
| LDL cholesterol | −0.004 (0.025) | 0.88 | — | |
| HDL cholesterol | 0.029 (0.027) | 0.27 | — | |
| Log triglycerides | −0.056 (0.025) | 0.023 | −0.071 (0.027) | 0.008 |
| Hypertension | −0.0003 (0.053) | 0.99 | — | |
| Diabetes mellitus | −0.082 (0.079) | 0.30 | — | |
| Cigarette smoking | −0.165 (0.096) | 0.09 | — | |
| Medications | ||||
| ACE inhibitors | −0.038 (0.061) | 0.53 | — | |
| ARBs | −0.053 (0.095) | 0.57 | — | |
| Beta-blockers | −0.018 (0.060) | 0.77 | — | |
| Calcium channel blockers | −0.009 (0.077) | 0.91 | — | |
| Diuretics | −0.006 (0.061) | 0.92 | — | |
| Hormone replacement | 0.320 (0.122) | 0.009 | 0.325 (0.122) | 0.008 |
| Statins | 0.084 (0.052) | 0.11 | 0.127 (0.058) | 0.027 |
Data are from the subset of individuals without known cardiovascular disease (N=1,612). The colony forming unit variable is square-root transformed. Coefficients (standard error) represent change in the dependent variable for an increase in the value of the covariates shown (by 1 standard deviation for continuous variables). In age- and sex- adjusted models, the association with age is adjusted for sex and the association with sex is adjusted for age.
When age- and sex-adjusted analyses were repeated using generalized estimating equations (accounting for 308 sibships: 221 sibships of 2 persons, 68 sibships of 3 persons, 13 sibships of 4 persons, and 6 sibships of 5 persons), results were similar (Supplemental Table 2). Results of multivariable-adjusted analyses performed using generalized estimating equations were also similar. The sex-specific, predicted 10-year coronary risk based on the Framingham Risk Score was inversely associated with CFU (P=0.01) (Figure 1). There was no significant interaction by sex (P for interaction=0.91). Only 10% of participants attending this examination had prior CVD, and CFU was not associated with prior CVD after adjustment for conventional risk factors (P=0.67). Estimated heritability was 2.2% in age- and sex-adjusted analyses, similar to the variation attributable to measured clinical covariates.
Figure 1.
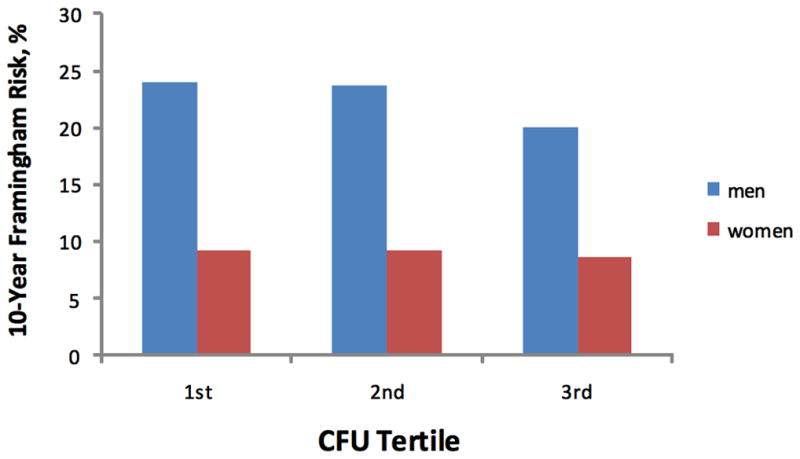
Percent risk of future cardiovascular disease, as determined by the Framingham Risk Score, by CFU tertile in men and women.
Genetic Correlates
The quantile-quantile plot from genome-wide association analyses (Figure 2) shows a deviation toward excess associations with low p-values, suggesting the presence of significant genetic association. The genomic inflation factor (lambda) was 1.027. Figure 3 shows the P values (-log base 10 scale) for the associations of the individual SNPs with CFU (using age- and sex-adjusted residuals of square-root transformed CFU), plotted against chromosomal position. The individual imputed variants with the strongest associations are listed in Table 3. Polymorphisms in the following 2 regions had genome-wide significant associations with CFU: rs6693017 (P=3.25×10−7) at chromosome position 1q41 (within or near the MOSC1 gene) and rs394352 (P=4.92×10−7) at chromosome position 6q26-q27 (within SLC22A3). SNPs at 3 additional loci had suggestive associations with CFU: 2p16.1 (near BCL11A), 7p14.1 (within POU6F2), and 19q13.12 (within HPN) (Supplemental Table 3). The SNPs near the MOSC1 gene were in linkage disequilibrium with one another (r2≥0.91, CEU HapMap release 22), as were SNPs near the SLC22A3 gene (r2≥0.81). We also repeated the genome-wide association study using age and sex as covariates in the models (instead of using age- and sex-adjusted residuals), as well as an analysis that additionally adjusted for all significant clinical correlates of CFU (log triglycerides, statin therapy, and hormone replacement therapy in addition to age and sex); these did not alter the associations with the above loci significantly, and beta coefficients and standard errors were largely unchanged (data not shown).
Figure 2.
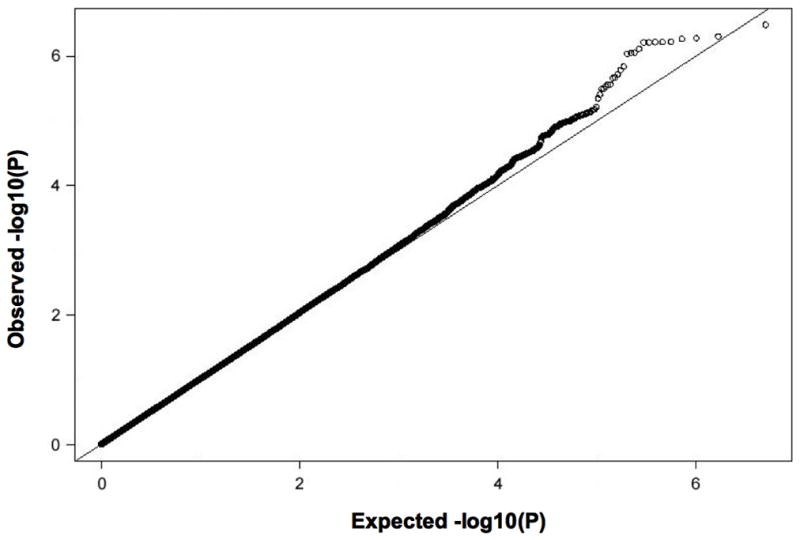
Quantile-quantile plot of −log(P value) of CFU association tests in the study sample.
Figure 3.

Distribution of P values for the association of individual SNPs with CFU, according to chromosome number and position.
Table 3.
SNPs Associated with Colony Forming Units After Adjustment for Age and Sex
| Chr | SNP | Position (bp) | Location relative to gene | MAF | A1–A2 | Allele for higher CFU | Observed to expected variance ratio | Beta (SE) | P value | Gene(s) at or near locus |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q41 | rs6693017 | 219043956 | Intron | 0.15 | T-G | G | 0.999 | 0.24 (0.05) | 3.25×10−7 | MOSC1 |
| rs17008835 | 219046052 | Intron | 0.16 | C-T | T | 1.000 | 0.23 (0.05) | 5.36×10−7 | ||
| rs7530493 | 219056323 | Downstream | 0.16 | G-T | T | 0.990 | 0.22 (0.05) | 1.44×10−6 | ||
| 6q26-q27 | rs402219 | 160709286 | Intron | 0.3 | A-G | G | 0.994 | 0.19 (0.04) | 6.01×10−7 | SLC22A, LPAL2, LPA |
| rs443043 | 160709531 | Intron | 0.3 | G-C | C | 0.994 | 0.19 (0.04) | 6.02×10−7 | ||
| rs1018234 | 160716048 | Intron | 0.3 | C-T | T | 0.995 | 0.19 (0.04) | 6.05×10−7 | ||
| rs316244 | 160716555 | Intron | 0.3 | G-A | A | 0.996 | 0.19 (0.04) | 6.07×10−7 | ||
| rs1510229 | 160725659 | Intron | 0.3 | C-T | T | 0.979 | 0.19 (0.04) | 5.22×10−7 | ||
| rs420038 | 160728138 | Intron | 0.3 | C-T | T | 0.997 | 0.19 (0.04) | 5.94×10−7 | ||
| rs394352 | 160728385 | Intron | 0.3 | C-T | T | 0.918 | 0.20 (0.04) | 4.92×10−7 | ||
| rs440962 | 160731144 | Intron | 0.3 | G-A | A | 0.994 | 0.19 (0.04) | 7.64×10−7 | ||
| rs446926 | 160733746 | Intron | 0.3 | A-G | G | 0.994 | 0.19 (0.04) | 8.70×10−7 | ||
| rs1510225 | 160738253 | Intron | 0.3 | C-T | T | 0.994 | 0.19 (0.04) | 8.85×10−7 | ||
SNP, single-nucleotide polymorphism; MAF, minor allele frequency;; A1, major allele; A2, minor allele; SE, standard error. The observed-to-expected variance ratio is a measure of imputation quality.
Several variants at the SLC22A3 locus that were associated with CFU were significantly associated with MI in the Framingham sample (P values ranging from 1.08×10−4 to 8.48×10−4; Table 4). The SNPs with the lowest P values for association with CFU were highly correlated with those having the lowest P values for association with MI (r2 0.579–0.961; Supplemental Table 4). Furthermore, the direction of effect for each allele was consistent for these SNPs: alleles associated with higher CFU were associated with lower risk for MI, and alleles associated with lower CFU were associated with higher MI risk. SNPs at the 1q41 locus were not associated with MI in Framingham.
Table 4.
SNPs Within or Near the SLC22A3-LPAL2-LPA Locus: Association With CFU and Myocardial Infarction in Framingham Heart Study
| SNP | Position (bp) | MAF | A1–A2 | MI risk allele* | Location Relative to Gene | Association with CFU | Association with MI* | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Beta (SE) | P value | HR (95% CI) | P value | |||||||
| SNPs with strongest associations with MI | rs569919 | 160687173 | 0.31 | C-T | C | Upstream | 0.15 (0.04) | 1.62 x10−4 | 0.71 (0.59–0.84) | 1.08×10−4 |
| rs394487 | 160698629 | 0.31 | C-T | C | Intronic | 0.16 (0.04) | 1.62 x10−5 | 0.74 (0.62–0.88) | 5.50×10−4 | |
| rs394468 | 160700902 | 0.31 | G-T | G | Intronic | 0.16 (0.04) | 1.22 x10−5 | 0.74 (0.62–0.87) | 4.94×10−4 | |
| rs377551 | 160701723 | 0.31 | G-A | G | Intronic | 0.16 (0.04) | 1.17 x10−5 | 0.73 (0.62–0.87) | 4.86×10−4 | |
| rs2661834 | 160770901 | 0.31 | C-T | C | Intronic | 0.17 (0.04) | 8.24 x10−6 | 0.75 (0.64–0.89) | 8.48×10−4 | |
| rs2063345 | 160780144 | 0.32 | A-G | A | Intronic | 0.17 (0.04) | 8.75 x10−6 | 0.75 (0.63–0.88) | 6.93×10−4 | |
| rs2174914 | 160783098 | 0.32 | G-C | G | Intronic | 0.17 (0.04) | 6.59 x10−6 | 0.74 (0.62–0.87) | 3.66×10−4 | |
| rs2457576 | 160785609 | 0.32 | G-C | G | Intronic | 0.17 (0.04) | 6.70 x10−6 | 0.74 (0.62–0.87) | 2.51×10−4 | |
| rs2457575 | 160788317 | 0.32 | A-G | A | Intronic | 0.17 (0.04) | 6.93 x10−6 | 0.74 (0.62–0.87) | 2.53×10−4 | |
| rs1567438 | 160799621 | 0.32 | T-C | T | Intronic | 0.16 (0.04) | 1.50 x10−5 | 0.73 (0.62–0.87) | 3.01×10−4 | |
| SNPs with strongest associations with CFU | rs402219 | 160709286 | 0.3 | A-G | A | Intronic | 0.19 (0.04) | 6.01×10−7 | 0.77 (0.65, 0.91) | 1.83×10−3 |
| rs443043 | 160709531 | 0.3 | G-C | G | Intronic | 0.19 (0.04) | 6.02×10−7 | 0.77 (0.65, 0.91) | 1.84×10−3 | |
| rs1018234 | 160716048 | 0.3 | C-T | C | Intronic | 0.19 (0.04) | 6.05×10−7 | 0.77 (0.65, 0.91) | 1.87×10−3 | |
| rs316244 | 160716555 | 0.3 | G-A | G | Intronic | 0.19 (0.04) | 6.07×10−7 | 0.77 (0.65, 0.91) | 1.88×10−3 | |
| rs1510229 | 160725659 | 0.3 | C-T | C | Intronic | 0.19 (0.04) | 5.22×10−7 | 0.76 (0.64, 0.90) | 1.41×10−3 | |
| rs420038 | 160728138 | 0.3 | C-T | C | Intronic | 0.19 (0.04) | 5.94×10−7 | 0.76 (0.65, 0.90) | 1.54×10−3 | |
| rs394352 | 160728385 | 0.3 | C-T | C | Intronic | 0.20 (0.04) | 4.92×10−7 | 0.75 (0.64, 0.90) | 1.20×10−3 | |
| rs440962 | 160731144 | 0.3 | G-A | G | Intronic | 0.19 (0.04) | 7.64×10−7 | 0.76 (0.64, 0.90) | 1.28×10−3 | |
| rs446926 | 160733746 | 0.3 | A-G | A | Intronic | 0.19 (0.04) | 8.70×10−7 | 0.76 (0.64, 0.90) | 1.21×10−3 | |
| rs1510225 | 160738253 | 0.3 | C-T | C | Intronic | 0.19 (0.04) | 8.85×10−7 | 0.76 (0.64, 0.90) | 1.20×10−3 | |
Analyses performed in a sample of 4,497 participants in FHS among whom 419 incident MI events occurred over 36 years of follow up. The HRs correspond to the minor allele; thus, HRs <1.0 denote that the major allele is associated with higher MI risk. SNP, single-nucleotide polymorphism; MAF, minor allele frequency; A1, major allele; A2, minor allele; CFU, colony forming unit; SE, standard error; MI, myocardial infarction; HR, hazards ratio.
SNPs in the upper part of the table are those with the strongest associations with MI, whereas SNPs in the lower part of the table are those with the strongest associations with CFU. Order of SNPs is by chromosomal position.
Discussion
In this study, we report the clinical and genetic correlates of early outgrowth CFUs in a community-based sample of predominantly healthy men and women. Our principal findings are three-fold. First, we observed modest associations between CFU and traditional cardiovascular risk factors. Lower CFU was related to higher overall risk factor burden, as reflected by the Framingham Risk Score, although this was largely attributable to the inverse relation of CFU with age. Second, we identified novel associations between CFU and genetic variants at several loci. Third, variants at one of these loci (SLC22A3) were not only associated with lower CFUs, but were also associated with higher MI risk. Taken together, our data suggest that variation in CFUs may represent a mechanism by which cardiovascular risk is modulated by genetic as well as acquired factors.
Interest in the determinants of variation in EPC phenotypes in humans stems from the previously reported association of EPC-related traits with cardiovascular risk, and the biological hypothesis that chronic depletion of EPC-like cells leads to an impaired capacity for endothelial repair. The present investigation employed a widely accepted method for identifying early outgrowth CFUs, and comprises the largest cohort of individuals with an assessment of circulating angiogenic-related traits studied to date. Other strengths of the study include the comprehensive ascertainment of cardiovascular risk factors, the focus on ambulatory individuals without acute illnesses that could lead to mobilization of pro-angiogenic cells,19, 20 and the availability of extensive genetic data.
Clinical correlates
We found relatively modest associations between early outgrowth CFU and conventional cardiovascular risk factors. In our study, lower CFU was observed in older individuals and women. Decreased number of cells considered to be EPCs with older age has been observed in multiple prior studies,12, 38 although underlying mechanisms remain unclear. Certain EPC phenotypes may be particularly susceptible to intrinsic senescence, as reflected by telomere shortening.39 However, the degree to which senescence affects CFUs is unknown.40
Reduced number of EPC-like cells in women has been reported in some studies12 but not others.41, 42 Studies that failed to find any effect of sex were considerably smaller than the present study and included younger women. The women in our study were almost entirely post-menopausal, suggesting the possibility of a sex hormone effect. Indeed, we found that hormone replacement therapy was associated with higher CFU,12 such that women taking HRT had comparable CFU numbers to men. Experimental studies suggest that estrogens may enhance EPC-related traits by reducing apoptosis of progenitor cells43 and stimulating endothelial nitric oxide synthase-dependent mobilization of progenitor cells.44 Therefore, estrogen withdrawal in post-menopausal women may lead to decreased CFU as well.
Consistent with prior experimental and clinical data pertaining to other EPC phenotypes,45, 46 we found that CFU was higher in individuals on statin medications. Notably, we also observed that CFU was inversely associated with triglycerides but not with other lipid traits such as LDL cholesterol. These findings support the hypothesis that one of the non-lipid benefits of statins in cardiovascular prevention could be their influence on circulating angiogenic cell number. Our results are consistent with a previously reported inverse association between triglycerides, but not LDL cholesterol, and an EPC phenotype that was assayed using a different culture method.12 Furthermore, experimental in vitro and in vivo data have identified mechanisms by which statins may affect EPC phenotypes in the absence of any lipid-lowering effects. For instance, statins have been shown to promote EPC differentiation and survival in vitro by modulating cell cycle regulation via the PI3K/Akt pathway;45 in a mouse model of MI, statins were also observed to promote EPC mobilization in an endothelial oxide synthase dependent fashion.46
With the exception of an association between CFU and triglycerides, CFU in our sample was not significantly associated with other metabolic traits, such as body mass index. CFU also was not related to the presence of diabetes, although our analysis of this association may have been underpowered. Despite an inverse relation with Framingham risk score, CFU was not associated with prevalent CVD in our sample; however, our ability to detect such an association may have been limited by the relatively low prevalence of CVD in our study cohort.
Genetic determinants
Prior investigations of genetic determinants have been limited to candidate gene studies, with particular focus on variants associated with CXC chemokine receptor 4 (CXCR4).47 In our genome-wide association data, significant relations of CFU with CXCR4 gene variants were not observed (P=0.277). On the other hand, we discovered common genetic variants at 2 novel loci, with P-values as low as 3.25×10−7 for association with CFU. While these loci need to be replicated in independent cohorts, they represent potentially novel mechanisms underlying variation in CFU number.
Interestingly, the SLC22A3-LPAL2-LPA locus is a replicated susceptibility locus for myocardial infarction.48 In our sample, variants in SLC22A3 were associated with both CFU (lowest P=4.9×10−7) and MI risk (lowest P=1.1×10−4). These data raise the possibility that variants at this locus influence MI risk at least partly through effects on angiogenic cell function. Although the variants in our study were within SLC22A3, multiple variants within or near SLC22A3, LPA, and LPAL2 were in linkage disequilibrium (r2>0.80). LPA codes for apolipoprotein(a), a component of lipoprotein(a) and a known risk factor for coronary disease.49, 50 LPAL2 encodes a structurally related isoform of apolipoprotein(a), and SLC22A3 encodes an organic cation transporter.51 The potential contribution of any of these proteins to the pathogenesis of cardiovascular disease remains unclear. The 1q41 locus contains MOSC1 (MOCO sulphurase C-terminal domain containing-1).51 Variants at this locus were not associated with MI.
Several lines of evidence link lower progenitor cell quantity and atherosclerosis. Experimental studies have demonstrated that bone marrow-derived EPC phenotypes can travel to and repair the endothelium at sites of intimal damage as well as stimulate new vessel growth,52 though a recent study found that bone marrow-derived or circulating cells made minimal contributions to endothelial cells in atherosclerotic plaque.9 In humans, several studies have reported an association between lower circulating CFU and prevalent CVD.17 In the present study, alleles at the SLC22A3-LPAL2-LPA locus associated with lower CFU were also related to MI with the expected direction of effect; in general, the top SNPs at this locus associated with lower CFU were also highly correlated with the top SNPs associated with increased risk of MI. These data are consistent with the hypothesis that lower circulating progenitors promote MI risk, although we cannot exclude the alternative scenario, that SLC22A3-LPAL2-LPA variants promote subclinical atherosclerosis, which then leads to lower CFU. Further studies are needed to test the hypothesis that reduced CFU promotes atherosclerosis susceptibility via reduced vasculogenic or vascular repair potential.
Limitations
Several limitations of our study merit consideration. Culture-based methods for identifying EPC phenotypes are intended to promote proliferation of cells with endothelial-specific lineage; however, these methods are variable in technique as well as their ability to isolate cells that can form de novo blood vessels.22 Although distinct from late outgrowth endothelial colonies that are able to differentiate into mature endothelial cells, as reported by Yoder et al.,22 the early outgrowth CFUs measured in this study have previously been shown to possess hematopoietic activity and their formation is dependent on known angiogenic cell populations such as monocytes and T cells.3 Notwithstanding their recognized angiogenic potential,40 however, CFUs likely comprise a heterogeneous population of cells with potentially distinct physiologic properties.22 Thus, further stratifying CFUs based on surface antigens, markers of senescence, and/or migratory capacity could provide additional insights. Because we studied an ambulatory, community-based cohort, the prevalence of prior CVD was low, limiting our ability to assess the cross-sectional relation between CFU number and CVD that has been reported by others. An important limitation of our genetic analyses is the lack of a replication sample, due in large part to the uniqueness of measuring CFUs in large samples with potential for genotyping. Although the association between the SLC22A3-LPAL2-LPA variants and MI risk has been replicated in other studies, the association of these variants with CFU also requires replication. EPC related traits have been assessed in very few large samples because they are difficult to measure and require specialized specimen handling. In future prospective cohorts, incorporation of specific, standardized protocols for obtaining samples suitable for endothelial progenitor assessment and other cell-based phenotyping appears warranted. Lastly, our sample was limited to white individuals of European ancestry, limiting the generalizability of these findings to other racial/ethnic groups.
Conclusion
We investigated the clinical and genetic determinants of CFU in a large, community-based cohort. We found modest correlations between CFU and cardiovascular risk factor burden, including the Framingham risk score. Furthermore, we identified common genetic variants that may potentially influence CFU variation. Variants at one of these loci, SLC22A3-LPAL2-LPA, were associated with both decreased CFU and increased risk for myocardial infarction. Together, these findings raise the possibility that lower CFU number contributes to increased cardiovascular risk, potentially through mechanisms distinct from traditional cardiovascular risk factors. Given the sometimes conflicting studies that may be found in the EPC literature,8 our data on a carefully characterized early outgrowth CFU phenotype in a large community sample, coupled to genetic analysis, provides a foundation for formulating additional hypotheses about the determinants and function of angiogenic cells. Additional clinical and mechanistic studies are necessary to explore further the role of specific CFU and angiogenic cell populations in both acquired and inherited cardiovascular risk.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported in part by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195), grant R01-HL083197 (TJW), and R01-HL93328 (RSV).
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Dimmeler S, Zeiher AM. Vascular repair by circulating endothelial progenitor cells: the missing link in atherosclerosis? J Mol Med. 2004;82:671–677. doi: 10.1007/s00109-004-0580-x. [DOI] [PubMed] [Google Scholar]
- 3.Sirker AA, Astroulakis ZM, Hill JM. Vascular progenitor cells and translational research: the role of endothelial and smooth muscle progenitor cells in endogenous arterial remodelling in the adult. Clin Sci (Lond) 2009;116:283–299. doi: 10.1042/CS20080001. [DOI] [PubMed] [Google Scholar]
- 4.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 5.Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, Masuda H, Silver M, Ma H, Kearney M, Isner JM, Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–637. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- 6.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 7.Kong D, Melo LG, Gnecchi M, Zhang L, Mostoslavsky G, Liew CC, Pratt RE, Dzau VJ. Cytokine-induced mobilization of circulating endothelial progenitor cells enhances repair of injured arteries. Circulation. 2004;110:2039–2046. doi: 10.1161/01.CIR.0000143161.01901.BD. [DOI] [PubMed] [Google Scholar]
- 8.Deb A, Patterson C. Hard luck stories: the reality of endothelial progenitor cells continues to fall short of the promise. Circulation. 2010;121:850–852. doi: 10.1161/CIR.0b013e3181d4c360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagensen MK, Shim J, Thim T, Falk E, Bentzon JF. Circulating endothelial progenitor cells do not contribute to plaque endothelium in murine atherosclerosis. Circulation. 2010;121:898–905. doi: 10.1161/CIRCULATIONAHA.109.885459. [DOI] [PubMed] [Google Scholar]
- 10.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 11.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 12.Xiao Q, Kiechl S, Patel S, Oberhollenzer F, Weger S, Mayr A, Metzler B, Reindl M, Hu Y, Willeit J, Xu Q. Endothelial progenitor cells, cardiovascular risk factors, cytokine levels and atherosclerosis--results from a large population-based study. PLoS ONE. 2007;2:e975. doi: 10.1371/journal.pone.0000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bielak LF, Horenstein RB, Ryan KA, Sheedy PF, II, Rumberger JA, Tanner K, Post WDMB, Shuldiner AR, Peyser PA. Circulating CD34+ cell count is associated with extent of subclinical atherosclerosis in asymptomatic amish men, independent of 10-year Framingham risk. Clin Med: Cardiology. 2009;3:1–8. doi: 10.4137/cmc.s2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy C, Kanaganayagam GS, Jiang B, Chowienczyk PJ, Zbinden R, Saha M, Rahman S, Shah AM, Marber MS, Kearney MT. Vascular dysfunction and reduced circulating endothelial progenitor cells in young healthy UK South Asian men. Arterioscler Thromb Vasc Biol. 2007;27:936–942. doi: 10.1161/01.ATV.0000258788.11372.d0. [DOI] [PubMed] [Google Scholar]
- 15.Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M, Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–1448. doi: 10.1016/j.jacc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 16.Tao J, Wang Y, Yang Z, Tu C, Xu MG, Wang JM. Circulating endothelial progenitor cell deficiency contributes to impaired arterial elasticity in persons of advancing age. J Hum Hypertens. 2006;20:490–495. doi: 10.1038/sj.jhh.1001996. [DOI] [PubMed] [Google Scholar]
- 17.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt-Lucke C, Rossig L, Fichtlscherer S, Vasa M, Britten M, Kamper U, Dimmeler S, Zeiher AM. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 19.Massa M, Rosti V, Ferrario M, Campanelli R, Ramajoli I, Rosso R, De Ferrari GM, Ferlini M, Goffredo L, Bertoletti A, Klersy C, Pecci A, Moratti R, Tavazzi L. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105:199–206. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]
- 20.Yamada M, Kubo H, Ishizawa K, Kobayashi S, Shinkawa M, Sasaki H. Increased circulating endothelial progenitor cells in patients with bacterial pneumonia: evidence that bone marrow derived cells contribute to lung repair. Thorax. 2005;60:410–413. doi: 10.1136/thx.2004.034058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoder MC. Platelet MPs obscure some EPC definitions. Blood. 2009;114:495–496. doi: 10.1182/blood-2009-04-218115. [DOI] [PubMed] [Google Scholar]
- 22.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timmermans F, Plum J, Yoder MC, Ingram DA, Vanderkerckhove B, Case J. Endothelial progenitor cells: identity defined? J Cell Mol Med. 2009;13:87–102. doi: 10.1111/j.1582-4934.2008.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107:1164–1169. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 25.Desai A, Glaser A, Liu D, Raghavachari N, Blum A, Zalos G, Lippincott M, McCoy JP, Munson PJ, Solomon MA, Danner RL, Cannon RO., 3rd Microarray-based characterization of a colony assay used to investigate endothelial progenitor cells and relevance to endothelial function in humans. Arterioscler Thromb Vasc Biol. 2009;29:121–127. doi: 10.1161/ATVBAHA.108.174573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prokopi M, Pula G, Mayr U, Devue C, Gallagher J, Xiao Q, Boulanger CM, Westwood N, Urbich C, Willeit J, Steiner M, Breuss J, Xu Q, Kiechl S, Mayr M. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood. 2009;114:723–732. doi: 10.1182/blood-2009-02-205930. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SJ, Zhang H, Wei YJ, Su WJ, Liao ZK, Hou M, Zhou JY, Hu SS. Adult endothelial progenitor cells from human peripheral blood maintain monocyte/macrophage function throughout in vitro culture. Cell Res. 2006;16:577–584. doi: 10.1038/sj.cr.7310075. [DOI] [PubMed] [Google Scholar]
- 28.Dawber TR, Meadors GF, Moore FE., Jr Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–281. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281–290. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- 30.Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 31.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 32.SAS/STAT User’s Guide, Version 8. Cary, NC: SAS Institute Inc; 2000. [Google Scholar]
- 33.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, Crawford G, Surti A, Guiducci C, Burtt NP, Parish S, Clarke R, Zelenika D, Kubalanza KA, Morken MA, Scott LJ, Stringham HM, Galan P, Swift AJ, Kuusisto J, Bergman RN, Sundvall J, Laakso M, Ferrucci L, Scheet P, Sanna S, Uda M, Yang Q, Lunetta KL, Dupuis J, de Bakker PI, O’Donnell CJ, Chambers JC, Kooner JS, Hercberg S, Meneton P, Lakatta EG, Scuteri A, Schlessinger D, Tuomilehto J, Collins FS, Groop L, Altshuler D, Collins R, Lathrop GM, Melander O, Salomaa V, Peltonen L, Orho-Melander M, Ordovas JM, Boehnke M, Abecasis GR, Mohlke KL, Cupples LA. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Team RDC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2006. [Google Scholar]
- 36.A framework for interpreting genome-wide association studies of psychiatric disorders. Mol Psychiatry. 2009;14:10–17. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- 37.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheubel RJ, Zorn H, Silber RE, Kuss O, Morawietz H, Holtz J, Simm A. Age-dependent depression in circulating endothelial progenitor cells in patients undergoing coronary artery bypass grafting. J Am Coll Cardiol. 2003;42:2073–2080. doi: 10.1016/j.jacc.2003.07.025. [DOI] [PubMed] [Google Scholar]
- 39.Kushner EJ, Van Guilder GP, Maceneaney OJ, Cech JN, Stauffer BL, Desouza CA. Aging and endothelial progenitor cell telomere length in healthy men. Clin Chem Lab Med. 2008 doi: 10.1515/CCLM.2009.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M, Rehman J. Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. Faseb J. 2009;23:1358–1365. doi: 10.1096/fj.08-110296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoetzer GL, MacEneaney OJ, Irmiger HM, Keith R, Van Guilder GP, Stauffer BL, DeSouza CA. Gender differences in circulating endothelial progenitor cell colony-forming capacity and migratory activity in middle-aged adults. Am J Cardiol. 2007;99:46–48. doi: 10.1016/j.amjcard.2006.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fadini GP, de Kreutzenberg S, Albiero M, Coracina A, Pagnin E, Baesso I, Cignarella A, Bolego C, Plebani M, Nardelli GB, Sartore S, Agostini C, Avogaro A. Gender differences in endothelial progenitor cells and cardiovascular risk profile: the role of female estrogens. Arterioscler Thromb Vasc Biol. 2008;28:997–1004. doi: 10.1161/ATVBAHA.107.159558. [DOI] [PubMed] [Google Scholar]
- 43.Strehlow K, Werner N, Berweiler J, Link A, Dirnagl U, Priller J, Laufs K, Ghaeni L, Milosevic M, Bohm M, Nickenig G. Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation. 2003;107:3059–3065. doi: 10.1161/01.CIR.0000077911.81151.30. [DOI] [PubMed] [Google Scholar]
- 44.Iwakura A, Luedemann C, Shastry S, Hanley A, Kearney M, Aikawa R, Isner JM, Asahara T, Losordo DW. Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation. 2003;108:3115–3121. doi: 10.1161/01.CIR.0000106906.56972.83. [DOI] [PubMed] [Google Scholar]
- 45.Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rossig L, Spyridopoulos I, Zeiher AM, Dimmeler S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res. 2003;92:1049–1055. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- 46.Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–1939. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 47.Xiao Q, Ye S, Oberhollenzer F, Mayr A, Jahangiri M, Willeit J, Kiechl S, Xu Q. SDF1 gene variation is associated with circulating SDF1alpha level and endothelial progenitor cell number: the Bruneck Study. PLoS ONE. 2008;3:e4061. doi: 10.1371/journal.pone.0004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M, Meitinger T, Wright BJ, Preuss M, Balmforth AJ, Ball SG, Meisinger C, Germain C, Evans A, Arveiler D, Luc G, Ruidavets JB, Morrison C, van der Harst P, Schreiber S, Neureuther K, Schafer A, Bugert P, El Mokhtari NE, Schrezenmeir J, Stark K, Rubin D, Wichmann HE, Hengstenberg C, Ouwehand W, Ziegler A, Tiret L, Thompson JR, Cambien F, Schunkert H, Samani NJ. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 49.Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 51.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;33:D54–58. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fadini GP, Agostini C, Sartore S, Avogaro A. Endothelial progenitor cells in the natural history of atherosclerosis. Atherosclerosis. 2007;194:46–54. doi: 10.1016/j.atherosclerosis.2007.03.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.