

Summary
A key characteristic of hematopoietic stem cells (HSCs) is the ability to self-renew. Genetic deletion of β-catenin during fetal HSC development leads to impairment of self-renewal while β-catenin is dispensable in fully developed adult HSCs. Whether β-catenin is required for maintenance of fully developed CML leukemia stem cells (LSCs) is unknown. Here, we use a conditional mouse model to show that deletion of β-catenin after CML initiation does not lead to a significant increase in survival. However, deletion of β-catenin synergizes with imatinib (IM) to delay disease recurrence after imatinib discontinuation and to abrogate CML stem cells. These effects can be mimicked by pharmacologic inhibition of β-catenin via modulation of prostaglandin signaling. Treatment with the cyclooxygenase inhibitor indomethacin reduces β-catenin levels and leads to a reduction in LSCs. In conclusion, inhibiting β-catenin by genetic inactivation or pharmacologic modulation is an effective combination therapy with imatinib and targets CML stem cells.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasia (MPN) characterized by massive proliferation of phenotypically normal mature myeloid cells. This disease is initiated by a reciprocal translocation of chromosomes 9 and 22 resulting in the generation of a constitutively active fusion kinase: BCR-ABL (Ben-Neriah et al., 1986; Druker, 2008). This oncogenic fusion is capable of transforming hematopoietic stem cells (HSCs) and is sufficient to initiate MPN in murine bone-marrow (BM) transplantation models (Daley et al., 1990). Targeted therapy using small molecule inhibitors of BCR-ABL such as imatinib (IM) (Druker et al., 2001; Druker et al., 1996) or second generation kinase inhibitors such as dasatinib or nilotinib (Kantarjian et al., 2010; Saglio et al., 2010) has revolutionized therapy for CML, however in the overwhelming majority of patients, the disease clone is not eliminated, an effect that has been attributed to a persistent leukemia stem cell (LSC) pool that is inherently resistant to these targeted therapies. LSCs in CML share immunophenotypic features with normal HSCs, reside in the BM and are resitant to IM treatment, despite inhibition of BCR-ABL (Corbin et al., 2010; Hu et al., 2009). Upon discontinuation of IM therapy, these LSCs are able to re-establish CML and to cause disease relapse (Savona and Talpaz, 2008).
A key characteristic of stem cells is their ability to self-renew. Several genes and signaling pathways control the fine balance between self-renewal and differentiation in HSCs and potentially also in LSC. One such pathway is the canonical Wnt-pathway (Jamieson et al., 2004; Majeti et al., 2009; Malhotra and Kincade, 2009; Muller-Tidow et al., 2004; Reya et al., 2003; Zhao et al., 2007). β-catenin, the pathway’s central effector molecule, is negatively regulated via phosphorylation by a multiprotein complex including APC, Axin, GSK-3β and casein kinase (Behrens et al., 1998; Rubinfeld et al., 1996). Several compounds interacting with this pathway in a variety of cancers are currently being investigated in pre-clinical studies (Chen et al., 2009; Huang et al., 2009; Peterson et al., 2009). Prostaglandin E2 is known to promote stabilization of β-catenin in colon cancer (Castellone et al., 2005) and can be modified by inhibition of its upstream regulator cyclooxygenase (COX). Recently, interference of prostaglandin-signaling has been shown to target the Wnt/β-catenin axis in HSCs (Goessling et al., 2009) and acute myeloid leukemia (AML) stem cells (Wang et al., 2010). Abrogation of β-catenin by the cyclooxygenase inhibitor indomethacin led to a 100-fold decrease in AML initiating cells in secondary recipients. Moreover, indomethacin treatment of fully developed, MLL-AF9 induced leukemia led to reduction of β-catenin levels and caused reduction of LSC frequency. These data indicate that certain subtypes of AML retain dependency on Wnt-signaling.
Recent studies have shed light on the impact of Wnt/β-catenin activity on development of BCR-ABL induced MPN in several CML mouse models. Deletion of β-catenin in HSC development (using vav-Cre) (Zhao et al., 2007) or concurrently with activation of BCR-ABL (by a retroviral fusion BCR-ABL-Cre) (Hu et al., 2009) in Ctnnb1fl/fl knockout mouse BM led to impaired leukemogenesis. These studies clearly indicate that β-catenin plays a role in the development of normal HSCs and BCR-ABL induced CML. However, β-catenin is not required for maintenance of normal self-renewal in fully developed HSCs, which prompts the question whether β-catenin is required for maintenance of BCR-ABL induced CML LSCs. This question is critical for therapeutic targeting LSC in chronic phase CML patients. Furthermore, given that all patients with CML are treated with tyrosine kinase inhibitors, it is important to determine the effects of pathway modulation in this context. We therefore aimed to investigate the impact of β-catenin modulation in established and IM treated BCR-ABL induced CML.
Results
Genetic deletion of β-catenin (Ctnnb1) reduces bone marrow and peripheral blood CML cells
In order to address whether β-catenin is required for LSC maintenance, we aimed to delete β-catenin in BM cells after the engraftment of BCR-ABL transduced stem cells in primary recipient mice. BM from mice with the following genotypes, Ctnnb1fl/fl Esr1-Cre+, Ctnnb1+/fl Esr1-Cre+, Ctnnb1+/+ Esr1-Cre+, were transduced with a retrovirus encoding human p210-BCR-ABL and GFP (MSCV-BCR-ABL-IRES-GFP) and injected into wildtype syngeneic recipient mice. Recipient mice were then followed for establishment of disease by assessment of GFP+ cells in the peripheral blood after BM transplantation. Upon confirmation of GFP+ cells (>3%) in peripheral blood, tamoxifen (TAM) was introduced via intraperitoneal injection (generally starting day +14). Mice were then followed for survival, and unexpectedly, recipient mice of Ctnnb1fl/fl Esr1-Cre+ (Ctnnb1-/-) BM showed similar survival to treated wildtype or untreated controls with all mice succumbing to a CML-like disease between 3-5 weeks after BM transplantation (Fig. 1A). There was no significant survival difference detectable between the different treatment cohorts, although, there was a trend for the Ctnnb1-/- group (p=.12). β-catenin excision was shown by PCR in Ctnnb1-/- recipients, demonstrating excision in the majority of GFP-sorted cells (Fig.S1). Upon analysis of moribund animals, we recognized that organ infiltration (especially hemorrhagic pulmonary infiltrates) were comparable in all treatment groups (Fig.1C). In contrast, there was a clear difference detectable with regard to BM and blood involvement. White blood counts were significantly lower in the Ctnnb1-/- (p=.0149*) group compared to TAM-treated WT controls (Fig.1B). BM samples of each mouse showed decreased GFP+ cells in Ctnnb1-/- mice (Fig.1D). This difference was noted in both the total GFP+ fraction as well as the GFP+/Sca-1+ fraction of individually analyzed BM samples from each group (Fig.1D). Significantly, it is this GFP+Sca-1+ population that is enriched for LSCs in that transplantation of these cells is sufficient to reconstitute MPN in secondary recipients (Hu et al., 2009). These data suggest that conditional β-catenin deletion after onset of BCR-ABL induced disease predominantly reduces more immature hematopoietic cells, including phenotypic LSCs, as compared to more mature cells that have infiltrated organs such as the lung.
Figure 1. Genetic deletion of β-catenin after engraftment of BCR-ABL positive disease leads to reduction of leukemia cells in blood and BM, but does not prolong survival of recipient mice.
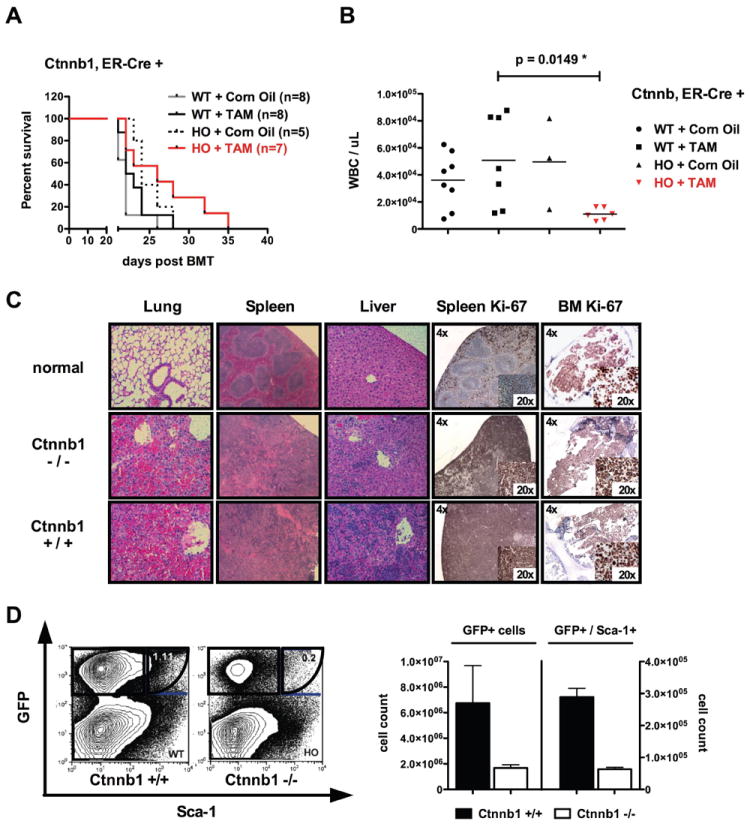
(A) After injection of 70,000 sorted GFP+ cells plus 1×106 supporter cells, no significant difference in survival was evident in primary recipient mice of homozygous floxed (HO) or wildtype (WT) BM cells. β-catenin (Ctnnb1) was excised after administration of Tamoxifen (TAM) and Corn Oil served as an ‘empty control’. (B) Blood counts of moribund animals display significant differences between recipients of homozygous (Ctnnb1fl/fl Cre+) (p=.0149*) and wildtype (Ctnnb1+/+ Cre+) controls. (C) BM analysis reveals reduction in GFP+ (~3 fold reduction in Ctnnb1-/- recipients) and GFP+Sca-1+ cells (~3fold reduction in Ctnnb1-/- recipient mice). The left panel demonstrates GFP % and the right panel absolute number of GFP+ cells. (D) Organ infiltration (predominantly lung infiltration) was the cause of death in the majority of recipient mice without differential infiltration patterns between the groups investigated (displayed are representative HE-stains of each genetic group). Loss of β-catenin did not influence proliferation (Ki67) of the majority of transformed cells in spleen and BM. Error bars indicate standard deviation (SD) of 3 cohorts investigated. Excision control PCR on sorted GFP+ bone marrow cells from moribund primary recipient mice is indicated in Supplemental Figure S1.
Combination of IM and β-catenin deletion results in delayed CML recurrence after IM discontinuation
Tyrosine kinase inhibitors such as imatinib (IM) are established as the first line therapy for patients with CML, resulting in high rates of hematologic, cytogenetic and molecular remissions. However, withdrawal of IM, even in patients that had achieved a complete molecular remission, results in relapsed disease in the majority of patients (Ross et al., 2010). This has been hypothesized to reflect a resistant and quiescent LSC population that can regenerate disease in the event of treatment interruption (Mahon et al., 2010; Ross et al., 2010; Rousselot et al., 2007). Based on this, we hypothesized that the combination of IM therapy, which targets most cells other than the LSC population, with Wnt/β-catenin pathway modulation, which should target more immature LSC-like cells, might delay relapse after IM withdrawal. Transplantation related reconstitution/engraftment of genetically modified cells is always an issue when investigating leukemia development. Therefore, our experiment (Figure 2) was specifically designed to take the transplantation related reconstitution/engraftment out of the equation. To address this, we examined whether activation or inhibition of Wnt/β-catenin signaling could have an effect on disease relapse after discontinuation of IM treatment. Wnt/β-catenin signaling is known to regulate the cell cycle in HSCs and constitutive activation is associated with loss of quiescence and stem cell exhaustion (Gothot et al., 1998; Kirstetter et al., 2006; Passegue et al., 2005; Scheller et al., 2006). To model constitutive activation of β-catenin we used a validated model of enhanced Wnt signaling, Apcmin mice (Lane et al., 2010). We assessed inactivation using Ctnnb1fl/fl Esr1-Cre+ mice, or the appropriate wildtype mice (Ctnnb1+/+ Esr1-Cre+, Ctnnb1+/+ Esr1-Cre-) as BM donors. 5-FU treated BM cells of donor mice (as indicated above) were infected with the p210-BCR-ABL retroviral construct and injected into syngeneic recipients. Following engraftment, IM treatment was initiated followed by TAM treatment to induce β-catenin deletion (Fig. 2A). After 3 weeks, IM treatment was discontinued and mice were monitored for disease progression. All mice were analyzed for disease burden after the first animal had to be sacrificed according to the animal facility guidelines. BM, spleen and peripheral blood were analyzed for signs of MPN. All wildtype animals investigated at that early time point had a significant amount of disease burden, so the first animal is not likely to be an outlier. White blood counts were significantly lower in animals where β-catenin was deleted as compared to wildtype-recipient controls (p=.048*), however, recipients of Apcmin BM showed similar levels compared to wildtype recipients (Fig.2B, left panel). Given the short time after IM discontinuation, no significant differences in spleen size were detected (Fig.2B, right panel). Investigating the BM, however, revealed greater than 100-fold reduction of GFP+ cells and GFP+Lin- cells in mice that received Ctnnb1fl/fl Esr1-Cre+ BM and that were treated with TAM (Fig.2C,D). Recipients of Apcmin BM showed similar levels of leukemic cells and a minor reduction in the lineage-negative (Lin-) population. These results indicate, that the combination of IM with genetic deletion of β-catenin reduces the amount of leukemic disease burden and delays disease relapse caused by persistent immature leukemia cells in vivo. Therefore, combined tyrosine kinase inhibition and β-catenin deletion was an effective combined therapy to delay disease progression.
Figure 2. Conditional deletion of β-catenin followed by withdrawal of IM treatment leads to delayed disease recurrence.
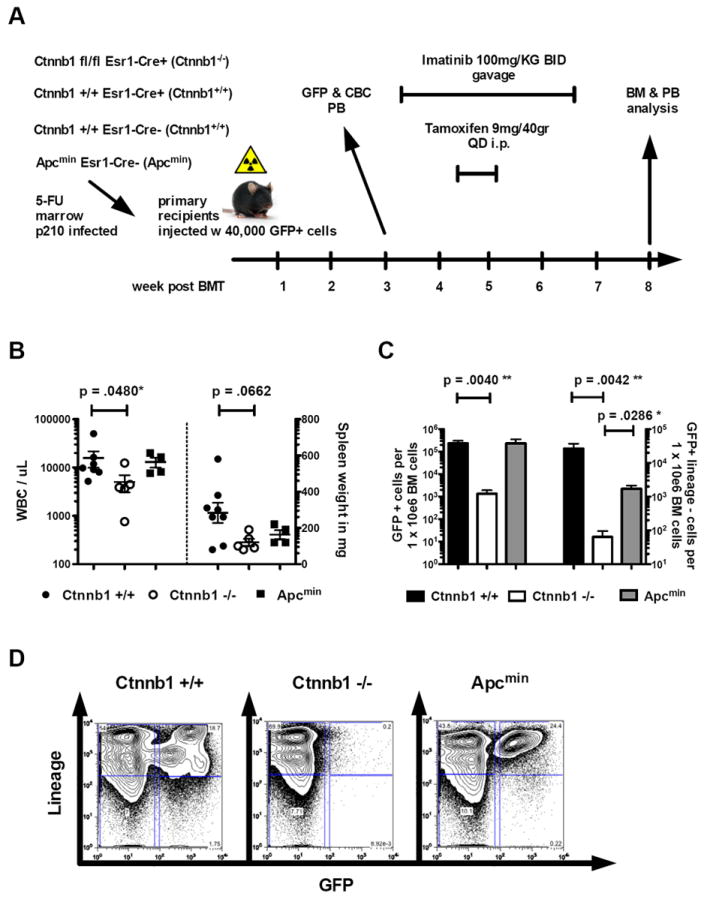
(A) Treatment schedule of primary recipient mice after transplantation with 40,000 GFP+ cells. (B-D) Endpoint analysis of white blood count (WBC), BM and spleen weight have been performed at disease onset in the first relapsed animal after discontinuation of treatment. At this point all remaining animals were sacrificed and analyzed. Disease burden (BCR-ABL transformed GFP+ cells) was analyzed as a correlate for relapse (‘cytogenetic or hematologic relapse after treatment discontinuation’) (B) WBCs are significantly reduced in recipients of Ctnnb1-/- BM, compared to Ctnnb1+/+ controls. Apcmin mice do not display any differences in WBC or spleen weight compared to Ctnnb1+/+ animals. (C, D) BM analysis reveals significant reduction of GFP+ cells and GFP+LSK in the Ctnnb1-/- recipient cohort. Apcmin mice show similar amounts of GFP+ cells as WT controls with a minor reduction in the GFP+Lin- compartment. (Error bars indicate standard deviation of WBCs n>4 per cohort displayed).
Combination of imatinib (IM) treatment and β-catenin deletion targets CML Leukemia Stem Cells
To determine whether combination of IM with genetic deletion of β-catenin could eliminate the LSC population and improve survival, we evaluated the ability of CML LSC to maintain disease in a serial BM transplantation assay, considered the most stringent test of self-renewal in vivo. Disease was induced by transduction of BM from mice with the following genotypes, Ctnnb1fl/fl Esr1-Cre+, Ctnnb1+/fl Esr1-Cre+, Ctnnb1+/+ Esr1-Cre+. Then we injected 70,000 GFP+ sorted cells together with 1×106 supporter cells in primary mice. Primary recipients were sacrificed after onset of disease and 150,000 LSC-enriched GFP+Sca-1+ cells were injected into lethally irradiated secondary recipients with 1×106 supporter cells as indicated above. IM treatment was initiated 7 days after transplantation into secondary recipient mice and continued for 21 days, combined with tamoxifen (TAM) administration days 14-18. Secondary recipients were sacrificed two days after discontinuation of IM treatment (day 30) for immunophenotypic analysis of BM and transplantation of unfractionated BM cells into tertiary recipients (Fig.3A). We analyzed BM for GFP+ or GFP- Lin-Sca-1+Kit+ (LSK) cells (Fig.3B, representative plot). The percentage and total numbers of GFP+Lin- and GFP+LSK (enriched for LSC activity) cells in the TAM treated animals showed a significant decrease compared to control treated animals (Fig.3C). When 1.5×106 whole BM cells isolated from treated secondary recipients were injected into lethally irradiated tertiary recipients, this resulted in significantly prolonged survival of recipients of Ctnnb1fl/fl Esr1-Cre+ (Ctnnb1-/-) BM (Fig.3D) compared to Ctnnb1+/+ Esr1-Cre+ (Ctnnb1+/+) controls (p=.0019**). Secondary recipients of heterozygous Ctnnb1+/fl Esr1-Cre+ (Ctnnb1+/-) BM showed a slight, but not significant reduction of GFP+LSK cells. When this BM was transplanted into tertiary recipients, these animals had a slight, but not significant survival benefit as compared to control (data not shown). In order to directly compare disease burden in tertiary recipient mice, we investigated the peripheral blood for GFP+ cells after the development of lethal disease in the first animal (Fig.3D, E). A significant difference in total number and percentage of GFP+ cells was evident at that time between recipient animals of Ctnnb1+/+ and Ctnnb1-/- BM (Fig.3E). As several mice developed disease in the Ctnnb1+/+ cohort but none of the Ctnnb1-/- became sick, we aimed to analyze the molecular disease burden as determined by BCR-ABL transcript detection in the peripheral blood by nested-RT-PCR. PCR-primers for the human p210 BCR-ABL construct were used together with murine Gapdh-controls. None out of 10 living animals that received Ctnnb1-/- BM had any b3a2-transcript detectable, while 2 out of 5 remaining animals that received Ctnnb1+/+ BM showed positivity for a BCR-ABL transcript (Fig.3F). The other 5 animals in the Ctnnb1+/+ cohort had already succumbed to MPN and BM and spleen infiltration by GFP+ (BCR-ABL+) cells had been confirmed.
Figure 3. Combination of genetic deletion of β-catenin with IM treatment leads to significant reduction of CML-LSC in a serial BM transplantation assay.
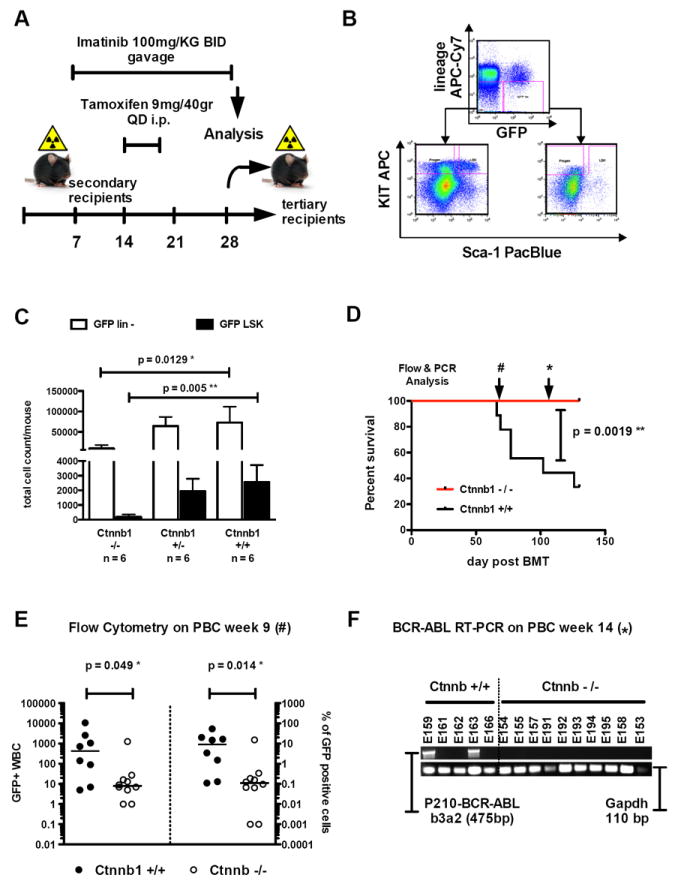
(A) Treatment schedule of secondary recipient mice after transplantation with 150,000 GFP+Sca-1+ cells plus supporter cells. (B) Representative analysis plot of GFP+Lin- and GFP+LSK compartments of secondary recipient mice. (C) Genetic deletion of β-catenin (Ctnnb1) leads to a significant decrease of GFP+Lin- cells (p=.0129*) and GFP+LSK (p=.005**) in secondary recipient mice when combined with imatinib (IM) treatment. Deletion of β-catenin in combination with IM led to a reduction of GFP+Lin- cells from 72988 in Ctnnb1+/+ recipient animals to 9231 in Ctnnb1-/- recipient animals (calculated as percentage of total harvested BM cells). These numbers correlated with the reduction of GFP+LSK-cells from 2560 to 194, respectively. Error bars indicate the standard deviation of n=6 mice investigated in the cohort displayed. (D) Transplantation of 1.5×106 whole BM cells leads to a significant survival advantage in tertiary recipients of Ctnnb1-/- BM when compared to Ctnnb1+/+ controls. Transplantation of higher cell numbers (6×106 whole BM cells) is displayed in Supplemental Figure S2. Analysis of disease burden at different time points by flow cytometry (E) or nested RT-PCR (F) showed a significant decrease in disease load in Ctnnb1-/- tertiary recipient mice. These results could be recapitulated using Mx1-Cre (Supplemental Figure S2 panels C-E).
We repeated the experiment using a higher cell number of unfractionated BM cells for injection into tertiary recipients. With 6×106 whole BM cells 1/5 mice developed GFP+MPN in the Ctnnb1-/- recipient group while all of the Ctnnb1+/+ recipients succumbed to disease (Fig.S2A). Moreover, all of the recipients of heterozygous floxed BM (Ctnnb1+/-) developed lethal MPN but with a slightly longer latency than the wildtype recipient control. Analysis of disease burden on day 36 after transplantation revealed significantly lower GFP+ cells in the peripheral blood of Ctnnb1-/- compared to wildtype and heterozygous controls (Fig.S2B). Although only 1/5 of animals in the Ctnnb1-/- (6×106 cells injected) cohort had succumbed to disease, we used transplants performed at limiting cell number to determine the estimated LSC frequency. With the two datapoints available (dilutions of 6×106 and 5×105) we estimate an approximate 35fold decrease in LSC. Taken together, our data shows a dramatic reduction in LSC frequency.
As there have been recent reports that highlight Esr1-Cre related toxicity on myelopoiesis (Higashi AY et al, J Immunol, 2009), we aimed to clarify the impact of β-catenin deletion on the reduction or disappearance of LSC in our model, we repeated the genetic model applied using the well characterized Mx1-Cre model (Fig. S2C-E). IM treatment was started on day 14 (100mg/KG per gavage BID). Mice were injected on days 15, 17 and 19 with pIpC at a dose of 12.5μg/gr body weight. After a treatment period of 21 days of IM, mice were sacrificed. We analyzed the BM (as indicated below) to determine the amount of immunophenotypic LSC (GFP+LSK-cells). This analysis showed an approximately 10-fold reduction in GFP+LSK cells. In order to determine the number of functional LSC, we transplanted dilutions of total BM (5 × 105 and 6 × 106 cells) into secondary recipient mice. Animals in the 6 × 106 Ctnnb1+/+ cohort developed lethal MPN within 45 days after transplantation, while only 25% of the Ctnnb1-/- mice died of MPN. When the graft was diluted to 5 × 105 cells, only 20% of the Ctnnb1+/+ but none of the mice with a Ctnnb1-/- genotype developed disease. This functional dataset using the established Mx1-Cre system suggests a ~10fold reduction of LSC (Fig. S2E).
Indomethacin is effective in modulating β-catenin levels and synergizes with IM in CML
To address whether modulation of Wnt/β-catenin signaling can be achieved pharmacologically in CML stem cells, we aimed to inhibit or enhance β-catenin levels in the human BCR-ABL positive cells line K562. While inhibition of β-catenin may contribute to reduction of LSC, stimulation of β-catenin activity could potentially bring LSC into cycle and therefore make them more susceptible to IM treatment (Scheller et al., 2006). Moreover, there is evidence, that inhibition of GSK3, which should activate β-catenin, is an effective approach used for AML (Holmes et al., 2008; Wang et al., 2008). As it is known that inhibition of prostaglandin signaling using COX2-inhibitors can decrease β-catenin level in HSCs and LSCs, we utilized indomethacin to inhibit and the GSK3-inhibitor BIO ((2’Z,3’E)-6-Bromoindirubin-3’-oxime) to stimulate Wnt/β-catenin pathway activity.
Treatment of BCR-ABL rearranged K562 cells with either indomethacin or IM led to reduction of β-catenin protein levels, but the combination of both drugs led to almost complete abrogation of β-catenin in vitro (Fig. 4A). In contrast, incubation with the GSK3β-inhibitor BIO caused pronounced accumulation of β-catenin. These stimulative BIO-effects could be partially reversed by co-incubation with indomethacin and IM. Next we developed a flow cytometry based assessment of β-catenin so that we could assess levels in small numbers of cells. When we assessed β-catenin levels in K562 cells treated with BIO and indomethacin, we found similar results to our immunoblot experiments (Figure 4B, left panel).
Figure 4. Modulation of prostaglandin signaling leads to alteration of β-catenin levels and influences colony formation.
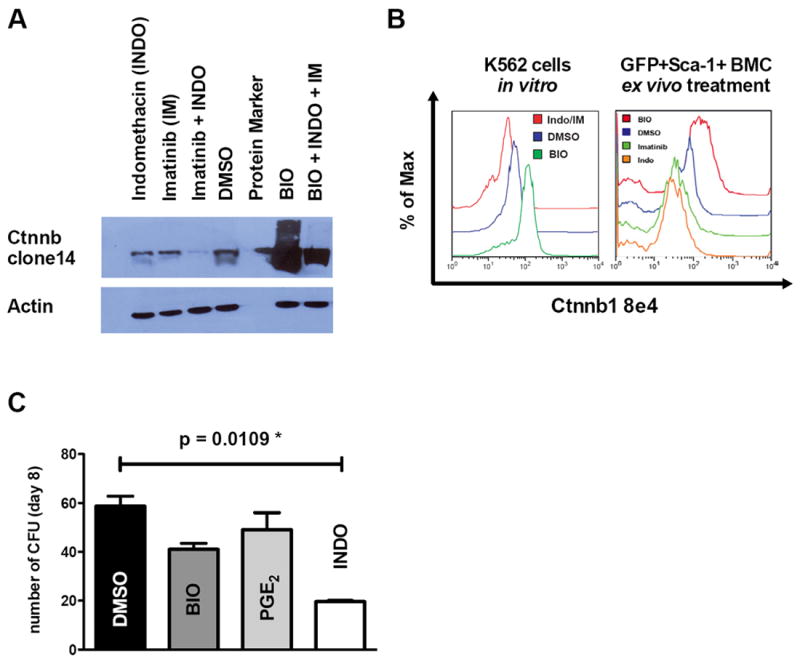
(A) Western blotting on whole cell lysates of K562 cells. Incubation of K562 cells with either IM (1μM) or indomethacin (40μM) led to reduction of β-catenin protein levels, while combined treatment revealed profound decrease of β-catenin. Increase of β-catenin levels were detectable upon GSK3β inhibition using BIO (0.5μM) and partially reversible by co-treatment with IM and indomethacin. (B) Using low cell numbers, these results could be validated using intracellular flow cytometry in K562 cells (left panel) and in GFP+Sca1+ leukemic cells from primary mice (right panel). (C). When plated in methylcellulose, indomethacin treated GFP+Sca1+ cells sorted from primary recipient mice (n=3 mice /group, error bars indicate SD) showed significantly impaired colony formation compared to DMSO, BIO or PGE2 treated cells (p=.0109). Evaluation of β-catenin modulation in CML-LIC derived from recipient mice after drug treatment is shown in Supplemental Figure S3.
As BIO and indomethacin proved to effectively modulate β-catenin in the cell culture model, and indomethacin even synergized with IM in β-catenin suppression, we aimed to confirm these modulatory effects and capacity in the LSC enriched population from mice with BCR-ABL induced MPN. Mouse BM cells were transduced with the BCR-ABL retrovirus after 5-FU treatment and injected in to primary (lethally irradiated) recipient mice as described above. Primary recipient mice were sacrificed on day 20 after transplantation, BM was harvested and sorted for GFP+Sca1+ cells. This LSC containing population was treated with either DMSO, IM, indomethacin or BIO. Flow cytometry revealed increased β-catenin levels in BIO treated GFP+Sca1+ cells, and a reduction of β-catenin could be detected in IM or indomethacin treated cells (Fig. 4B, right panel). Additionally, we treated GFP+Sca1+ cells ex vivo with BIO, PGE2, indomethacin or DMSO and then plated them in methylcellulose. This revealed decreased colony formation upon indomethacin treatment while enhancement of β-catenin using BIO or PGE2 did not show any significant change in colony formation (Fig. 4C). When we tested primary recipient mice for induction of cell cycle by GSK3 inhibition, we found that administration of BIO led to induction of cell cycle in both GFP-negative LSK but even more pronounced in GFP-positive LSK (Fig. S3A). As we aimed to link indomethacin treatment with effects on β-catenin levels in CML-LIC, we performed an ELISA-based assay for detection of cAMP-levels on GFP+Sca-1+ cells. Low levels of cAMP were detected in the DMSO treated controls, however, treatment with indomethacin lowered the cAMP-levels in the GFP+Sca-1+ leukemia initiating fraction (as indicated by increase in absorbance; Fig. S3B). Although this is not a functional assay, decrease of cAMP levels by indomethacin treatment in the leukemia initiating population links the functional data to the mechanism of PGE2 stimulation of HSCs, which was through stimulation of cAMP levels and PKA phosphorylation of β-catenin (Gossling et al., Cell, 2009). These data show that indomethacin works together with IM to suppress β-catenin in vitro and in vivo prompting further assessment of the combination.
Imatinib (IM) plus indomethacin prolongs survival in a serial murine BM transplantation model
As inhibition of PGE2-signaling suppressed β-catenin levels in CML cells, we aimed to elucidate whether co-treatment with indomethacin and IM could effectively reduce the number of LSC and could lead to delayed onset of disease in a serial transplantation approach. As described above, 150,000 GFP+Sca-1+ cells isolated from mice with a primary BCR-ABL induced CML were injected into secondary recipients along with supporter BM cells. These mice were treated with IM (d7-28) and indomethacin or vehicle control (d14-18, d21-25) (Fig.5A). After 28 days, all mice were sacrificed and analyzed in detail (Fig.5B). Normal spleen size and white blood counts of treated secondary recipients indicated efficacy of IM only and of IM plus indomethacin treatment (Fig.S4A, B). Strikingly, we observed a significant reduction of GFP+LSK cells in the indomethacin plus IM co-treated animals compared to IM only controls (Fig.5C). However, the numbers of normal LSK cells (GFP-, derived from supporter BM) were not affected by any co-treatment (data not shown).
Figure 5. Pharmacologic inhibition of PGE2-signaling in combination with IM treatment leads to reduction of LSCs in a serial BM transplantation assay in vivo.
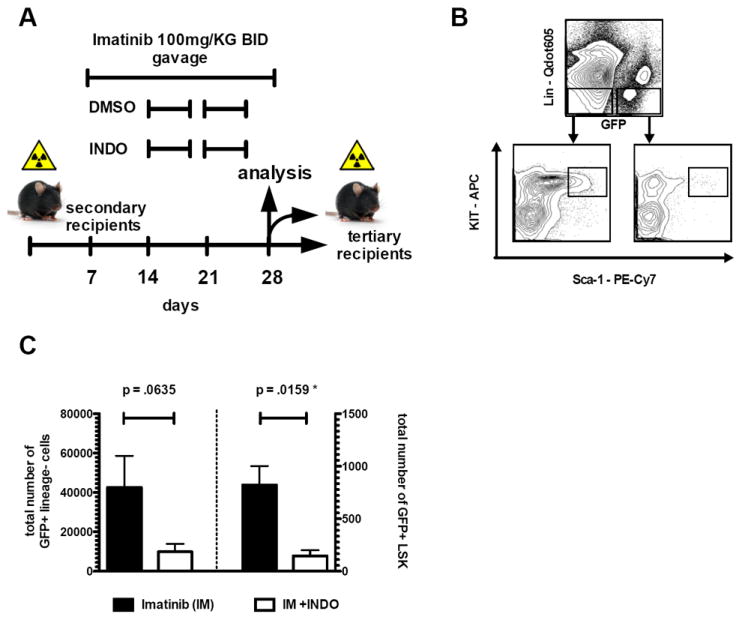
(A) Treatment schedule of secondary recipient mice (n ≥ 8 mice/cohort) injected with 150,000 GFP+Sca-1+ cells out of primary recipient mice. (B) Representative analysis plot of GFP+Lin- and GFP+LSK compartments of secondary recipient mice after treatment. (C) Co-treatment of secondary recipient mice with IM and indomethacin leads to reduction of GFP+Lin- cells (p=.0635) and to significant depletion of GFP+LSK cells (p=.0159) in the BM. GFP+Lin- cells were reduced from 42,560 (IM only group) to 9,867 (IM+INDO treated cohort). This translated into a significant reduction of GFP+LSK cells from 823 to 146, respectively. (Error bars indicate SD of GFP positive cells; n>4 per cohort investigated). Evaluation of secondary recipients after drug combination therapy is displayed in Supplemental Figure S4.
To investigate the functional relevance of this reduction, we injected 2×106 whole BM cells into tertiary recipient mice and monitored these animals for onset of disease. All tertiary recipients of the IM only treated BM developed lethal disease within 36 days after BM transplantation. However, recipients of IM plus indomethacin co-treated BM, showed delayed onset of disease and a significantly prolonged survival of up to 62 days after transplantation (Fig.6A, p<.0001***). In contrast to previous reports that the deletion of β-catenin using vav-Cre before transformation with BCR-ABL had resulted in development of acute lymphoblastic leukemia rather than CML, (Zhao et al., 2007) we observed that both treatment groups developed morphologically and immunophenotypically similar MPN (Fig.6B, C), despite delayed onset of disease in the indomethacin plus IM treated group. As activation of canonical Wnt signaling in our genetic model (using Apcmin mice) had not shown any significant reduction in disease burden, we also aimed to elucidate whether pharmacological stimulation of β-catenin could have an effect on LSC frequency or survival of tertiary recipient mice. Stimulation of β-catenin by co-treatment with the GSK3β inhibitor BIO resulted in a minor decrease of GFP+Lin- and GFP+LSK cells in secondary animals (Fig.S4C). However, this did not translate into any survival benefit of tertiary recipients (Fig.S5).
Figure 6. Prostaglandin-signaling is relevant as a target in CML-LSC and has prognostic relevance in human CML.
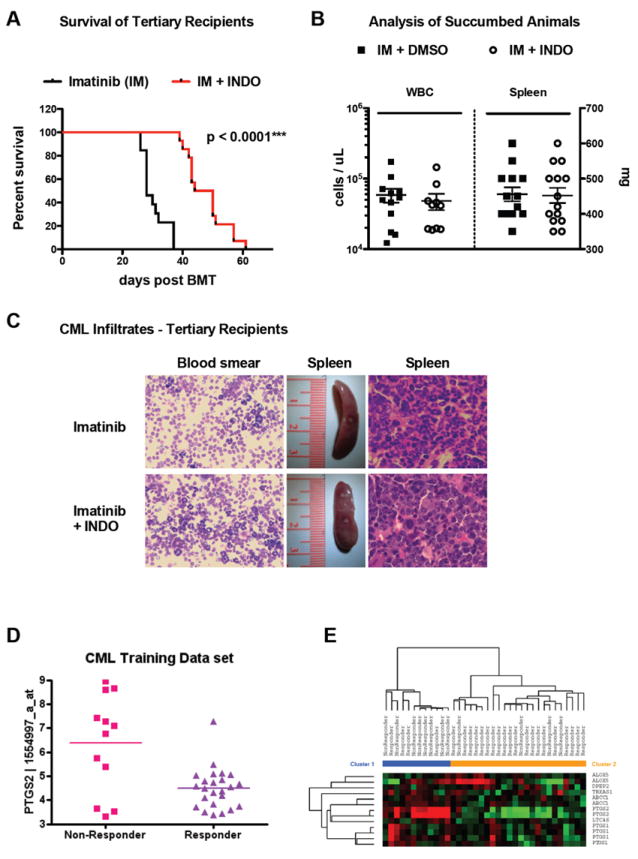
(A-C) Pharmacologic inhibition of PGE2-signaling in combination with IM treatment leads to delayed onset of disease in tertiary recipient mice. Injection of 1.5×106 whole BM cells leads to delayed onset of disease in tertiary recipients of IM plus indomethacin treated BM cells in comparison to IM only treated controls (A). Endpoint analysis of moribund animals revealed no difference in disease phenotype in the different treatment arms with regard to white blood count, spleen weight (B) or peripheral blood/organ involvement (C). Error bars indicated standard deviation of all mice (n>13) investigated in both cohorts investigated. Gene Expression Profiling of CML patient derived CD34+ cell samples (D, E) reveals a prognostically relevant PGE2-signature. Expression levels of prostaglandin synthetase 2 correlate with molecular response in the dataset investigated (p=0.0004, McWeeney et al. 2010). Moreover, sample clustering based on the “REACTOME_PROSTANOID_HORMONES” MSigDB gene set correlates also with response/non-response in this dataset (p=0.0067) (E). Figure S5 shows survival of tertiary recipient mice after combination of imatinib and BIO in secondary donor animals.
In order to correlate these findings of our mouse model with human data, we analyzed a previously published dataset that investigated CD34+ cells from IM-naive chronic-phase CML patients to predict responses upon IM treatment (McWeeney et al., 2010). Interestingly, supervised analysis comparing responders versus non-responders revealed the prostaglandin-endoperoxide synthase (PTGS2, also called cyclooxygenase-2) among the top candidates. PTGS2 expression levels were significantly lower in the ‘responder’ cohort (p=0.0067, Fig. 6D), therefore confirming, that inhibition of PTGS2/COX2 may translate into molecular responses (and therefore targeting LSC). Moreover, we found a Molecular Signatures Database (MSigDB) gene set defined by genes down-/up-regulated in CD4+ T-lymphocytes after stimulation with prostaglandin E2 (MSigDB gene set CHEMNITZ_RESPONSE_TO_PROSTAGLANDIN_E2, M2214) to be significantly enriched in the responder versus non-responder associated gene expression pattern (p=0.0284). Finally, clustering of the CML samples based on only genes involved in prostanoid hormones (MSigDB gene set REACTOME_PROSTANOID_HORMONES, M14714) correlated with response/non-response, and thus showed decreased expression levels of prostaglandin related genes to be correlated with treatment response (p=0.0067, Fig. 6E). These data provide evidence for an approach to target CML stem cells through the inhibition of Wnt/β-catenin signaling in combination with tyrosine kinase inhibitors by using a widely available and affordable clinical therapeutic (indomethacin).
Discussion
The Wnt/β-catenin pathway has been shown to be necessary for HSC development, but is not necessary for HSC maintenance in fully developed stem cells (Koch et al., 2008; Zhao et al., 2007). Also, β-catenin has been shown to be necessary for development of LSC in a mouse model of BCR-ABL induced MPN (Hu et al., 2009; Zhao et al., 2007). However, these data do not address the critical question as to whether β-catenin is required for maintenance of LSCs after the establishment of BCR-ABL induced disease. This is a critical question that will help guide development of therapeutics that target the Wnt/β-catenin pathway in patients with CML who are maintained on tyrosine kinase inhibitors. We hypothesized that inhibition of canonical Wnt-signaling could potentially affect the maintenance of IM-resistant CML stem cells, thereby providing synergy when used in conjunction with IM treatment. To address this question, we conditionally inactivated β-catenin after engraftment of BCR-ABL transformed HSCs/LSCs. Although we and others have had extensive experience with the hematopoietic specific Mx1-Cre recombinase, the induction of cre-expression in this model is induced by an interferon response through the injection of pI:pC in mice. Interferon was the standard therapy (alone or in combination with cytarabine) for CML for almost a decade and is known to induce complete cytogenetic remissions (Guilhot et al., 1997). Therefore, using Mx1-Cre in a conditional β-catenin knockout model could potentially confound our results through the anti-leukemic effects of elevated interferon levels. We therefore chose in the first set of experiments a tamoxifen (TAM)-inducible estrogen-receptor Cre recombinase. However, as there have been recent reports that highlight Esr1-Cre related toxicity on myelopoiesis (Higashi AY et al, J Immunol, 2009), we repeated our experiments using the Mx1-Cre system with consistent results. A caveat using the retroviral transduction model for BCR-ABL is the requirement of cell division for retroviral integration. Therefore, only dividing HSCs will be transformed by the oncogene. However, by two weeks after transplantation of these transformed pre-leukemic stem cells, a notable fraction of GFP+ LSK remains in G0 in cell cycle analysis before Esr1-Cre or Mx1-Cre mediated excision of β-catenin was initiated. Upon IM monotherapy an increasing fraction of stem cells remains quiescent in the BM. This indicates – at least in part – that also a temporarily quiescent fraction of stem cells can be affected by genetic deletion of β-catenin.
Unexpectedly, conditional deletion of β-catenin did not result in significant prolongation of survival in primary recipient mice and all recipients died due to organ infiltration. However, in the peripheral blood and BM, a clear reduction of GFP+ cells was detectable. Thus, these data support the tenet that deletion of β-catenin may result in reduction of LSCs, but may spare the differentiated progenitors, most likely due to a lack of dependence on β-catenin signaling for survival of this population. Furthermore, these results may reflect differences in the experimental design compared to previously reported – when β-catenin was deleted in early development (Zhao et al., 2007) or simultaneously to transformation with BCR-ABL using a fusion retrovirus vector (Hu et al., 2009). In our model differentiated progenitor cells had already infiltrated different organ sites prior to β-catenin deletion. These data demonstrate that therapeutic approaches which largely target LSC will need to be combined with therapeutics that predominately target progenitors (like Imatinib) in order to abrogate the detrimental effects of the high progenitor burden.
In order to target LSC populations and to simulate a clinically relevant setting for patients with CML that targets residual disease in tyrosine kinase inhibitor treated patients, we combined IM treatment with genetic modulation of β-catenin. Based on previous studies, there was rationale to test β-catenin loss of function in combination with IM. However, there is growing evidence that activation of Wnt signaling results in alteration of HSCs. Wnt/β-catenin activation by stabilization of β-catenin has been shown to lead to HSC-exhaustion due to cell cycle entry and consecutive apoptosis after initial expansion of the HSC pool (Scheller et al., 2006). Less pronounced activation using a GSK3β inhibitor led to increase of the HSC pool and enhanced repopulation capacity (Trowbridge et al., 2006). These data suggested that activation of Wnt signaling might lead to increased proliferation of the LSC pool and therefore increase susceptibility to IM treatment. However, it was unclear whether activation or inhibition of this pathway would be most beneficial in the context of IM therapy. Thus, we decided to assess either loss-of function or constitutive activation models of the Wnt/β-catenin pathway. We induced CML by transformation of murine stem- and progenitor cells with the human p210-BCR-ABL oncogene. IM treatment was initiated after engraftment of transformed cells and confirmed with either genetic deletion (by administration of tamoxifen in Ctnnb1fl/fl Esr1-Cre+ BMC) or activation (Apcmin BM) of Ctnnb1. While activation of β-catenin seemed to leave the disease initiating population largely unaffected, deletion of β-catenin led to reduction of LSCs as displayed by delayed and reduced recurrence of disease. The specific effect of β-catenin deletion on LSC self-renewal was manifest by an inability of Ctnnb1-/- marrow to maintain disease through subsequent generation of recipients in a serial BM transplantation assay. Genetic deletion in combination with IM treatment led to abrogation of LSCs in secondary (treated) recipients and delayed disease kinetics in tertiary recipient mice. Not only did we observe delayed onset of disease, there was also a 10-35 fold reduction in LSC frequency from secondary mice transplanted with β-catenin depleted leukemia compared to WT controls, demonstrating a striking and potent effect on LSC that might be utilized for clinical gain.
As targeted therapies are currently developed for a variety of solid tumors (Alvarez et al., 2010; Roock et al., 2010) and leukemia (Fischer et al., 2010; Mason et al., 2009), we aimed to investigate, whether canonical Wnt signaling could become a ‘druggable’ target in patients with minimal residual disease-CML. Activation of canonical Wnt signaling is known to be an important oncogeneic event in solid tumors, especially in colon carcinomas (Behrens, 2000; Oving and Clevers, 2002) and long-term treatment with COX-inhibitors/non-steroidal-anti-inflammatory drugs (NSAID) has been found to reduce the relative risk for colon carcinoma development (Gupta and Dubois, 2001; Rothwell et al., 2010b). Recently, reduction of tumor incidence has been reported for long-term use of the COX-inhibitor acetylsalicylic acid (aspirin) for a variety of cancers, providing provocative evidence that dysregulated Wnt signaling may be a common feature of many malignancies (Rothwell et al., 2010a). In the hematopoietic system, perturbations in the COX-Prostaglandin-Wnt cascade have been shown to have important effects on HSC- and LSC-function (Goessling et al., 2009; Wang et al., 2010). Stimulation of Wnt signaling with PGE2 has been shown to enhance HSC homing, survival and proliferation (Frisch et al., 2009; Hoggatt et al., 2009) (Goessling et al., 2009). Conversely, reduction of β-catenin levels using the COX-inhibitor indomethacin significantly decrease MLL-AF9 LSC frequency (Wang et al., 2010). To elucidate the role of pharmacological β-catenin modulation in this CML model, we investigated both, stimulation (using the GSK3β inhibitor BIO) or inhibition (using the COX-inhibitor indomethacin) of β-catenin in combination with IM treatment in vivo. As indicated by small spleen sizes and normal white blood counts, imatinib was an effective therapy against transformed progenitors and differentiated cells in secondary recipients. Given the difficulty with limiting dilution transplantation experiments in this model we used survival of tertiary recipient mice as an indicator of LSC function. This experimental design has been used as an alternative for assessment of LSC function in CML and AML LSC models (Hu et al., 2009; Zhao et al., 2007). Using this serial transplantation assay, we did not observe biologically significant results on LSC function by the combination of BIO (leading to Wnt activation) and IM. However, our results indicate, that reduction of β-catenin level using indomethacin leads to delayed onset of disease (in tertiary recipients) as a result of LSC reduction in the secondary recipient mice. This finding is consistent with analyses of gene-expression datasets that were previously published on CD34+ cells of CML patients and have been correlated with response to IM treatment. Patients that show enrichment for prostaglandin-signaling related signatures show inferior molecular responses upon IM therapy.
In summary, we have demonstrated that targeting β-catenin by genetic deletion or drug treatment is an effective combination therapy with IM, and eradicates LSC that remain after IM treatment. This leads to a delayed recurrence of CML after IM discontinuation providing support for the notion that targeting LSC will be an effective combination approach with tyrosine kinase inhibitors in this and perhaps other diseases. Recent reports indicate that adult HSCs do not require fully active β-catenin for maintenance (Koch et al., Blood 2006) (Luis et al., 2011) and our data show that that pharmacologic inhibition of β-catenin leads to reduction of GFP+ (BCR-ABL transformed) LSK cells thus suggesting a therapeutic index. However, this difference has not been addressed yet in a human setting. Thus, establishing a therapeutic index between normal HSCs and LSCs in human CML following β-catenin modulation and IM treatment will be a crucial prerequisite for Wnt-signaling inhibitors in clinical trials. These findings prompt an important question as to why CML-LSCs are dependent upon β-catenin whereas normal HSCs are less dependent on this pathway. The answer to this question will be the focus of much future work, and is likely to point to further vulnerabilities in cancer stem cells that can be targeted for therapeutic purposes. Finally, these data provide a rational template and preclinical model for the development and utilization of β-catenin-targeted therapies to eradicate LSCs in patients with CML.
Experimental Procedures
Bone-marrow (BM) infection & transplantation
Murine BM transplantation was performed as previously described (Hu et al., 2009). Donor mice (C57BL/6, Charles River Labs, Wilmington, MA) were injected with 150mg/KG 5-FU i.v. and BM was harvested on day 5. BM cells were infected by co-localization of virus supernatant (containing IL-3, IL-6 and SCF) (Peprotech, Rocky Hill, NJ) with mouse BM cells on retronectin-coated plates. Infection was repeated after 24 hours. One day after second infection, GFP+ cells were sorted using a FACSAria™ and used for transplantation. For transplantation, GFP+ BM cells were transplanted via lateral tail vein injection into lethally irradiated (1100cGy, split-dose) 4-6 week old C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) in the presence of 1 million freshly isolated erythrocyte-lysed ‘supporter’ BM cells. For serial transplantation experiments BM of primary recipient mice was harvested and 150,000 GFP+Sca-1+ sorted cells were injected into secondary recipients. Tertiary recipient mice were injected with whole BM cells, harvested from secondary recipients after treatment was discontinued.
Drug treatment of mice
Imatinib was dissolved in PBS solution at a stock solution of 100mg/ml and administered by oral gavage twice daily at a single dose of 100mg/kg. Indomethacin and BIO (6-Bromoindirubin-3’-oxime), (both Sigma, St.Louis, MO, USA) were diluted in DMSO and administered by i.p.-injection at a dose of 4mg/kg/day and 50μg/kg/day, respectively. DMSO was injected as a negative control.
Methylcellulose Colony Formation Assay
For investigation of colony formation in methylcellulose, BM cells of primary CML mice were harvested on day 21 after BMT and sorted for GFP+Sca-1+ cells. Cells were treated ex vivo with either BIO (0.5μM), PGE2 (10μM, Sigma, St.Louis, MO, USA) or indomethacin (40μM) for 48 hours and 1000 cells were plated subsequently in 1.1mL of methylcellulose (M3234, Stem Cell Technologies), supplemented with cytokines (IL-3, IL-6, SCF). Colony numbers were counted on day 8 after plating.
Flow Cytometry
For immunophenotype analysis, peripheral blood cells, BM or spleen cells were resuspended in PBS/1% FBS after erythrocyte lysis (PharmLyse™, BD Pharmingen, San Diego, CA). Sorting and analysis of LSK-cells or Sca-1+cells was performed as previously described (Krivtsov et al., 2009). Intracellular flow cytometry was performed as described previously (Kalaitzidis and Neel, 2008) using a primary antibody against Ctnnb1 (Clone 8e4, A.G. Scientific, SanDiego, CA) followed by a secondary Alexa594 labeled antibody (A31623, Invitrogen, Carlsbad) and analyzed using a LSRII™ (Becton-Dickinson) cytometer. Analysis was performed using FlowJo™ software (Treestar, Ashland, OR).
Western Blotting
Western Blot experiments were performed as described previously (Heidel et al., 2006) using antibodies for total β-catenin (BD Transduction Lab, No. 61054).
PCR Assays
For qualitative detection of BCR-ABL, a nested-PCR assay was performed using 200μL of whole blood. RNA was prepared using TRIzol™ Reagent (Invitrogen, Carlsbad, CA). cDNA was transcribed from 1μg of RNA using the Tetro cDNA Synthesis Kit (Bioline, Taunton, MA). 40 cycles of PCR were conducted. Primers are indicated in the supplement. This resulted in amplification of a 475bp fragment (b3a2 breakpoint) of the human p210 BCR-ABL construct.
Statistics and Analysis of Gene Expression Data
For survival analysis, Kaplan-Meier curves were plotted using GraphPad Prism™ version 5.00 (GraphPad Software, SanDiego, CA). Differences between survival distributions were analyzed using the logrank test. Statistical analyses were performed using Student t test (normal distribution) or Mann-Whitney U test (when normal distribution was not given). P less than .05 was considered statistically significant (p<.05 indicated as *, p<.01 indicated as ** and p<0.001 indicated as ***).
Published gene expression data (GSE14671) (McWeeney et al., 2010) and curated gene sets (M2214 and M14714) were downloaded from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and the Molecular Signatures Database (MSigDB v3.0, http://www.broadinstitute.org/gsea/msigdb/), respectively. Gene expression data were normalized and analyzed as previously reported (Luck et al., 2011).
Supplementary Material
Highlights.
β-catenin is required for the maintenance of CML stem cells
β-catenin deletion suppresses CML recurrence after imatinib withdrawal
β-catenin deletion synergizes with imatinib to target CML stem cells
COX-inhibitors reduce β-catenin levels in CML stem cells and are synergistic with imatinib
Acknowledgments
This work was supported by a Mildred-Scheel grant (DKH D/08/00661, Deutsche Krebshilfe to F.H.H.) and an NCI grant (# CA66996 to S.A.A.). S.A.A. is a Leukemia and Lymphoma Society Scholar, and L.B. was supported in part by the German Research Foundation (Heisenberg-Stipendium BU 1339/3-1).
Footnotes
The authors have no financial conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez RH, Valero V, Hortobagyi GN. Emerging targeted therapies for breast cancer. J Clin Oncol. 2010;28:3366–3379. doi: 10.1200/JCO.2009.25.4011. [DOI] [PubMed] [Google Scholar]
- Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci. 2000;910:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. discussion 33-25. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. 2009;23:1446–1454. doi: 10.1038/leu.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2010 doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch BJ, Porter RL, Gigliotti BJ, Olm-Shipman AJ, Weber JM, O’Keefe RJ, Jordan CT, Calvi LM. In vivo prostaglandin E2 treatment alters the bone marrow microenvironment and preferentially expands short-term hematopoietic stem cells. Blood. 2009;114:4054–4063. doi: 10.1182/blood-2009-03-205823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothot A, van der Loo JC, Clapp DW, Srour EF. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92:2641–2649. [PubMed] [Google Scholar]
- Guilhot F, Chastang C, Michallet M, Guerci A, Harousseau JL, Maloisel F, Bouabdallah R, Guyotat D, Cheron N, Nicolini F, et al. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French Chronic Myeloid Leukemia Study Group. N Engl J Med. 1997;337:223–229. doi: 10.1056/NEJM199707243370402. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, Brandts C, Serve H, Roesel J, Giles F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444–5455. doi: 10.1182/blood-2009-01-201335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes T, O’Brien TA, Knight R, Lindeman R, Shen S, Song E, Symonds G, Dolnikov A. Glycogen synthase kinase-3beta inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells. 2008;26:1288–1297. doi: 10.1634/stemcells.2007-0600. [DOI] [PubMed] [Google Scholar]
- Hu Y, Chen Y, Douglas L, Li S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009;23:109–116. doi: 10.1038/leu.2008.262. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Kalaitzidis D, Neel BG. Flow-cytometric phosphoprotein analysis reveals agonist and temporal differences in responses of murine hematopoietic stem/progenitor cells. PLoS One. 2008;3:e3776. doi: 10.1371/journal.pone.0003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, Moiraghi B, Shen Z, Mayer J, Pasquini R, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7:1048–1056. doi: 10.1038/ni1381. [DOI] [PubMed] [Google Scholar]
- Koch U, Wilson A, Cobas M, Kemler R, Macdonald HR, Radtke F. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008;111:160–164. doi: 10.1182/blood-2007-07-099754. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Wang Y, Feng Z, Armstrong SA. Gene expression profiling of leukemia stem cells. Methods Mol Biol. 2009;538:231–246. doi: 10.1007/978-1-59745-418-6_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SW, Sykes SM, Al-Shahrour F, Shterental S, Paktinat M, Lo Celso C, Jesneck JL, Ebert BL, Williams DA, Gilliland DG. The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood. 2010;115:3489–3497. doi: 10.1182/blood-2009-11-251728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck SC, Russ AC, Botzenhardt U, Paschka P, Schlenk RF, Dohner H, Fulda S, Dohner K, Bullinger L. Deregulated apoptosis signaling in core-binding factor leukemia differentiates clinically relevant, molecular marker-independent subgroups. Leukemia. 2011 doi: 10.1038/leu.2011.154. [DOI] [PubMed] [Google Scholar]
- Luis TC, Naber BA, Roozen PP, Brugman MH, de Haas EF, Ghazvini M, Fibbe WE, van Dongen JJ, Fodde R, Staal FJ. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell. 2011;9:345–356. doi: 10.1016/j.stem.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- Majeti R, Becker MW, Tian Q, Lee TL, Yan X, Liu R, Chiang JH, Hood L, Clarke MF, Weissman IL. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U S A. 2009;106:3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell. 2009;4:27–36. doi: 10.1016/j.stem.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason KD, Khaw SL, Rayeroux KC, Chew E, Lee EF, Fairlie WD, Grigg AP, Seymour JF, Szer J, Huang DC, Roberts AW. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–2041. doi: 10.1038/leu.2009.151. [DOI] [PubMed] [Google Scholar]
- McWeeney SK, Pemberton LC, Loriaux MM, Vartanian K, Willis SG, Yochum G, Wilmot B, Turpaz Y, Pillai R, Druker BJ, et al. A gene expression signature of CD34+ cells to predict major cytogenetic response in chronic-phase chronic myeloid leukemia patients treated with imatinib. Blood. 2010;115:315–325. doi: 10.1182/blood-2009-03-210732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Tidow C, Steffen B, Cauvet T, Tickenbrock L, Ji P, Diederichs S, Sargin B, Kohler G, Stelljes M, Puccetti E, et al. Translocation products in acute myeloid leukemia activate the Wnt signaling pathway in hematopoietic cells. Mol Cell Biol. 2004;24:2890–2904. doi: 10.1128/MCB.24.7.2890-2904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LF, Turbiak AT, Giannola DM, Donato N, Showalter HD, Fearon ER, Talpaz M. Wnt-Pathway Directed Compound Targets Blast Crisis and Chronic Phase CML Leukemia Stem Progenitors. Blood (ASH Annual Meeting Abstracts) 2009;114 [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Roock WD, Vriendt VD, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2010 doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA, Slader C, Field C, Dang P, Filshie RJ, et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010;24:1719–1724. doi: 10.1038/leu.2010.185. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2010a doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010b;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, Blanchet O, Marit G, Gluckman E, Reiffers J, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007;109:58–60. doi: 10.1182/blood-2006-03-011239. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, Pasquini R, Clark RE, Hochhaus A, Hughes TP, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8:341–350. doi: 10.1038/nrc2368. [DOI] [PubMed] [Google Scholar]
- Scheller M, Huelsken J, Rosenbauer F, Taketo MM, Birchmeier W, Tenen DG, Leutz A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7:1037–1047. doi: 10.1038/ni1387. [DOI] [PubMed] [Google Scholar]
- Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature. 2008;455:1205–1209. doi: 10.1038/nature07284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, Lagoo A, Reya T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.