

Abstract
The dual-specificity mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) inactivates MAP kinases by dephosphorylation. Here we show that the proinflammatory cytokine interleukin (IL)-17A induces adult mouse primary cardiac fibroblast (CF) proliferation and migration via IL-17 receptor A//IL-17 receptor C-dependent MKP-1 suppression, and activation of p38 MAPK and ERK1/2. IL-17A mediated p38 MAPK and ERK1/2 activation is inhibited by MKP-1 overexpression, but prolonged by MKP-1 knockdown. IL-17A induced miR-101 expression via PI3K/Akt, and miR-101 inhibitor reversed MKP-1 down regulation. Importantly, MKP-1 knockdown, inhibition of p38 MAPK by SB 203580, inhibition of ERK1/2 by U0126 and PD 98059, or overexpression of dominant negative MEK1, each markedly attenuated IL-17A-mediated CF proliferation and migration. Similarly, IL-17F and IL-17A/F heterodimer that also signal via IL-17RA/IL-17RC, stimulated CF proliferation and migration. These results indicate that IL-17A stimulates CF proliferation and migration via Akt/miR-101/MKP-1-dependent p38 MAPK and ERK1/2 activation. These studies support a potential role for IL-17 in cardiac fibrosis and adverse myocardial remodeling.
Keywords: Cytokines, interleukins, cardiac fibrosis, myocardial remodeling, migration, proliferation, signal transduction
1. Introduction
Inflammatory mechanisms are integral components of myocardial injury, fibrosis, and adverse remodeling. The pro-inflammatory cytokine interleukin (IL)-17 belongs to a new family of cytokines that share no homology with other known interleukins, and plays a critical role in various autoimmune and inflammatory diseases [1–3]. This structurally distinct cytokine family consists of at least 6 ligands from A to F, and 5 receptors from IL-17R A–E. While IL-17 receptors are expressed in both immune and non-immune cells, the IL-17 ligands are expressed mostly in immune cells, especially those of the Th17 lineage [1–3]. We previously reported that primary human cardiac fibroblasts express both IL-17RA and IL-17RC, and IL-17A stimulates matrix metalloproteinase (MMP)-1 expression via p38 MAPK- and ERK1/2-dependent C/EBPβ, NF-κB, and AP-1 activation [4]. Interestingly, fibroblasts also express IL-17A [5, 6]. This suggests both autocrine and paracrine IL-17 signaling in cardiac fibroblasts.
Recently, a critical role for IL-17A in postmyocarditis cardiac remodeling and the progression to dilated cardiomyopathy has been reported [7]. Those authors demonstrated that while IL-17A is dispensable for the development of myocarditogenic peptide-induced myocarditis in mice, its deficiency reduced expression of proinflammatory cytokines IL-6, TNF-α, and IL-1β in the heart, attenuated inflammatory cell recruitment, reduced interstitial myocardial fibrosis, downregulated expressions of MMPs 2 and 9, and delayed progression to dilated cardiomyopathy [7]. Moreover, administration of IL-17A neutralizing antibodies after the onset of myocarditis markedly inhibited cardiac fibrosis and preserved ventricular function [8], suggesting that IL-17A is critical in cardiac fibrosis, adverse remodeling and dilated cardiomyopathy. Of note, IL-17A has been shown to be obligatory in bleomycin and IL-1β-induced pulmonary fibrosis [9].
Fibroblasts, the most abundant cell type in the adult mammalian heart, reside in the interstitium between contracting cardiomyocytes, and provide structural support by regulating extracellular matrix deposition and turnover [10]. However, following injury or infection, fibroblasts express a wide variety of proinflammatory cytokines, chemokines and adhesion molecules, enhance extracellular matrix degradation via MMP production, and migrate and proliferate, leading ultimately to cardiac fibrosis and adverse remodeling [10]. Since IL-17 induces MMP expression [4], and since IL-17A null mice express attenuated MMP expression and reduced fibrosis following myocarditogenic peptide-induced myocarditis and CVB3 infection [7, 8], and as fibroblast proliferation and migration are critical in cardiac fibrosis, we hypothesized that IL-17 exerts mitogenic and migratory effects on fibroblasts, and determined the underlying molecular mechanisms. Supporting our hypothesis, we report for the first time that IL-17A is a potent inducer of fibroblast proliferation and migration, and its mitogenic and migratory effects are dependent on Akt/miR-101-mediated MAPK phosphatase-1 (MKP-1) suppression and activation of the mitogen-activated protein kinases p38 and ERK1/2. These results further support a role for IL-17 in cardiac fibrosis and adverse cardiac remodeling.
2. Materials and Methods
2.1. Materials
The materials used in this report are detailed in ‘Supplementary methods’ section.
2.2. Isolation of cardiac fibroblasts (CF)
All animal studies were approved by the Institutional Care and Use Committee at the University of Texas Health Science Center in San Antonio, and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Fibroblasts were isolated from the hearts of 8–10-wk-old male C57Bl/6 mice as detailed in ‘Supplementary methods’.
2.3. Adeno and lentiviral infection
Infection of CF with adeno and lenti viruses is detailed in ‘Supplementary methods’. At the indicated MOI, viral infection had no off-target effects, and failed to modulate CF adherence, shape and viability (trypan blue-dye exclusion; data not shown).
2.4. Transfections
CF were transfected with miR-101 inhibitor using the Neon® transfection system (MPK-5000, Invitrogen). CF were microporated (pulse voltage: 1300 volts, pulse width: 20 ms, pulse number: 2, tip type: 10 μl) with 50 nM of the miR-101a inhibitor and incubated for 24 h. CF showed transfection efficiency of 49% with only 7% cell death as determined using the pEGFP-N1 vector. IL-17RA and IL-17RC were targeted using specific siRNA (40 nM), and non-targeting siRNA served as a control.
2.5. mRNA and micro RNA expression
DNA-free total RNA was prepared using the RNAqueous®-4PCR kit (Ambion). RNA quality was assessed by capillary electrophoresis using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). All RNA samples used for quantitative PCR had RNA integrity numbers greater than 9.0 (scale = 1–10) as assigned by default parameters of the Expert 2100 Bioanalyzer software package (v2.02). MMP-2 and MMP-9 expressions were quantified by RT-qPCR using Taqman probes. IL-17RA and IL-17RC mRNA expression were analyzed by RT-PCR. miR-101 expression was analyzed by Northern blot analysis using the antisense oligonucleotide (5′ to 3′) TTCAGTTATCACAGTACTGTA/3Starfire described previously [11]. In brief, 25 μg of RNA were separated on 12.5% urea-polyacrylamide gels under denaturing conditions, transferred to GeneScreen Plus membranes (PerkinElmer, Waltham, MA), and UV-cross-linked. Radiolabeling of miRNA antisense oligonucleotides was performed with a StarFire labeling kit (IDT, Coralville, IA) according to the manufacturer’s instructions. Prehybridization, hybridization, and washing of membranes are detailed in ‘Supplementary methods’.
2.6. Protein expression
Preparation of whole cell homogenates, immunoblotting, detection of the immunoreactive bands by enhanced chemiluminescence (ECL Plus; GE Healthcare), and their quantification by densitometry are detailed in ‘Supplementary methods’. MMP-2 and MMP-9 activation by gelatin zymography, and activation and activities of Akt, p38 MAPK, and ERK1/2 are also detailed in ‘Supplementary methods’.
2.7. Cell proliferation
CF proliferation was analyzed by [3H]TdR and BrdU incorporation assays, and are detailed in ‘Supplementary methods’.
2.8. Cell migration
CF migration was quantified using BD BioCoat Matrigel invasion chambers and 8.0-μm pore polyethylene terephthalate membranes with a thin layer of Matrigel basement membrane matrix. The assay is detailed in ‘Supplementary methods’.
2.9. Cell death detection
Cell death was analyzed by a photometric enzyme immunoassay (Cell Death Detection ELISAPLUS kit, Roche Applied Science). The nitric oxide donor SNAP served as a positive control.
2.10. Statistical analysis
Results are expressed as means ± S.E. For statistical analysis we used analysis of variance followed by an appropriate post hoc multiple comparison test (Tukey method). Data were considered statistically significant at P < 0.05.
3. Results
3.1. Interleukin-17A stimulates cardiac fibroblast (CF) proliferation
Proliferation and migration of resident fibroblasts contribute to reparative and reactive fibrosis, and adverse cardiac remodeling. Using 3H-TdR incorporation as a marker of CF proliferation, we found that IL-17A induces CF proliferation in a time- (Fig. 1A) and dose (Fig. 1B)-dependent manner. BrdU incorporation confirmed these results (data not shown). Consistent with these mitogenic effects, IL-17A induced phosphorylation of the protein synthesis markers p70 S6 kinase and ribosomal S6 protein (Fig. 1C). Importantly, pre-incubation with anti-IL-17A antibodies significantly attenuated IL-17A induced CF proliferation (Fig. 1D) and the protein synthesis markers (Fig. 1E). Since IL-17A is known to signal via IL-17RA/IL-17RC heterodimer [12], we investigated their expression in CF using RT-PCR. Our results show that CF express both IL-17RA and IL-17RC at basal conditions (Fig. 1F), and their knockdown markedly attenuates IL-17A-mediated CF proliferation (Fig. 1G; knockdown of was confirmed by RT-PCR and is shown on the right). Since IL-17A is known to induce other proinflammatory and pro-mitogenic cytokines [7, 13], we next examined whether IL-17A’s mitogenic effects could be mediated by other cytokines. Pre-incubation with either anti-IL-18 or TNF-α neutralizing antibodies failed to significantly modulate IL-17A-induced CF proliferation (Supplementary Fig. S1). Of note, these antibodies significantly inhibited respective cytokine-induced NF-κB DNA binding activity in CF (Supplementary Fig. S1), demonstrating their efficacy. These results indicate that IL-17A is a potent inducer of CF proliferation, and its pro-mitogenic effects are independent of other proinflammatory cytokines (Fig. 1).
Fig. 1. Interleukin-17A stimulates cardiac fibroblast (CF) proliferation.

A, Time-dependent effect of IL-17A on CF proliferation. CF at 70–80% confluence, were made quiescent by incubating in medium supplemented with 0.5% BSA for 48h. The quiescent CF were treated with recombinant murine IL-17A (10 ng/ml) and proliferation assays (incorporation of 3H-thymidine into DNA) carried out at the indicated times *P < at least 0.05 vs. untreated (n=12). B, Dose-dependent effects of IL-17A. Quiescent CF were treated with the indicated concentrations of IL-17A for 48 h then analyzed as in A for proliferation. *P < 0.05, **P < 0.001 vs. untreated (n=12). C, IL-17A induces markers of protein synthesis in CF. Quiescent CF treated with IL-17A (10 ng/ml for up to 3 h) were analyzed for phospho-ribosomal S6 protein and p70 S6 kinase by immunoblotting using cleared whole cell homogenates. A representative of 3 independent experiments is shown. D, Specificity of IL-17A. Quiescent CF were treated with IL-17A neutralizing antibodies (10 μg/ml for 1 h) prior to IL-17A (10 ng/ml) addition. Normal IgG served as a control. Cell proliferation at 48 h was determined as in A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A + control IgG (n=12). E, IL-17A neutralization attenuates IL-17A-induced protein synthesis markers. Quiescent CF were treated as in D, but for 2 h, were analyzed as in C (n=3). F, CF express IL-17RA and IL-17RC mRNA. Total RNA from CF were analyzed for IL-17R expression by RT-PCR (n=3). G, Knockdown of IL-17RA and IL-17RC inhibit IL-17A-induced CF proliferation. CF transfected with receptor-specific or non-targeting control siRNA (40 nM for 48 h; knockdown was confirmed by RT-PCR and is shown on the right) were treated with IL-17A (10 ng/ml for 48 h). Cell proliferation was analyzed as in A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A + control siRNA (n=12).
3.2. IL-17A stimulates cardiac fibroblast proliferation via p38 MAPK and ERK1/2
Mitogen-activated protein kinases (MAPK) play critical roles in cell proliferation and migration [14]. Results show IL-17A induced time dependent p38 MAPK phosphorylation (Fig. 2A, left hand panel) and kinase activation (Fig. 2A, right hand panel), and these effects were markedly attenuated by SB 203580, but not PD 98059 or the JNK inhibitor SP 600125 (Fig. 2B). Similarly, IL-17A induced time dependent ERK1/2 phosphorylation (Fig. 2C, left hand panel) and kinase activation (Fig. 2C, right hand panel), and these effects were markedly attenuated by PD 98059, but not SB 203580 or SP 600125 (Fig. 2D). Treatment with U0126 and infection with Ad.dnMEK1 also inhibited IL-17A mediated ERK/1/2 phosphorylation and kinase activity (Supplementary Fig. S2). Importantly, the p38 MAPK inhibitor SB 203580 and the ERK1/2 inhibitor PD 98059 markedly attenuated IL-17A-induced CF proliferation (Fig. 2E). Similar inhibition was observed when CF were treated with the MEK1/2 inhibitor U0126 or infected with Ad.dnMEK1 prior to IL-17A addition (Fig. 2F). The effect of the JNK inhibitor SP 600125, however, was relatively modest (Fig. 2G). These results indicate that IL-17A induces CF proliferation via p38 MAPK and ERK1/2 activation (Fig. 2).
Fig. 2. IL-17A stimulates cardiac fibroblast proliferation via p38 MAPK and ERK1/2.

A, IL-17A activates p38 MAPK. Quiescent CF treated with IL-17A (10 ng/ml) were analyzed for total and phospho-p38 MAPK by immunoblotting (n=3) and p38 MAPK kinase activity using immunecomplex kinase assays (right hand panel, n=3). B, SB 203580 inhibits IL-17A-induced p38 MAPK activation. Quiescent CF were treated with the indicated MAPK inhibitors prior to IL-17A (10 ng/ml) addition. p38 MAPK activation was analyzed by immunoblotting (n=3). C, IL-17A activates ERK1/2. Quiescent CF treated as in A were analyzed for total and phospho-ERK1/2 by immunoblotting (left hand panel; n=3) and ERK1/2 kinase activity using immunecomplex kinase assays (right hand panel, n=3). D, PD 98059 inhibits IL-17A-induced ERK1/2 activation. Quiescent CF were treated with the indicated MAPK inhibitors prior to IL-17A (10 ng/ml) addition. ERK1/2 activation was analyzed by immunoblotting (n=3). E, IL-17A induces CF proliferation via p38 MAPK and ERK1/2. Quiescent CF treated with the p38 MAPK inhibitor SB 203580 or the ERK1/2 inhibitor PD 98059 prior to IL-17A (10 ng/ml for 48 h) addition. CF proliferation was analyzed as in Fig. 1A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A + DMSO (n=12). F, IL-17A induces CF proliferation via MEK. Quiescent CF treated with the MEK inhibitor U0126 (20 μM in DMSO for 1 h) or infected with Ad.dnMEK1 (MOI 100 for 24 h) prior to IL-17A (10 ng/ml for 48 h) addition. CF proliferation was analyzed as in Fig. 1A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A + Ad.GFP (n=12). G, Effects of the JNK inhibitor on IL-17A-induced CF proliferation were modest. Quiescent CF treated with the JNK inhibitor SP 600125 prior to IL-17A (10 ng/ml for 48 h) addition were analyzed for proliferation as in Fig. 1A. *P < 0.001 vs. untreated, †P < 0.05 vs. IL-17A + DMSO (n=12).
3.3. IL-17A inhibits MKP-1
MKP-1 (also known as dual specificity protein phosphatase (DUSP)-1) dephosphorylates and inactivates p38 MAPK and ERK1/2 [15]. Since IL-17A rapidly activated p38 MAPK and ERK1/2 (Fig. 2), we next investigated whether IL-17A inhibits MKP-1. MKP-1 is present in the CF under basal conditions, and treatment with IL-17A markedly attenuated its levels for up to 3 h (Fig. 3A). However, MKP-1 levels returned to basal levels at later time periods, and were somewhat higher than basal conditions at 4 and 6 h (Fig. 3A). Further, while knockdown of MKP-1 prolongs (Fig. 3B; densitometric analysis from three independent experiments is summarized in the lower panel. Knockdown of MKP-1 was confirmed by immunoblotting and is shown on the right), adenoviral transduction of wild type MKP-1 inhibits both basal and IL-17A induced p38 MAPK (Fig. 3C; overexpression of MKP-1 was analyzed by immunoblotting, and is shown on the right). Similar effects were observed on ERK1/2 phosphorylation (Fig. 3D and 3E). Importantly, while ectopic expression of wild type MKP-1 significantly attenuated, MKP-1 knockdown moderately increased IL-17A induced CF proliferation (Fig. 3F), indicating that IL-17A induces CF proliferation via MKP-1 inhibition (Fig. 3).
Fig. 3. IL-17A inhibits MKP-1.

A, IL-17A inhibits MKP-1 expression. Quiescent CF treated with IL-17A (10 ng/ml) were analyzed for MKP-1 protein levels by immunoblotting. *P < 0.05 vs. untreated (n=3). B, Knockdown of MKP-1 prolongs p38 MAPK activation. CF transduced with MKP-1 shRNA by lentiviral infection (MOI 5 for 48 h) were treated with IL-17A (10 ng/ml). p38 MAPK activation was analyzed as in Fig. 2D. Knockdown of MKP-1 is shown on the right. *P < at least 0.01 vs. untreated (n=3). C, Forced expression of wild type MKP-1 inhibits p38 MAPK activation. CF were infected with Ad.MKP-1 (MOI 10 for 24 h) prior to IL-17A (10 ng/ml) addition. MKP-1 levels were analyzed by immunoblotting. Expression of MKP-1 following Ad.MKP-1 infection is shown on the right. *P < 0.01 vs. untreated (n=3). D, Knockdown of MKP-1 prolongs ERK1/2 activation. CF treated as in B were analyzed for ERK1/2 activation as in Fig. 2F. *P < at least 0.01 vs. untreated (n=3). E, Forced expression of wild type MKP-1 inhibits ERK1/2 activation. CF treated as in C were analyzed for ERK1/2 activation. Expression of MKP-1 following Ad.MKP-1 infection is shown on the right. *P < 0.01 vs. untreated (n=3). F, MKP-1 regulates CF proliferation. CF treated as in B and C, but for 48 h with IL-17A, were analyzed for proliferation as in Fig. 1A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A, §P < 0.05 vs. IL-17A + non-targeting shRNA (n=12).
3.4. IL-17A suppresses MKP-1 via Akt-dependent miR-101a induction
miR-101a has recently been shown to target MKP-1 expression in LPS-treated mouse macrophages, and Akt plays a role in miR-101 induction [16]. Since IL-17A induced CF proliferation (Fig. 1A), and since Akt is a pro-survival factor [17], we investigated whether IL-17A activates Akt and induces miR-101 in CF in an Akt-dependent manner. IL-17A induced time-dependent Akt phosphorylation (Fig. 4A) and kinase activity (Fig. 4B) in CF, effects that were significantly inhibited by pre-treatment with the pharmacological inhibitor Akti-X or by adenoviral transduction of dnAkt1 (Fig. 4C). Further, the PI3K inhibitor wortmannin or forced expression of dnPI3Kp85, both inhibited IL-17A-induced Akt activation (Fig. 4D). We next investigated whether IL-17A induces miR-101 via Akt. Relative expression of miR-101 was analyzed by Northern blotting, U6 snRNA served as a loading control. IL-17A induced a time-dependent increase of miR-101a mRNA in CF (Fig. 4E), an effect significantly inhibited by the PI3K inhibitors and adenoviral transduction of mutant PI3Kp85 and Akt1 (Fig. 4F). Further, the miR-101a inhibitor reversed IL-17A mediated MKP-1 suppression (Fig. 4G) and attenuated CF proliferation (Fig. 4H). Together, these results indicate that IL-17A induces CF proliferation via PI3K/Akt/miR-101a-mediated MKP-1 suppression (Fig. 4).
Fig. 4. IL-17A suppresses MKP-1 via PI3K/Akt-dependent miR-101a induction.

A, B, IL-17A activates Akt. Quiescent CF treated with IL-17A (10 ng/ml) were analyzed for phospho-Akt (Thr308) by immunoblotting (A; n=3) and Akt kinase activity by immune complex kinase assay (B; n=3). C, IL-17A induced Akt activation is inhibited by AktI-X and Ad.dnAkt1. CF were treated with AktI-X (2.5 μM in water for 1 h) or infected with Ad.dnAkt1 (MOI 100 for 24 h) prior to IL-17A (10 ng/ml for 1 h) addition. Akt activation was analyzed by immunoblotting as in A (n=3). D, IL-17A activates Akt via PI3K. CF were treated with wortmannin (250 nM in DMSO for 1 h) or infected with Ad.dnPI3Kp85 (MOI 100 for 24 h) prior to IL-17A (10 ng/ml for 1 h) addition. Akt activation was analyzed by immunoblotting as in A (n=3). E, IL-17A induces miR-101 expression. Quiescent CF treated with IL-17A (10 ng/ml) were analyzed for miR-101a expression by Northern blotting. U6 served as a loading control (n=3). F, IL-17A induces miR-101 expression via PI3K/Akt-dependent signaling. Quiescent CF treated as in C and D, but for 30 min with IL-17A (10 ng/ml), were analyzed for miR-101a expression by Northern blotting (n=3). G, miR-101 inhibitor reverses IL-17A-induced MKP-1 suppression. CF transfected with miR-101 inhibitor (50 nM for 24 h) and treated with IL-17A (10 ng/ml for 1 h) were analyzed for MKP-1 expression by immunoblotting. miScript Inhibitor with no homology to any mammalian gene served as a negative control (n=3). H, IL-17A induces CF proliferation via Akt and miR-101. CF infected with Ad.dnAkt1 (MOI 100 for 24 h) or transfected with miR-101 inhibitor (50 nM for 24 h) were treated with IL-17A (10 ng/ml for 48 h), and then analyzed for proliferation as in Fig. 1A. *P < 0.001 vs. untreated, †P < 0.01 vs. IL-17A ± Ad.GFP or negative control (n=12).
3.5. IL-17A induces cardiac fibroblast migration
Since IL-17A induced CF proliferation via miR-101/MKP-1-dependent p38 MAPK and ERK1/2 activation (Figs. 1–4), we next investigated whether IL-17A also induces CF migration. Using the Matrigel invasion assay, we found that IL-17A induced CF migration, an effect significantly inhibited by IL-17A neutralizing antibodies, AktI-X, SB 203580, PD 98059, miR-101a mimic and MKP-1 knockdown (Fig. 5), indicating that IL-17A induces CF proliferation and migration via a common signal transduction pathway (Fig. 5).
Fig. 5. IL-17A induces cardiac fibroblast migration via Akt, miR-101, MKP-1, p38 MAPK and ERK1/2.

Cultured CF were trypsinized, re-suspended in medium containing 0.5% BSA, and layered on Matrigel basement membrane matrix-coated filters. CF were treated with AktI-X (2.5 μM in water for 1 h), SB 203580 (1 μM in DMSO for 30 min), or PD 98059 (10 μM in DMSO for 1 h) prior to IL-17A (10 ng/ml) addition. CF transfected with miR-101 inhibitor (50 nM for 24 h) or infected with lentiviral MKP-1 shRNA (MOI 5 for 48 h) were similarly layered on Matrigel basement membrane matrix-coated filters and then treated with IL-17A. The lower chambers also contained similar levels of IL-17A. Plates were incubated at 37°C for 12 h to allow cell migration. Specificity of IL-17A was verified by incubating the cells with IL-17A-neutralizing antibodies (10 μg/ml) for 1 h before IL-17A addition. DMSO, GFP shRNA, miScript negative control, and normal mouse IgG1 served as specific controls. Cells migrating to the other side of the membrane were quantified using MTT assay. *P < 0.001 vs. untreated; †P < 0.01 vs. IL-17A (n=6).
3.6. IL-17A induced MMP expression is inhibited by MKP-1
Following injury or inflammation in the heart, invasion by CF is known to follow ECM destruction by MMPs. Since IL-17A induced CF migration (Fig. 5), we next investigated whether IL-17A induces MMP expression. Indeed IL-17A induced mRNA and protein expression, and activity of MMP-2 (Fig. 6A, left hand panel; protein levels in the upper panel, activity in the culture supernatant by zymography, lower panel) and MMP-9 (Fig. 6A, right hand panel; protein levels in the upper panel, activity in the culture supernatant by zymography, lower panel) in CF, and both SB 203580 and PD 98059 markedly attenuated their expression (Fig. 6B; MMP-2, left hand panel; MMP-9, right hand panel). Mimicking the effects of MAPK inhibitors, forced expression of wild type MKP-1 by adenoviral transduction blunted MMP expression (Fig. 6C). Importantly, the broad-spectrum MMP inhibitors marimastat and GM 6001 both markedly attenuated IL-17A-induced CF migration (Fig. 6D), indicating that IL-17A induces MMP expression and CF migration via MKP-1 (Fig. 6).
Fig. 6. IL-17A induced MMP expression is inhibited by MKP-1.

A, IL-17A induces MMP expression. Quiescent CF treated with IL-17A (10 ng/ml) were analyzed for MMP-2 (left hand) and MMP-9 (right hand) mRNA expression (RT-qPCR; 18S served as an invariant control), protein levels in cleared cell lysates (immunoblotting; αTubulin served as a loading control), and activity (zymography) using culture supernatants. *P < 0.001 vs. untreated (n=6). B, The MAPK inhibitors attenuate IL-17A-induced MMP expression. Quiescent CF were treated with the p38 MAPK inhibitor SB 203580 (1 μM in DMSO for 30 min) or the ERK1/2 inhibitor PD 98059 (10 μM in DMSO for 1 h) prior to IL-17A (10 ng/ml) addition. MMP-2 (left hand) and MMP-9 (right hand) expressions were analyzed by RT-qPCR and immunolotting (insets). *P < 0.001 vs. untreated; †P < at least 0.05 vs. IL-17A (n=6). C, Forced expression of MKP-1 inhibits IL-17A-mediated MMP induction. CF were infected with Ad.MKP-1 (MOI 10 for 24 h) prior to IL-17A (10 ng/ml) addition. MMP expression was analyzed by RT-qPCR. *P < 0.001 vs. untreated; †P < at least 0.05 vs. IL-17A (n=6). D, Broad-spectrum MMP inhibitors attenuate IL-17A induced CF migration. Quiescent CF were treated with marimastat (10 mM in DMSO) or GM 6001 (10 μM in DMSO for 15 min) prior to IL-17A addition (10 ng/ml) were analyzed for cell migration as in Fig. 5. *P < 0.001 vs. untreated; †P < at least 0.05 vs. IL-17A (n=6).
3.7. IL-17F and IL-7A/F are equally potent in inducing CF proliferation and migration
Since IL-17F and IL-17A/F heterodimer also signal via IL-17RA and IL-17RC complex [2, 12], and since CF express both the receptors, we next investigated their effects on CF proliferation and migration. Indeed, both IL-17F and IL-17A/F heterodimer induced significant proliferation (Fig. 7A) and migration (Fig. 7B) of CF. These results indicate that all three known ligands of IL-17RA/IL-17RC complex induce CF proliferation and migration (Fig. 7).
Fig. 7. IL-17F and IL-7A/F induce cardiac fibroblast proliferation and migration.

A, IL-17F and IL-17A/F induce CF proliferation. Quiescent CF were treated (10 ng/ml) with IL-17F or IL-17A/F, and analyzed for proliferation as in 1A. *P < 0.001 vs. untreated (n=12). B, IL-17F and IL-17A/F induce CF migration. CF were treated with IL-17F or IL-17A/F as in 5A and then analyzed for migration after 12 h. *P < at least 0.01 vs. untreated (n=12).
4. Discussion
Here we demonstrate for the first time that IL-17A is a direct and potent inducer of mouse primary cardiac fibroblast proliferation and migration. We further show that IL-17A signals via IL-17RA/IL-17RC complex, suppresses the dual-specificity phosphatase MKP-1, and activates the mitogen-activated protein kinases p38 MAPK and ERK1/2. Moreover, while overexpression of wild type MKP-1 blunted, its knockdown prolonged IL-17A mediated p38 MAPK and ERK1/2 activation. IL-17A induced miR-101 expression via PI3K/Akt, and miR-101 inhibitor reversed MKP-1 down regulation. Importantly, inhibition of p38 MAPK and ERK1/2 markedly attenuated IL-17A-induced CF proliferation and migration (Fig. 8). Interestingly, JNK, which is well known to play a role in cytokine signaling [18], only moderately attenuated IL-17A effects. We also show that, similar to IL-17A, IL-17F and IL-17A/F heterodimer stimulate CF proliferation and migration. These results support a potential role for IL-17 in cardiac fibrosis and adverse myocardial remodeling.
Fig. 8. Schema showing the signal transduction pathways involved in IL-17A mediated primary cardiac fibroblast proliferation and migration.
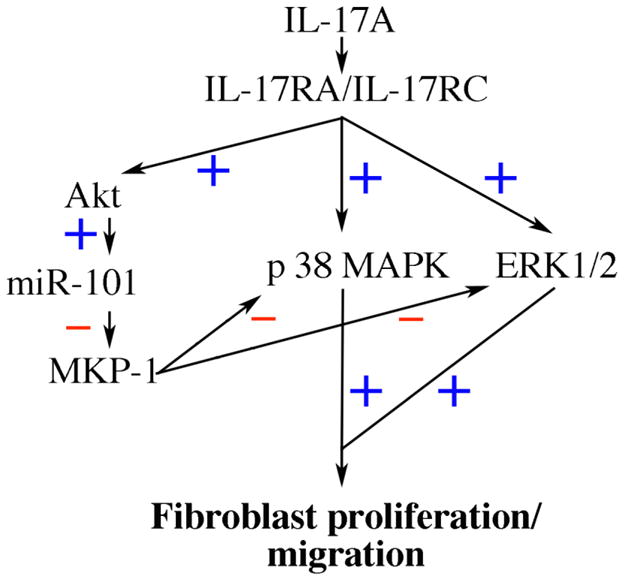
IL-17A signals via IL-17RA/IL-17RC heterodimer, and induces CF proliferation and migration via Akt/miR-101/MKP-1-dependent p38 MAPK and ERK1/2 activation.
The serine-threonine protein kinases p38 MAPK and ERK1/2 phosphorylate both cytoplasmic and nuclear targets that are critical in cytokine signaling, cell proliferation and migration. While upstream MAP kinase kinases (MKK) activate MAPKs by phosphorylating critical serine and threonine residues leading to their activation, the dual-specificity phosphatases dephosphorylate MAP kinases and block the sometimes deleterious effects of chronic activation of downstream signaling pathways. Our data indicate that IL-17A markedly attenuated MKP-1 levels in CF. Of note, MKP-1 has equal substrate specificity towards p38 MAPK and ERK1/2 [19, 20].
We also show that while IL-17A induces time dependent phosphorylation of both p38 MAPK and ERK1/2, forced expression of wild type MKP-1 blocked their activation, whereas MKP-1 knockdown prolonged p38 MAPK and ERK1/2 activation. Interestingly, while MKP-1 levels were significantly inhibited at 1 h following IL-17A treatment, its levels returned to basal levels by 3 h, and in fact were higher at later time periods, suggesting an induction mechanism. Since ERK1/2 transcriptionally regulates MKP-1 [21], it is possible that inhibition of MKP-1 initially results in enhanced MAPK activation, and activated MAPKs in turn induce MKP-1 as a negative feedback mechanism. Of note, neither knockdown nor MKP-1 overexpression significantly modulated cell viability following IL-17A treatment (data not shown).
We also found that IL-17A suppresses MKP-1 via microRNA (miR)-101 induction. Recently, miR-101 has been shown to directly target MKP-1 to regulate LPS-mediated MAPK activation in RAW264.7 macrophage cells [16]. Those authors identified a conserved miR-101 response element (MRE) in MKP-1 3′untranslated region (UTR) across species. Our results show that while forced expression of miR-101 mimic attenuated, miR-101 inhibitor restored MKP-1 expression, indicating that IL-17A-induced miR-101 binds a specific MRE in MKP-1 3′ UTR, and thus represses or degrades MKP-1 mRNA. Further, we also show that IL-17A activates PI3K-dependent Akt in fibroblasts, and inhibition of PI3K and Akt markedly attenuated IL-17A-induced miR-101 expression, and fibroblast proliferation and migration.
Our study also indicates that IL-17A is a potent inducer of fibroblast invasion, and that invasion follows extracellular matrix degradation. IL-17A enhances the expression levels and activation of the type IV collagenases MMP-2 and MMP-9, and the broad-spectrum MMP inhibitors marimastat and GM 6001 both inhibit IL-17A-induced fibroblast migration and proliferation. IL-17A is also known to induce other MMPs. We previously reported that IL-17A induces MMP-1 in human cardiac fibroblasts via MAPK activation [4]. Supporting our in vitro results, it has been previously demonstrated that the β-adrenergic receptor agonist isoproterenol induces IL-17 mRNA expression in the failing rat heart in vivo, and IL-17A induces MMP-2 and MMP-9 mRNA expression in neonatal rat cardiac fibroblasts in vitro [22]. Since IL-17A knockout mice display reduced inflammation, fibrosis and MMP expression in myocarditogenic peptide-induced myocarditis [7], our results support the hypothesis that IL-17A is a critical mediator in cardiac fibrosis and adverse remodeling. Studies are in progress to determine this possibility in post-ischemic myocardial injury and remodeling in vivo using IL-17A and IL-17RA null mice.
Similar to IL-17A, both IL-17F and IL-17A/F heterodimer induced fibroblast proliferation and migration. Though we did not investigate the underlying molecular mechanisms, we hypothesize that both IL-17F and IL-17A/F activate signaling pathways that are similar to that described here for IL-17A, as these ligands also signal via the IL-17R complex composed of IL-17RA and IL-17RC [2]. Gaffen and colleagues have previously reported that this receptor complex physically associates with the adapter molecule Act1 or CIKS through SEFIR/SEFIX domains, leading to the activation of multiple and distinct signal transduction pathways mediated through unique receptor subdomains, including activation of ERK1/2 and p38 MAPK [2, 12]. One of the pathways mediated by the IL-17R complex is activation of NF-κB and AP-1 [4, 23, 24], and both these nuclear transcription factors are critical for MMP induction.
In summary, our studies demonstrate that IL-17A suppresses the dual phosphatase MKP-1 via Akt/miR-101 induction, activates p38 MAPK and ERK1/2, and induces cardiac fibroblast proliferation and migration. We have demonstrated that IL-17’s effects are direct, and are not mediated by other cytokines. However, it has been previously reported that IL-17A synergizes with TNF-α and other proinflammatory cytokines in exerting its biological effects. Since myocardial fibrosis and adverse remodeling are complex disease processes, and are mediated by multiple cytokines, it is reasonable to speculate that IL-17A acts in concert with other cytokines.
Our results show that IL-17A, IL-17F, and IL-17A/F all induce migration and proliferation of primary adult cardiac fibroblasts that are important mediators in adverse cardiac remodeling. IL-17 induced MKP-1 suppression leads to MAP kinase activation, the induction of multiple MMPs that contribute to adverse remodeling, and the activation of several transcription factors, including NF-κB and AP-1 that induce other proinflammatory cytokines, chemokines, and adhesion molecules, thus further propagating myocardial inflammation and injury. IL-17 is a potential therapeutic target in cardiac fibrosis and adverse myocardial remodeling.
Supplementary Material
Highlights.
Interleukin-17 is a potent inducer of fibroblast migration and proliferation.
IL-17A inhibits the mitogen-activated protein kinase-1 via miR-101 induction.
IL-17A induces miR-101 via PI3K/Akt signaling.
IL-17A activates p38 MAPK and ERK1/2.
IL-17F and IL-17A/F heterodimer mimic IL-17A effects.
Acknowledgments
This work was supported by the Veterans Affairs Office of Research and Development- Biomedical Laboratory Research and Development Service Award 1IO1BX000246 and the NHLBI Grant HL-86787 to BC. PD is a supported by HL-70241 and HL-80682. The contents of this report do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- BrdU
bromodeoxyuridine
- CF
cardiac fibroblasts
- CMV
cytomegalo virus
- DMSO
dimethylsulfoxide
- dn
dominant negative
- DUSP
dual specificity protein phosphatase
- ERK
extracellular signal-regulated protein kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- IL
interleukin
- IL-17R
interleukin-17 receptor
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MKP-1
MAPK inhibitor-1
- MMP
matrix metalloproteinase
- miR
micro RNA
- MRE
microRNA responsive element
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenoltetrazolium bromide
- PI3K
phosphatidylinositol-3 kinase
- siRNA
small interference RNA
- shRNA
small hairpin RNA
- S6K
S6 kinase
- TdR
thymidine
- TNF
tumor necrosis factor
- wt
wild type
Footnotes
Conflict of interest: There are no commercial affiliations or conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Iwakura Y, Ishigame H, Saijo S, Nakae S. Immunity. 34(2):149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Gaffen SL. Nat Rev Immunol. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian Y, Kang Z, Liu C, Li X. Cell Mol Immunol. 7(5):328–333. doi: 10.1038/cmi.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, Barnes JL, Chandrasekar B. Am J Physiol Heart Circ Physiol. 2007;293(6):H3356–3365. doi: 10.1152/ajpheart.00928.2007. [DOI] [PubMed] [Google Scholar]
- 5.Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Am J Physiol Heart Circ Physiol. 2008;294(5):H2078–2087. doi: 10.1152/ajpheart.01363.2007. [DOI] [PubMed] [Google Scholar]
- 6.LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Am J Physiol Cell Physiol. 2007;292(5):C1799–1808. doi: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 7.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, Gabrielson K, Iwakura Y, Rose NR, Cihakova D. Circ Res. 106(10):1646–1655. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 8.Fan Y, Weifeng W, Yuluan Y, Qing K, Yu P, Yanlan H. Virol J. 8:17. doi: 10.1186/1743-422X-8-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA. J Exp Med. 207(3):535–552. doi: 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeisberg EM, Kalluri R. Circ Res. 107(11):1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakrabarty A, Tranguch S, Daikoku T, Jensen K, Furneaux H, Dey SK. Proc Natl Acad Sci U S A. 2007;104(38):15144–15149. doi: 10.1073/pnas.0705917104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho AW, Shen F, Conti HR, Patel N, Childs EE, Peterson AC, Hernandez-Santos N, Kolls JK, Kane LP, Ouyang W, Gaffen SL. J Immunol. 185(2):1063–1070. doi: 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. J Immunol. 1998;160(7):3513–3521. [PubMed] [Google Scholar]
- 14.Wang Y. Circulation. 2007;116(12):1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keyse SM. Curr Opin Cell Biol. 2000;12(2):186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 16.Zhu QY, Liu Q, Chen JX, Lan K, Ge BX. J Immunol. 185(12):7435–7442. doi: 10.4049/jimmunol.1000798. [DOI] [PubMed] [Google Scholar]
- 17.Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M. Physiol Rev. 91(3):1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weston CR, Davis RJ. Curr Opin Cell Biol. 2007;19(2):142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Franklin CC, Kraft AS. J Biol Chem. 1997;272(27):16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- 20.Abraham SM, Clark AR. Biochem Soc Trans. 2006;34(Pt 6):1018–1023. doi: 10.1042/BST0341018. [DOI] [PubMed] [Google Scholar]
- 21.Brondello JM, Brunet A, Pouyssegur J, McKenzie FR. J Biol Chem. 1997;272(2):1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- 22.Feng W, Li W. Exp Mol Pathol. 88(2):299–304. doi: 10.1016/j.yexmp.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Patel DN, King CA, Bailey SR, Holt JW, Venkatachalam K, Agrawal A, Valente AJ, Chandrasekar B. J Biol Chem. 2007;282(37):27229–27238. doi: 10.1074/jbc.M703250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U. J Biol Chem. 286(15):12881–12890. doi: 10.1074/jbc.M110.199547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.