

Abstract
We report analysis of N-glycans derived from disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma by microchip electrophoresis with laser-induced fluorescence detection. Serum samples in 10-μL aliquots are enzymatically treated to cleave the N-glycans that are subsequently reacted with 8-aminopyrene-1,3,6-trisulfonic acid to add charge and a fluorescent label. Separations at 1250 V/cm and over 22 cm yielded efficiencies up to 700,000 plates for the N-glycans and analysis times under 100 s. Principal component analysis (PCA) and analysis of variance (ANOVA) tests of the peak areas and migration times are used to evaluate N-glycan profiles from native and desialylated samples and to determine differences among the four sample groups. With microchip electrophoresis, we are able to distinguish the three patient groups from each other and from disease-free individuals.
Keywords: microfluidics, microchip electrophoresis, N-glycans, glycan profiling, disease-state monitoring, Barrett's esophagus, high-grade dysplasia, esophageal adenocarcinoma
Over the past twenty years, the incidence of esophageal adenocarcinoma has increased at a rate greater than any other malignancy in many countries, including the U.S.1 Fewer than 20% of patients are expected to survive past three years after being diagnosed with this condition.2 Survival rates are poor because most patients demonstrate locally advanced disease by the time they are diagnosed. Moreover, a noninvasive method to screen for esophageal adenocarcinoma is currently not available. Over a decade ago, correlation between cancer and specific glycan structures was suggested for clinical prognosis.3,4 Cancerous cells shed surface proteins and protein fragments with altered glycans into the blood stream and can affect serum protein glycosylation. Glycomic profiling of human blood serum may have tremendous clinical value due to the potential for early and reliable diagnosis and non-invasive sample collection.
Esophageal adenocarcinoma of the distal esophagus represents 60–90% of all esophageal cancers5 and is thought to develop from a premalignant lesion, a condition known as Barrett's esophagus. The onset of Barrett's esophagus is usually associated with long term damage from the abnormal reflux of stomach acid into the esophagus, i.e., gastroesophageal reflux disease. Barrett's esophagus is observed when the normal squamous cell epithelium of the esophagus is replaced by a columnar cell epithelium, which resembles the cells lining the intestine, as a response to gastroesophageal reflux. This metaplastic transformation is usually observed in the distal esophagus but can occur over the whole length of the esophagus. Patients with Barrett's esophagus are 30 to 125-times more likely to develop esophageal adenocarcinoma than the general population.6 Malignancy development is a gradual progression at the cellular level where the metaplastic columnar cells are replaced with dysplastic, immature cells, and Barrett's esophagus with high-grade dysplasia is considered a precursor to invasive adenocarcinoma. A greater understanding of the biochemistry associated with disease progression may help to more efficiently identify individuals at risk of developing esophageal adenocarcinoma.
With adequate sensitivity and rapid sampling capabilities, mass spectrometry (MS) is common in biomedical and clinical applications of glycan analysis.7,8 MS-based methods are effective in glycan profiling to determine the alterations associated with several cancers, including breast cancer,9–13 ovarian cancer,14,15 prostate cancer,16,17 hepatocellular carcinoma,18 colon cancer,19 pancreatic cancer,20,21 and esophageal adenocarcinoma.22 Although very powerful, methods based purely on mass spectrometry cannot resolve glycan structural isomers, which may provide additional insight into the pathways and mechanisms of cancer progression. Moreover, MS-based glycan analysis is expensive and sometimes considered too sophisticated for routine clinical studies. Capillary electrophoresis coupled with laser-induced fluorescence provides excellent resolution of glycan structures and their isomers and tremendous sensitivity.8 Our group and others have used capillary8,23–26 and microchip27–32 electrophoresis for glycan analysis. To date, most of these microchip-based systems have employed relatively short separation channels (<10 cm) and modest electric field strengths (<500 V/cm). These devices provide sufficient resolution to separate and compare major components in a mixture, e.g., glycans associated with liver disease.32 With longer separation channels (>20 cm) and higher electric field strengths (>1000 V/cm), microfluidic devices are able to rapidly and efficiently separate N-glycans derived from model glycoproteins, e.g., ribonuclease B and fetuin, and from clinically relevant serum samples, e.g., breast cancer patients and ovarian cancer patients.30,31 Efficient separations on capillary- or chip-based instruments permit both low and high abundance N-glycans to contribute to the statistical analysis for disease-state differentiation.
In this study, we use microchip electrophoresis to analyze N-glycans derived from 10-μL aliquots of serum from disease-free individuals and patients suffering from Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma. Samples are arbitrarily labeled to conceal the physical condition of the patients to avoid bias in sample preparation and analysis. Serum samples are enzymatically treated to cleave the N-glycans that are subsequently labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS). Microfluidic devices with 22-cm long separation channels and asymmetrically tapered, 180° turns produce separation efficiencies up to 700,000 plates and analysis times less than 100 s. Principal component analysis (PCA) and analysis of variance (ANOVA) tests of both native and desialylated N-glycan samples are able to differentiate the four sample groups.
Experimental Section
Materials
We purchased acetic acid, acetone, acetonitrile, ammonium bicarbonate, ammonium persulfate, citric acid, dimethylsulfoxide (DMSO), 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES), β-mercaptoethanol, γ-methacryloxypropyltrimethoxy-silane (MAPTOS), methanol, potassium phosphate monobasic, sodium cyanoborohydride, sodium tetraborate, N,N,N',N'-tetramethylethylenediamine (TEMED), trifluoroacetic acid, and tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) from Sigma-Aldrich Co.; acrylamide from Bio-Rad Laboratories, Inc.; ammonium hydroxide from J.T. Baker; high-purity 8-aminopyrene-1,3,6-trisulfonic acid (APTS) from Beckman Coulter, Inc.; hydrogen peroxide from Macron Fine Chemicals; peptide-N-glycosidase F (PNGase F) of Chryseobacterium menigosepticum (EC 3.2.2.18) from Northstar BioProducts; sodium hydroxide from Fisher Scientific; sialidase from ProEnzyme, Inc.; Microposit MF-319 developer from Rohm and Haas Electronic Materials; 353NDT Epoxy from Epoxy Technology; chromium etchants 8002-A and 1020 and buffered oxide etchant from Transene Co., Inc.; B270 mask blanks and cover plates from Telic Co.; C18 Sep-Pak® cartridges from Waters Corp.; and activated carbon cartridges and 1000-Da cutoff cellulose dialysis tubes from Harvard Apparatus.
Serum Samples
This study was approved by the Indiana University institutional review board. Serum samples were collected from patients with documented Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma. Serum samples collected from sex and age matched healthy volunteers were used as the disease-free samples. From disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma, we analyzed 14, 7, 15, and 12 native samples and 15, 9, 15, and 15 desialylated samples, respectively.
Sample Preparation
Human blood serum samples (10-μL aliquots) were prepared as described previously.30 The samples were initially lyophilized and resuspended in 100 μL of 25 mM ammonium bicarbonate. N-Glycans were cleaved from the serum glycoproteins and glycoprotein fragments with PNGase F according to previously established procedures.33 The enzymatically released N-glycans were diluted to 1 mL with water. C18 Sep-Pak® cartridges and activated carbon cartridges were then used to isolate the N-glycans from peptides and proteins. A portion of the released N-glycans were lyophilized, resuspended in 10 mM phosphate buffer (pH 6), and desialylated by adding 0.2 μL of sialidase and incubating at 37°C for 6 h. All native and desialylated samples were dried with a vacuum CentriVap Concentrator (Labconco Corp.) prior to labeling with APTS34,35 to impart charge on the N-glycans for electrophoresis and permit fluorescence detection. Sample containers were arbitrarily labeled to conceal the physical conditions of the patients to prevent bias in sample preparation and data analysis.
Microfluidic Device Fabrication
Microfluidic devices were fabricated by standard photolithography, wet chemical etching, and cover plate bonding, as described previously.30 B270 glass substrates coated with 120 nm of Cr and 530 nm of AZ1518 photoresist were exposed to 200 mJ/cm2 UV radiation through a photogenerated mask (HTA Photomask) on a mask aligner (205S, Optical Associates, Inc.). The photoresist was developed for 2 min in MF-319 developer and rinsed with water. The channel pattern was subsequently transferred to the chromium layer by etching in chromium etchant 8002-A. The glass substrates were etched in buffered oxide etchant until the channels were 15-μm deep. A stylus-based profiler (Dektak 6M, Veeco Instruments, Inc.) was used to measure the depths and widths of the channels. After etching was complete, the channel widths of straight channels and turns were 86 μm and 29 μm, respectively. Holes were sandblasted at the ends of the channels with a sandblaster (AEC Air Eraser, Paasche Airbrush Co.). The photoresist layer was removed by rinsing with acetone, and the chromium layer was removed by etching in chromium etchant 1020. For the bonding procedure, etched substrates and cover plates were hydrolyzed in a solution of NH4OH, H2O2, and H2O (2:1:2), sonicated in H2O, brought into contact with each other, dried at 90°C for 2 h, ramped to 550°C in a furnace, and annealed for 10 h. Short segments of glass tubing (6-mm o.d. × 4-mm i.d. × 6-mm tall) were affixed over the sandblasted holes with 353NDT epoxy.
Microchannel Coating
The microchannels were coated with linear poly(acrylamide) to minimize electroosmotic flow and prevent analyte adsorption by a slight modification of Hjertén's method.36 Microchannels were cleaned sequentially with 1.0 M sodium hydroxide, water, and methanol for 20 min each, filled with a solution of 45 μL MAPTOS dissolved in 1.5 mL methanol with 0.02 M acetic acid, and allowed to react for 45 min. The microchannels were rinsed with methanol and water for 15 min each to remove the residual silane solution. The microchannels were filled with an aqueous solution containing 2.4% (w/w) acrylamide, 1.0 μL/mL TEMED, and 1.0 mg/mL ammonium persulfate and allowed to react for 2 h. The microchannels were flushed with water for 20 min and filled with buffer.
Microchip Electrophoresis
The microfluidic device with the 22-cm long serpentine separation channel and asymmetrically tapered, 180° turns37–39 is depicted in Figure 1. For electrophoretic separations, a fast-slewing high voltage power supply (0–10 kV), developed in-house, was used to apply potentials to the sample, buffer, and waste reservoirs. A commercial high voltage power supply (0–30 kV; CZE 1000R, Spellman High Voltage Electronics Corp.) was used to apply the potential at the analysis reservoir. The high voltage outputs were controlled through an analog output board (PCI-6713, National Instruments Corp.) by a program written in LabVIEW 8.0 (National Instruments Corp.). Samples were introduced into the analysis channel by standard pinched injections,40 and 1 mM phosphate and 20 mM HEPES buffer (pH 6.8) was used for electrophoretic separations.
Figure 1.
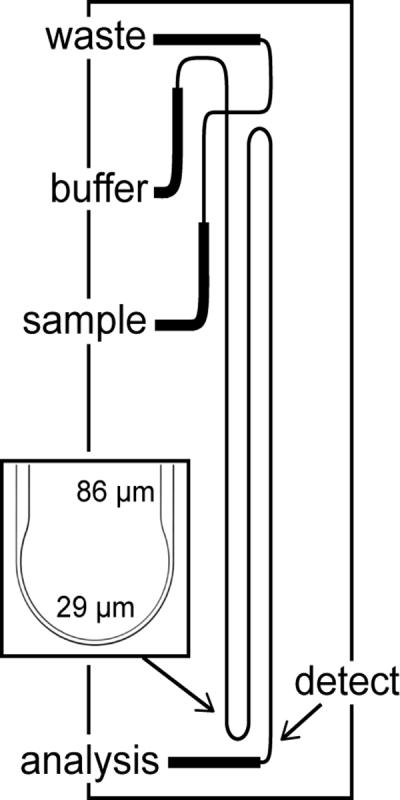
Schematic of the microfluidic device with a serpentine channel used for N-glycan analysis. The analysis channel is 22-cm long from the cross intersection to the detection point indicated by the arrow and has two asymmetrically tapered, 180° turns. The inset is a bright-field image of an asymmetrically tapered, 180° turn with taper ratio 3. The straight channel width and turn width are 86 μm and 29 μm, respectively.
An inverted optical microscope (TE-2000U, Nikon, Inc.) configured for epifluorescence was equipped with a 20× objective and HQ FITC filter cube (Chroma Technology Corp.) to monitor the separations. The 488-nm line of an argon ion laser (Melles Griot, Inc.) was attenuated to 1.0 mW with neutral density filters and focused to a spot in the analysis channel 22-cm downstream from the cross intersection. The fluorescence signal was spatially filtered with a 600-μm pinhole, detected with a photomultiplier tube (H5783-01, Hamamatsu Corp.), amplified by a low-noise current preamplifier (SR570, Stanford Research Systems, Inc.), and recorded with a multifunction data acquisition board (PCI-6032E, National Instruments Corp.) and the program written in LabVIEW. The sampling frequency was 100 Hz.
Data Analysis
To compensate for small fluctuations in the migration times (i.e., <2% relative standard deviation (RSD)), C Stats software developed in-house aligned all electropherograms to a reference electropherogram, and after peak alignment, the average variation in the migration times was <0.5%. The reference electropherogram simply needed to have a reasonable signal-to-noise (S/N) ratio to obtain proper alignment. After alignment, the C Stats software extracted the areas for all peaks from each sample. The average areas of individual peaks were obtained from three replicate electropherograms and were normalized to the total area of all peaks from the averaged electropherogram. The average peak areas for the same peak from all samples were used to calculate a sample average and standard deviation for all peaks. Prior to principal component analysis (PCA), individual peak areas were scaled by subtracting the sample average and dividing by the sample standard deviation. This scaling prevented peaks with a high S/N ratio from dominating the analysis and permitted all peaks to contribute fairly. PCA was performed with MarkerView (Applied Biosystems), which allowed visualization of multivariate information, and was supervised with prior knowledge of the sample groups. For the pairwise comparisons, Microsoft Excel was used to perform single-variable analysis of variance (ANOVA). To calculate separation efficiencies, selected peaks from the electropherograms were fitted with a Gaussian function in OriginPro 8.5 (OriginLab Corp.).
Results and Discussion
Microchip Electrophoresis of N-Glycans
N-Glycans were derived from 10-μL aliquots of serum, labeled with APTS, and analyzed by microchip electrophoresis with laser-induced fluorescence detection. The microfluidic devices with 22-cm long separation channels shown in Figure 1 were operated at 1250 V/cm electric field strength and provided analysis times under 100 s and separation efficiencies up to 700,000 plates. A typical electropherogram of native N-glycans derived from a patient with esophageal adenocarcinoma is shown in Figure 2. To compare the various samples, we used the N-glycan peaks with migration times between 55 and 100 s. Components that arrive prior to 55 s include structures associated with the labeling reaction and low molecular weight N-glycans, which are partially removed during dialysis and, consequently, are difficult to quantify. Figure 2b shows an expanded region along the migration window of interest and the separation resolution obtained for several lower intensity peaks. In this migration window, electropherograms of native and desialylated N-glycans derived from the blood serum of disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma are shown in Figures 3 and 4, respectively. Overall, the electropherograms had similar features and comparable signal intensities. Although some information is lost after removal of sialic acids from glycan structures by sialidase, the glycan profiles are simpler (i.e., have a fewer number of peaks), and the fluorescence signal is higher. From electropherograms similar to Figure 4, components A–E had separation efficiencies from 500,000 to 700,000 plates for slowly migrating components (E) to faster migrating components (A), respectively. Migration time reproducibilities were <2% RSD prior to peak alignment and <0.5% after peak alignment with the C Stats software. Separation field strengths greater than 1250 V/cm led to greater variability in the migration times and lower confidence in the alignment of the electropherograms, which is a critical step for the statistical analysis discussed below.
Figure 2.

(a) Electropherogram of a native N-glycan sample derived from the blood serum of a patient with esophageal adenocarcinoma. N-glycans with migration times from 55 to 110 s are used for the disease-state analysis. (b) Expanded region of the electropherogram in panel (a) with peaks 11, 23, 26, and 33 labeled. The electric field strength is 1250 V/cm, the separation length is 22 cm, and the N-glycans are labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS).
Figure 3.

Electropherograms of the native N-glycan samples derived from the blood serum of a disease-free individual and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma. The electric field strength is 1250 V/cm, and the separation length is 22 cm.
Figure 4.

Electropherograms of desialylated N-glycan samples derived from the blood serum of a disease-free individual and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma. Separation performance is evaluated with components A–E. The electric field strength is 1250 V/cm, and the separation length is 22 cm.
Principal Component Analysis (PCA) of Electropherograms
PCA is a chemometric tool commonly used to capture the variables that have the largest variance in given data sets in order to establish differences among sample groups. These variables are the principal components and are orthogonal to each other.41,42 We evaluated PCA for the 25- and 50-most intense peaks from the electropherograms of the native N-glycan samples, and the principal component scores plots are illustrated in Figure 5a–b, respectively. For the PCA with the 25-most intense peaks, the four sample groups are distinguished to some degree by their first and second principal component (PC1 and PC2) scores. However, the differentiation improves considerably when the 50-most intense peaks from the native samples are considered (Figure 5b), and the four sample groups are easily distinguished.
Figure 5.

Principal component analysis (PCA) scores plots for the (a) 25- and (b) 50-most intense peaks from native N-glycan samples derived from the serum of disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma.
A similar trend is observed for the PCA scores plots for the desialylated N-glycan samples in Figure 6. When the 50-most intense peaks for the desialylated samples are considered in Figure 6a, the disease-free and esophageal adenocarcinoma samples are well differentiated, but there is significant overlap for the Barrett's and high-grade dysplasia sample groups. The PCA scores plot shows complete differentiation of all the sample groups when the 75-most intense peaks from the desialylated N-glycan samples are included in the analysis, as shown in Figure 6b.
Figure 6.

Principal component analysis (PCA) scores plots for the (a) 50- and (b) 75-most intense peaks from desialylated N-glycan samples derived from the serum of disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma.
Differentiation among the sample groups is expected to improve as more N-glycan peaks are considered. Moreover, the non-biased mechanism of PCA is able to recognize the patterns among the normalized peak areas of the different sample groups, which provides better differentiation and clustering. In all cases, the disease-free sample group is well differentiated from the esophageal adenocarcinoma sample group, which indicates significant differences in their respective glycomic profiles. Of note, 50–60 N-glycan peaks are typically present in detectable quantities in most samples. Consequently, PCA with the 75-most intense peaks for the desialylated samples may not be reliable over larger sample sets. The number of N-glycan peaks used to generate the PCA plots, e.g., 25, 50, and 75, is arbitrarily chosen to demonstrate the general trend of improved differentiation as the number of peaks, i.e., amount of information, is increased.
Relative Peak Areas of Native and Desialylated N-Glycans
To determine which components contributed most to the differences among the sample groups, we used analysis of variance (ANOVA) tests to find statistically significant differences for the normalized peak areas of the N-glycans. The peaks considered had a high S/N ratio, a uniform peak shape, and adequate resolution from neighboring peaks. For example, see peaks 11, 23, 26, and 33 in Figure 2b. The average peak areas for several native and desialylated N-glycan peaks that compare disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma are shown in Figure 7. The p-values generated from the ANOVA tests for the native and desialylated peaks that compare the different sample groups are listed in Table 1. The pairwise comparisons revealed 13 native and 12 desialylated N-glycan peaks with p-values ≤0.12 and showed major quantitative differences among the four sample groups. We expected to find more quantitative differences in the native N-glycan peaks than the desialylated N-glycan peaks, but these differences may be obscured due to peak overlap caused by the greater complexity of the native N-glycan samples.
Figure 7.

Average peak areas for the (a) native and (b) desialylated N-glycan peaks reflecting the overall changes among disease-free individuals and patients with Barrett's esophagus, high-grade dysplasia, and esophageal adenocarcinoma. Peak numbers are assigned relative to the peak areas from reference electropherograms. Migration times and p-values for the native and desialylated N-glycan peaks are listed in Table 1. Error bars are +σ.
Table 1.
Migration Times, Peak Numbers, and p-Values from the Analysis of Variance (ANOVA) Tests for the Native and Desialylated N-Glycan Samples Derived from the Blood Serum of Disease-Free Individuals and Patients with Barrett's Esophagus, High-Grade Dysplasia, and Esophageal Adenocarcinoma.
| Native |
Desialvlated |
|||||
|---|---|---|---|---|---|---|
| samples | time (s) | peak | p-valuea | time (s) | peak | p-valuea |
| disease-free vs. Barrett's esophagus | ||||||
| 66.02 | 26 | 0.0794 | 94.10 | 4 | 0.00385 | |
| 74.77 | 29 | 0.121 | 85.95 | 12 | 0.0419 | |
| 88.31 | 5 | 0.0859 | ||||
| disease-free vs. high-grade dysplasia | ||||||
| 90.02 | 10 | 0.0652 | 85.95 | 12 | 0.0181 | |
| 94.10 | 4 | 0.0215 | ||||
| 91.73 | 2 | 0.0970 | ||||
| 69.98 | 20 | 0.120 | ||||
| disease-free vs. esophageal adenocarcinoma | ||||||
| 71.82 | 23 | 0.0125 | 57.12 | 42 | 0.0678 | |
| 60.55 | 8 | 0.0178 | 87.00 | 3 | 0.0966 | |
| Barrett's esophagus vs. high-grade dysplasia | ||||||
| 73.40 | 6 | 0.0886 | 88.31 | 5 | 0.0824 | |
| 61.33 | 3 | 0.100 | ||||
| 74.77 | 29 | 0.115 | ||||
| 66.02 | 26 | 0.118 | ||||
| Barrett's esophagus vs. esophageal adenocarcinoma | ||||||
| 67.43 | 11 | 0.0320 | 94.10 | 4 | 0.0313 | |
| 60.55 | 8 | 0.0344 | 90.44 | 8 | 0.109 | |
| high-grade dysplasia vs. esophageal adenocarcinoma | ||||||
| 76.24 | 33 | 0.0182 | ||||
| 60.55 | 8 | 0.116 | ||||
p-values ≤ 0.12 are shown.
In summary, N-glycans derived from blood serum samples are easily resolved by microchip electrophoresis, and statistical analysis of the glycan profiles permits differentiation of disease-states. Principal component analysis and analysis of variance tests for native and desialylated N-glycan samples independently confirmed the differences in the glycosylation patterns among the four sample groups. These results suggest that microfluidic devices have the potential to be used as a simple and robust method for routine analysis of clinical samples. Presently, these methods are limited by the need for structure identification. Future studies will involve the development of glycan standards and mass spectrometric analysis to elucidate the glycan structures that are associated with disease. Also, the contribution of structural isomers to disease-state differentiation may provide additional insight into the biological mechanisms of cancer progression.
Acknowledgements
This work was supported in part by NIH U01 CA128535. The authors thank the Indiana University Nanoscale Characterization Facility for use of its instruments.
References
- (1).Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. CA-Cancer J. Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- (2).Fiorica F, Di Bona D, Schepis F, Licata A, Shahied L, Venturi A, Falchi AM, Craxi A, Camma C. Gut. 2004;53:925–930. doi: 10.1136/gut.2003.025080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hakomori S. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- (4).Kobata A. Glycoconjugate J. 1998;15:323–331. doi: 10.1023/a:1006961532182. [DOI] [PubMed] [Google Scholar]
- (5).Devesa SS, Blot WJ, Fraumeni JF. Cancer. 1998;83:2049–2053. [PubMed] [Google Scholar]
- (6).Blot WJ, Devesa SS, Fraumeni JF. JAMA-J. Am. Med. Assoc. 1993;270:1320–1320. [PubMed] [Google Scholar]
- (7).Mechref Y, Novotny MV. Chem. Rev. 2002;102:321–369. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- (8).Mechref Y, Novotny MV. J. Chromatogr. B. 2006;841:65–78. doi: 10.1016/j.jchromb.2006.04.049. [DOI] [PubMed] [Google Scholar]
- (9).Kirmiz C, Li BS, An HJ, Clowers BH, Chew HK, Lam KS, Ferrige A, Alecio R, Borowsky AD, Sulaimon S, Lebrilla CB, Miyamoto S. Mol. Cell. Proteomics. 2007;6:43–55. doi: 10.1074/mcp.M600171-MCP200. [DOI] [PubMed] [Google Scholar]
- (10).Kyselova Z, Mechref Y, Kang P, Goetz JA, Dobrolecki LE, Sledge GW, Schnaper L, Hickey RJ, Malkas LH, Novotny MV. Clin. Chem. 2008;54:1166–1175. doi: 10.1373/clinchem.2007.087148. [DOI] [PubMed] [Google Scholar]
- (11).Abd Hamid UM, Royle L, Saldova R, Radcliffe CM, Harvey DJ, Storr SJ, Pardo M, Antrobus R, Chapman CJ, Zitzmann N, Robertson JF, Dwek RA, Rudd PM. Glycobiology. 2008;18:1105–1118. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- (12).Goetz JA, Mechref Y, Kang P, Jeng MH, Novotny MV. Glycoconjugate J. 2009;26:117–131. doi: 10.1007/s10719-008-9170-4. [DOI] [PubMed] [Google Scholar]
- (13).Pierce A, Saldova R, Abd Hamid UM, Abrahams JL, McDermott EW, Evoy D, Duffy MJ, Rudd PM. Glycobiology. 2010;20:1283–1288. doi: 10.1093/glycob/cwq090. [DOI] [PubMed] [Google Scholar]
- (14).An HJ, Miyamoto S, Lancaster KS, Kirmiz C, Li BS, Lam KS, Leiserowitz GS, Lebrilla CB. J. Proteome Res. 2006;5:1626–1635. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- (15).Saldova R, Royle L, Radcliffe CM, Hamid UMA, Evans R, Arnold JN, Banks RE, Hutson R, Harvey DJ, Antrobus R, Petrescu SM, Dwek RA, Rudd PM. Glycobiology. 2007;17:1344–1356. doi: 10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- (16).Tabares G, Radcliffe CM, Barrabes S, Ramirez M, Aleixandre RN, Hoesel W, Dwek RA, Rudd PM, Peracaula R, de Llorens R. Glycobiology. 2006;16:132–145. doi: 10.1093/glycob/cwj042. [DOI] [PubMed] [Google Scholar]
- (17).Kyselova Z, Mechref Y, Al Bataineh MM, Dobrolecki LE, Hickey RJ, Vinson J, Sweeney CJ, Novotny MV. J. Proteome Res. 2007;6:1822–1832. doi: 10.1021/pr060664t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Goldman R, Ressom HW, Varghese RS, Goldman L, Bascug G, Loffredo CA, Abdel-Hamid M, Gouda I, Ezzat S, Kyselova Z, Mechref Y, Novotny MV. Clin. Cancer Res. 2009;15:1808–1813. doi: 10.1158/1078-0432.CCR-07-5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vercoutter-Edouart AS, Slomianny MC, Dekeyzer-Beseme O, Haeuw JF, Michalski JC. Proteomics. 2008;8:3236–3256. doi: 10.1002/pmic.200800151. [DOI] [PubMed] [Google Scholar]
- (20).Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. J. Proteome Res. 2006;5:1792–1802. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- (21).Zhao J, Qiu WL, Simeone DM, Lubman DM. J. Proteome Res. 2007;6:1126–1138. doi: 10.1021/pr0604458. [DOI] [PubMed] [Google Scholar]
- (22).Mechref Y, Hussein A, Bekesova S, Pungpapong V, Zhang M, Dobrolecki LE, Hickey RJ, Hammoud ZT, Novotny MV. J. Proteome Res. 2009;8:2656–2666. doi: 10.1021/pr8008385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu JP, Shirota O, Wiesler D, Novotny M. Proc. Natl. Acad. Sci. U. S. A. 1991;88:2302–2306. doi: 10.1073/pnas.88.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nashabeh W, El Rassi Z. J. Chromatogr. A. 1992;600:279–287. doi: 10.1016/0021-9673(92)85559-c. [DOI] [PubMed] [Google Scholar]
- (25).Kakehi K, Susami A, Taga A, Suzuki S, Honda S. J. Chromatogr. A. 1994;680:209–215. doi: 10.1016/0021-9673(94)80069-3. [DOI] [PubMed] [Google Scholar]
- (26).Archer-Hartmann SA, Sargent LM, Lowry DT, Holland LA. Anal. Chem. 2011;83:2740–2747. doi: 10.1021/ac103362r. [DOI] [PubMed] [Google Scholar]
- (27).Dang FQ, Zhang LH, Jabasini M, Kaji N, Baba Y. Anal. Chem. 2003;75:2433–2439. doi: 10.1021/ac034110a. [DOI] [PubMed] [Google Scholar]
- (28).Callewaert N, Contreras R, Mitnik-Gankin L, Carey L, Matsudaira P, Ehrlich D. Electrophoresis. 2004;25:3128–3131. doi: 10.1002/elps.200406020. [DOI] [PubMed] [Google Scholar]
- (29).Dang FQ, Kakehi K, Nakajima K, Shinohara Y, Ishikawa M, Kaji N, Tokeshi M, Baba Y. J. Chromatogr. A. 2006;1109:138–143. doi: 10.1016/j.chroma.2005.11.133. [DOI] [PubMed] [Google Scholar]
- (30).Zhuang Z, Starkey JA, Mechref Y, Novotny MV, Jacobson SC. Anal. Chem. 2007;79:7170–7175. doi: 10.1021/ac071261v. [DOI] [PubMed] [Google Scholar]
- (31).Zhuang Z, Mitra I, Hussein A, Novotny MV, Mechref Y, Jacobson SC. Electrophoresis. 2011;32:246–253. doi: 10.1002/elps.201000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Vanderschaeghe D, Szekrenyes A, Wenz C, Gassmann M, Naik N, Bynum M, Yin HF, Delanghe J, Guttman A, Callewaert N. Anal. Chem. 2010;82:7408–7415. doi: 10.1021/ac101560a. [DOI] [PubMed] [Google Scholar]
- (33).Mechref Y, Novotny MV. Anal. Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- (34).Evangelista RA, Liu MS, Chen FTA. Anal. Chem. 1995;67:2239–2245. [Google Scholar]
- (35).Evangelista RA, Chen FTA, Guttman A. J. Chromatogr. A. 1996;745:273–280. [Google Scholar]
- (36).Hjerten S. J. Chromatogr. 1985;347:191–198. [Google Scholar]
- (37).Griffiths SK, Nilson RH. Anal. Chem. 2001;73:272–278. doi: 10.1021/ac000936q. [DOI] [PubMed] [Google Scholar]
- (38).Molho JI, Herr AE, Mosier BP, Santiago JG, Kenny TW, Brennen RA, Gordon GB, Mohammadi B. Anal. Chem. 2001;73:1350–1360. [Google Scholar]
- (39).Ramsey JD, Jacobson SC, Culbertson CT, Ramsey JM. Anal. Chem. 2003;75:3758–3764. doi: 10.1021/ac0264574. [DOI] [PubMed] [Google Scholar]
- (40).Jacobson SC, Hergenröder R, Koutny LB, Warmack RJ, Ramsey JM. Anal.Chem. 1994;66:1107–1113. [Google Scholar]
- (41).Hotelling H. J. Educ. Psychol. 1933;24:417–441. [Google Scholar]
- (42).Musumarra G, Barresi V, Condorelli DF, Scire S. Biol. Chem. 2003;384:321–327. doi: 10.1515/BC.2003.037. [DOI] [PubMed] [Google Scholar]