

Abstract
The Bowman-Birk inhibitor (BBI) is a soybean-derived serine protease inhibitor with anti-inflammatory properties. Experimental autoimmune encephalomyelitis (EAE) serves as an animal model of the central nervous system (CNS) inflammatory disorder multiple sclerosis (MS). EAE is mediated by Th1 and Th17 cells which migrate into the CNS and initiate inflammation directed against myelin components, resulting in CNS pathology and neurological clinical deficit. We have shown previously that oral treatment with BBI delays onset of EAE and reduces its severity. These beneficial effects were associated with an increase in IL-10 secretion by immune cells of BBI-treated mice. It is not known, however, whether this was a causal relationship or simply an epiphenomenon. In the present study we provide evidence that BBI regulates CD4+ T cell immune responses in EAE. BBI administration delayed the onset of EAE and reduced its severity in an IL-10-dependent manner, as BBI-mediated suppression of EAE was abrogated in IL-10 knockout mice. The beneficial effects were accompanied by reduced IFN-γ, IL-17 and increased IL-10 production, as well as increased Foxp3 expression. CD4+ T cells were the major source of IL-10 in the periphery and in the CNS during BBI treatment. Furthermore, BBI-treated mice had reduced numbers of infiltrated cells in the CNS, including Th17 cells, as compared with PBS-treated control animals. In conclusion, our data provide clear evidence for the essential role of IL-10 in BBI-mediated suppression in EAE, and indicate that BBI may be a promising candidate for the development of a novel MS therapy.
Keywords: Bowman-Birk inhibitor, experimental autoimmune encephalomyelitis, interleukin-10, Th17, Tregs
1. INTRODUCTION
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) characterized by progressive demyelination of the brain and spinal cord and accumulation of neurological deficit (Keegan and Noseworthy, 2002). Experimental autoimmune encephalomyelitis (EAE) is an animal model of MS used to study its pathogenic mechanisms and test potential therapies (Cruz-Orengo et al., 2011). In EAE, myelin-specific CD4+ T lymphocytes migrate into the CNS and mediate neuronal demyelination and damage, similar to that in MS patients (Axtell et al., 2010).
The Bowman-Birk inhibitor (BBI) is a soybean-derived protease inhibitor comprised of 71 amino acid residues, 7 disulfide bonds and with a molecular weight of 8 kDa. BBI can withstand boiling water temperature, is resistant to a wide pH range and proteolytic enzymes of the gastrointestinal tract; it is orally bioavailable, and non-allergenic (Marin-Manzano et al., 2009; Park et al., 2007; Losso et al., 2008). BBI is functionally a double-headed serine protease inhibitor that inhibits both trypsin- and chymotrypsin-like proteases (Birk,Y., 1985). Thus far, several human serine proteases, associated with function of inflammatory cells, have been shown to be highly sensitive to inhibition by BBI, including elastase, alpha-chymotrypsin, chymase and cathepsin G (Ware et al.,1997; Larionova et al., 1993). It is likely that there are additional, yet unknown, serine proteases susceptible to inhibition by BBI. BBI is effective suppressor of carcinogenesis (Daly et al., 2006), and has anti-inflammatory effects (Qi et al., 2005). Our previous studies indicated that BBI reduces inflammation in EAE and attenuates neuronal loss, making it a potential candidate for oral therapy in MS (Touil et al., 2008). However, the mechanism by which BBI ameliorates EAE is currently unclear.
IL-10 was first described as a factor produced by Th2 cells, which inhibits cytokine production by Th1 cells (Fiorentino et al., 1991). It has been demonstrated that IL-10 inhibits the production of a wide range of T-cell cytokines by affecting antigen presentation and co-stimulation mediated by antigen-presenting cells (APCs) (de Waal Malefyt et al., 1991). IL-10 also plays an important role in the regulation of autoimmune injury in EAE, as evidenced by increased susceptibility of IL-10-/- mice to EAE (Bettelli et al., 1998), whereas mice over-expressing IL-10 are highly resistant to EAE induction (Cua et al., 1999). Also, further studies have shown that increased IL-10 levels in spinal cord correlate with EAE remission, and exogenous administration of IL-10 effectively ameliorated EAE when targeted directly to the CNS (Croxford et al., 2001). Similarly, lower production of IL-10 in humans appears to be a risk factor for MS, as accumulative data have shown that MS patients had lower IL-10-secreting T cell frequency (Vandenbark et al., 2001) than controls. Overall, experimental findings indicate that IL-10 has an important disease suppressor function in both EAE and MS.
Regulatory T cells (Tregs) are a CD4+ T cell subset with potent immune-suppressive activities that are critical for maintaining self-tolerance and immune homeostasis (Fontenot et al., 2005). Tregs are activated by interactions with APCs, then transmigrate to inflammation sites and draining lymphoid organs where they suppress T-cell activation (Mahnke et al., 2003). Defects in Treg function invariably result in autoimmunity and inflammatory diseases. (Sakaguchi et al., 2008; Shevach, 2000). Tregs play an important protective role in EAE based upon the finding that mice depleted of Treg cells exhibit an increased susceptibility to EAE (Akirav et al., 2009), whereas adoptive transfer of Tregs reduces EAE incidence (Stephens et al., 2009). In addition, mice expressing a transgenic T cell receptor specific for myelin basic protein (MBP) and lacking recombination-activating genes (RAGs) spontaneously develop EAE due to the absence of Tregs (Matejuk et al., 2003).
The goal of the present study was to investigate potential immunological mechanisms underlying the effects of BBI in EAE. Here, we show that BBI significantly delayed EAE onset and reduced its clinical severity. EAE suppression was associated with the reduced numbers of immune cells that infiltrated into the CNS during disease, enhanced production of IL-10 and increased numbers of CD4+CD25+Foxp3+ Tregs. Most important, we show that IL-10 production is essential for a beneficial effect of BBI in EAE, as evidenced by the inability of BBI to suppress EAE development in IL-10-/- mice.
2. MATERIAL AND METHODS
2.1. Mice
C57BL/6 and IL-10-/- mice were purchased from the Jackson Laboratory. Experimental mice were housed in the animal facility at the Thomas Jefferson University for at least one week before inclusion in experiments. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
2.2. Reagents
The Bowman–Birk inhibitor (BBI) was purchased from Aldrich Sigma (St. Louis, MO). The following antibodies for flow cytometry were from BD Biosciences: anti-CD4 (RM4-5), anti-CD25 (clone PC-61), anti-IFN-γ (XMG1.2), anti-IL-17A (TC11-18H10), anti-IL-10 (AF-417-NA) and anti-CD16/32 (2.4G2). Anti-Foxp3 (FJK-16s) was from eBioscience. DuoSet ELISA kit used to quantify IL-10 was from R&D Systems.
2.3. Induction of EAE and BBI treatment
Active EAE was induced in female C57BL/6 mice as previously described (El-Behi et al., 2009). Briefly, female 8- to 10-week-old wild-type or IL-10-/- mice were immunized subcutaneously at two sites on the back with 180 μg MOG35–55 in complete Freund’s adjuvant containing Mycobacterium tuberculosis strain H37Ra (5 mg/ml; Difco). Mice were injected intraperitoneally on days 0 and 2 with 200 ng pertussis toxin (Difco) in PBS. Mice immunized with MOG35-55 were treated daily with BBI (1 mg/day or 3 mg/day) dissolved in PBS or PBS by oral gavage from the day of immunization.
2.4. Isolation of CNS-infiltrating cells
EAE mice were sacrificed, and brains and spinal cords were removed and pooled after transcardial perfusion with PBS. Tissues were mechanically dissociated through a 100-μm strainer and washed with PBS. The resultant pellet was fractionated on a 60–30% Percoll step gradient by centrifugation at 300g for 20 min. Infiltrating mononuclear cells were collected from the interface and washed, then were counted and analyzed by flow cytometry as described below.
2.5. Flow cytometry
For intracellular staining, cells were stimulated for 4 h with PMA (50 ng/ml; Sigma) and ionomycin (500 ng/ml; Sigma) and were treated with GolgiPlug (1 μg per 1 × 106 cells; BD Pharmingen). In the staining procedure, Fc receptors on cells were first blocked with anti-CD16-CD32 (2.4G2; BD Pharmingen) and surface and intracellular staining was done according to manufacturer’s instructions for staining with Fix & Perm reagents (Caltag Laboratories). Data were acquired on a FACSAria (BD Biosciences) and analyzed with FlowJo software (TreeStar).
2.6. ELISA
Splenocytes and lymph nodes cells were cultured with MOG35-55 for 3 days; thereafter supernatants were harvested and analyzed for IL-10 by ELISA kits according to the manufacturer’s instructions.
2.7. Evaluation of blood-brain barrier permeability
A 2% solution of Evans Blue in normal saline (4 ml/kg of body weight) was injected intraperitoneally. The stain was allowed to circulate for 24 hrs. Afterwards, mice were transcardially perfused with 50 ml PBS, and brain tissue was removed, homogenized in PBS and centrifuged (30 min, 15,000 rcf, 4 °C). The supernatant was collected and an equal amount of 50% trichloroacetic acid was added, incubated overnight at 4 °C, and then centrifuged (30 min, 15,000 rcf, 4 °C). Evans Blue concentration was measured at 610 nm and determined using the standard curve. Results are presented as the ratio between μg of Evans Blue/g of tissue from mice with EAE and naïve mice.
2.8. Real-time PCR
Total RNA was prepared from mononuclear cells isolated from the CNS of mice with EAE treated with BBI or PBS, using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized with a reverse transcription kit (Applied Biosystems). Primer pairs for quantitative real-time PCR were from Applied Biosystems. Gene expression was analyzed by TaqMan real-time PCR (Applied Biosystems). Ribosomal 18S RNA was used as an endogenous control in all experiments. Error bars indicate SEM values calculated from -ΔΔCt values from triplicate PCR reactions, according to Applied Biosystems protocols.
2.9. Statistics
The two-tailed Student’s t test and the χ2 test were used to analyze the significance of results. Additionally, analysis of variance (ANOVA) was used for comparison of average clinical scores. P values of less than 0.05 were considered significant.
RESULTS
BBI suppresses EAE and induces IL-10 production
To investigate the role of BBI in the development of EAE, C57BL/6 mice were immunized with encephalitogenic MOG35-55 peptide and treated with 1 or 3 mg/mouse/day of BBI from day 0 p.i. BBI treatment significantly delayed the onset of EAE and reduced severity of clinical EAE (Table 1), with the higher BBI dose (3 mg) being more effective (Fig. 1A). However, once BBI-treated mice started to develop clinical EAE they continued to accumulate clinical deficit at a pace similar to control mice, indicating that BBI primarily delayed disease onset, but did not have a substantial effect on disease progression thereafter. In agreement with our previous findings, inflammatory cells infiltrated the CNS of BBI-treated mice in significantly lower numbers than with PBS-treated mice (Fig. 1B). This was true for all three time points p.i. analyzed, and, rather unexpectedly, at day 21 p.i. when disease severity in BBI- and PBS-treated mice became similar, indicating that inflammatory infiltration does not necessarily always correlate with observed clinical deficit. Despite similar clinical scores at day 21 p.i. mice treated with 3 mg/day of BBI had more inflammatory cells in their CNS than those treated with 1 mg/day of BBI. A possible explanation for this discrepancy is that disease onset having occurred on different days, these two treatment groups were in different phases of disease development, with mice in 3 mg/day group still being in the phase when influx of peripheral cells into the CNS is more vigorous than in the 1 mg/day group.
Table 1.
BBI suppresses EAE
| Treatment | Incidence | Day of onset | Peak severity |
|---|---|---|---|
| PBS | 11/11 (100%) | 12.6 ± 1.0 | 3.9 |
| 1 mg BBI | 10/10 (100%) | 14.7 ± 1.1* | 2.9* |
| 3 mg BBI | 3/4 (75%) | 17.0 ± 1.7* | 2.4* |
C57BL/6 mice were immunized with MOG35-55 and treated with BBI (1 mg/day, or 3 mg/day) or PBS by oral gavage from the day of immunization until the end of the experiment; mice were scored daily for clinical disease.
p < 0.05 vs. PBS control group. Data are representative of three independent experiments.
Figure 1. BBI suppresses EAE in C57BL/6 mice.
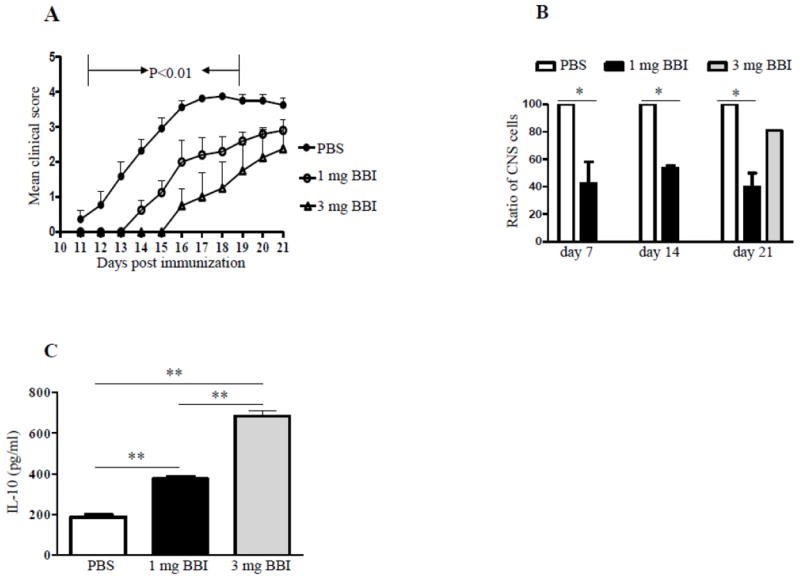
(A) C57BL/6 mice were immunized with MOG35-55 and treated with BBI (1 mg/day, n=10 or 3 mg/day, n=4) or PBS (n=11) by oral gavage from the day of immunization until the end of the experiment; mice were scored daily for clinical disease. (B) On days 7, 14 and 21 p.i., mice were sacrificed and cells from CNS isolated and counted. Data are shown as percentage relative to the cell number obtained from PBS-treated mice. (C) On day 21 p.i., draining LN cells were harvested and stimulated with anti-CD3/CD28 for 3 days; thereafter supernatants were analyzed for IL-10 by ELISA. P values refer to the comparison between BBI- vs. PBS-treated groups. Data are representative of three independent experiments.
In agreement with our previous findings (Touil et al., 2008), BBI treatment resulted in significantly increased IL-10 production by draining lymph node cells harvested on day 21 p.i. and stimulated with anti-CD3/28 (Fig.1C). Cells of mice treated with 3 mg/day of BBI produced more IL-10 than those of mice treated with 1 mg/day of BBI, which correlates with stronger overall EAE suppression mediated by a higher BBI dose. Whether the higher BBI dose enhanced IL-10 expression of BBI in T cells, promoted development of more IL-10 producing T cells, or retarded their egress from lymph nodes, thus increasing their proportion there, remains unclear.
BBI-mediated protection against EAE is abated in IL-10-/- mice
We have shown that BBI induces IL-10 production (Touil et al., 2008) in a dose-dependent manner (Fig. 1C), suggesting that IL-10 may play a role in BBI-mediated protection against EAE. We next used IL-10-/- mice to test that possibility. To this end, IL-10-/- and wild-type (WT) control mice were immunized for EAE induction and treated daily with BBI or PBS by oral gavage. As expected, treatment of WT mice with BBI delayed the onset of clinical disease, while in IL-10-/- mice, BBI had no effect (Fig. 2A), demonstrating that IL-10 plays an essential role in BBI-mediated suppression of EAE. After delayed disease onset, BBI-treated WT mice continued to accumulate clinical deficit until day 24.p.i. when their disease became as severe as in PBS-treated mice. These data indicate that BBI does not permanently suppress EAE, but rather substantially delays disease development.
Figure 2. BBI-mediated suppression of EAE is abated in IL-10-/- mice.
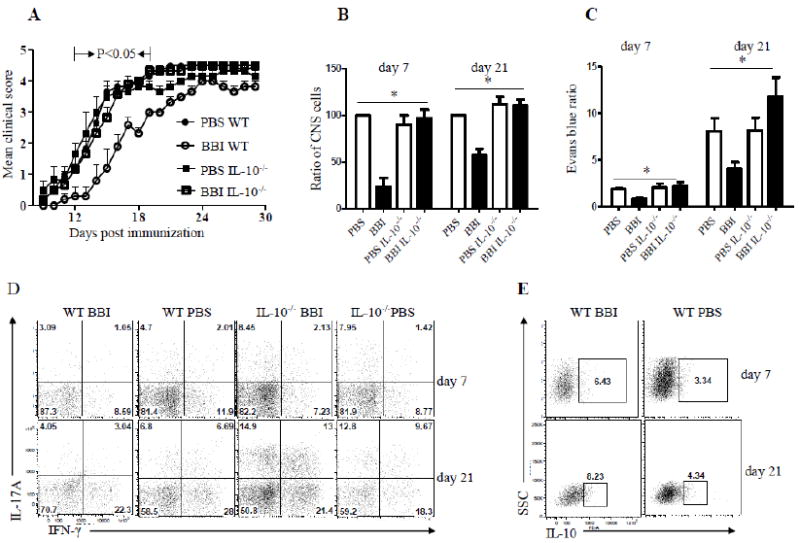
(A) WT and IL-10-/- mice on C57BL/6 background were immunized with MOG35-55 and treated with 1 mg/day of BBI or PBS (n=12 in each group) by oral gavage from the day of immunization until the end of the experiment. Mice were scored daily for clinical disease. (B) WT and IL-10-/- mice (n=12 in each group) were immunized with MOG35-55 and treated with BBI (1 mg/day) or PBS by oral gavage from the day of immunization. Mice were sacrificed on days 7 and 21 p.i.; cells from CNS were isolated and counted. Data are shown as a percentage relative to the cell number obtained from WT PBS-treated mice. (C) The integrity of BBB was evaluated by measurement of Evans blue content in brains at days 7 and day 20 p.i. (n=6 per group). Results are presented as a ratio between background levels obtained from naïve mice injected with Evans blue and mice with EAE. Values represent the mean ± S.E.M. (D) Mononuclear cells were isolated from spinal cords on days 7 and 21 p.i., stained and analyzed by flow cytometry for IL-17A and IFN-γ production. CD4+ T cells are shown. (E) Mononuclear cells were isolated from spinal cords on days 7 and 21 p.i., stained and analyzed by flow cytometry for IL-10 production. CD4+ T cells are shown. P value refers to comparison between BBI-treated WT mice and other groups. Data are representative of two independent experiments.
We next sought to investigate whether IL-10 is required for the reduction of CNS inflammatory infiltration typically induced by BBI treatment. As shown in Fig. 2B, treatment with BBI significantly reduced infiltration of inflammatory cells into the CNS of WT mice, with approximately 50% reduction in their numbers at both time points analyzed. In contrast, there was no difference in CNS infiltration between PBS-treated and BBI-treated IL-10-/- mice (Fig. 2B), confirming that IL-10 production is necessary for anti-inflammatory effects of BBI in EAE. Total numbers of mononuclear cells isolated on days 7 and 21 p.i. from the CNS of IL-10-/- mice treated with PBS and BBI, and PBS-treated WT mice were similar (data not shown).
To test whether IL-10 plays a role in the maintenance of blood-brain barrier (BBB) integrity by BBI in EAE mice, we used Evans blue dye. As shown in Fig. 2C, BBI-treated EAE mice had a significantly lower content of Evans blue in the brain when compared with PBS-treated mice. In contrast, BBI-treated IL-10-/- mice had somewhat increased levels of Evans blue in the brain compared to IL-10-/- mice treated with PBS. These results were in agreement with clinical disease severity and the extent of inflammatory infiltration, further supporting the conclusion that IL-10 is necessary for beneficial effects of BBI in EAE.
EAE is mediated by encephalitogenic T cells that produce inflammatory cytokines such as IL-17 (Th17) and IFN-γ (Th1) (El-behi et al. 2010). Thus, we used flow cytometry to analyze the effects of BBI on the numbers of IL-17- and IFN-γ-producing CD4+ T cells among cells that infiltrated the CNS. BBI-treated mice displayed a substantial decrease in the proportions of both IL-17-producing Th17 cells and IFN-γ producing Th1 cells as compared with those of PBS-treated control animals, whereas in IL-10-/- mice, BBI treatment did not have such an effect at two time points analyzed (Fig. 2D). On the other hand, BBI-treated mice had a higher proportion of IL-10-producing cells in the CNS compared to PBS-treated mice (Fig. 2E). Taken together, these findings suggest that BBI limits the infiltration of inflammatory cells into the CNS, reducing Th17 and Th1 responses in the CNS during EAE development, and that IL-10 is required for EAE suppression by BBI.
CD4+ T cells are the major source of IL-10 following BBI treatment
Our previous and current data demonstrate that BBI enhances IL-10 production by immune cells (Touil et al., 2008). To define the cellular source of IL-10, we harvested mononuclear cells from spleen and LNs on day 7 p.i. and stimulated them with MOG35-55. Cells derived from LNs of BBI-treated mice produced significantly higher levels of IL-10 compared to those from PBS-treated mice, while splenic cells showed no difference (Fig. 3A). Next, we asked whether CNS-infiltrating cells of BBI-treated mice express more IL-10 than those of PBS-treated mice. BBI-treated mice had largely increased IL-10 mRNA levels expressed by mononuclear cells isolated from the CNS compared to PBS-treated animals (Fig. 3B). Flow cytometric analyses of mononuclear cells from the CNS, spleen and LNs showed higher proportions of CD4+IL-10+ cells among CD4+ cells isolated from all three organs of BBI-treated mice (Fig. 3C). Furthermore, CD4+ T cells appear to be the main producers of IL-10, as a negligible fraction of CD4- cells stained for IL-10. Collectively, these data show that BBI treatment increased the proportion of IL-10-producing CD4+ T cells.
Figure 3. CD4+ T cells are the main source of IL-10 following BBI treatment.
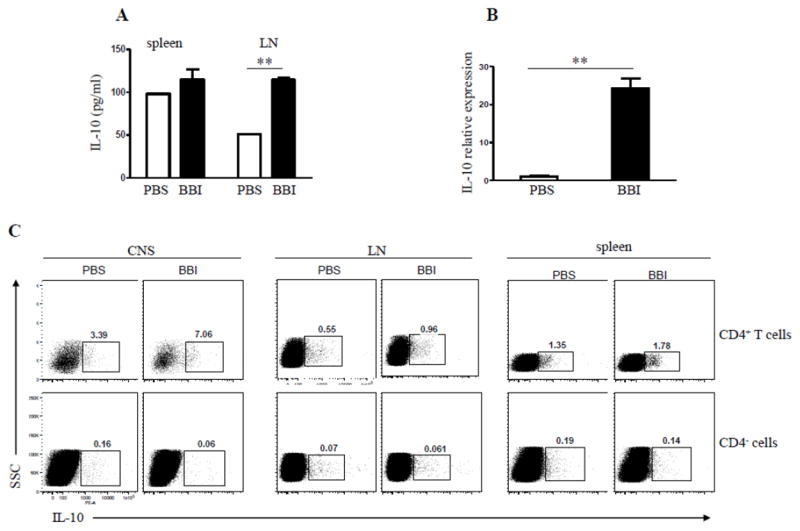
C57BL/6 mice were immunized with MOG35-55 and treated daily with BBI (1 mg/day) or PBS by oral gavage from day 0 p.i. and sacrificed on day 7 p.i. (A) LN cells were stimulated with MOG35-55 for 3 days, and concentrations of IL-10 measured in the supernatants by ELISA. (B) C57BL/6 mice (n=6 per group) were immunized with MOG35-55 and treated daily with BBI (1 mg/day) or PBS by oral gavage from the day of immunization. Mice were sacrificed on day 14 p.i.; mononuclear cells were isolated from the CNS and used to isolate mRNA. mRNA levels of IL-10 were determined by quantitative real-time PCR. Data are representative of three independent experiments. (C) Spinal cord, splenic and LN cells of immunized mice that had been treated for 14 days p.i. with BBI were isolated and analyzed for intracellular production of IL-10 by flow cytometry. ** p < 0.01. Data are representative of three independent experiments.
BBI treatment increases proportion of CD4+CD25+Foxp3+ Tregs
It has been shown that Tregs in the CNS produce IL-10 and suppress EAE (McGeachy et al., 2005; Rynda-Apple et al., 2010). To test if BBI alters Treg numbers, mice were immunized with MOG35-55 and treated with BBI or PBS. On day 14 p.i., cells isolated from the CNS, spleen and lymph nodes were evaluated for Treg phenotype by flow cytometry. As shown in Fig. 4A, BBI-treated mice had markedly higher proportions of CD4+CD25+Foxp3+ Tregs in the CNS and in the spleen compared to PBS-treated mice, whereas the increase among LNs cells was less pronounced. Analysis of IL-10 production among Foxp3+CD4+ cells from BBI-treated mice showed an increase in their intracellular IL-10 levels (Fig. 4B). These data show that BBI increases both the proportion of Tregs among CD4+ T cells and simultaneously promotes their IL-10 production.
Figure 4. BBI treatment induces CD4+CD25+ Foxp3+ Tregs.
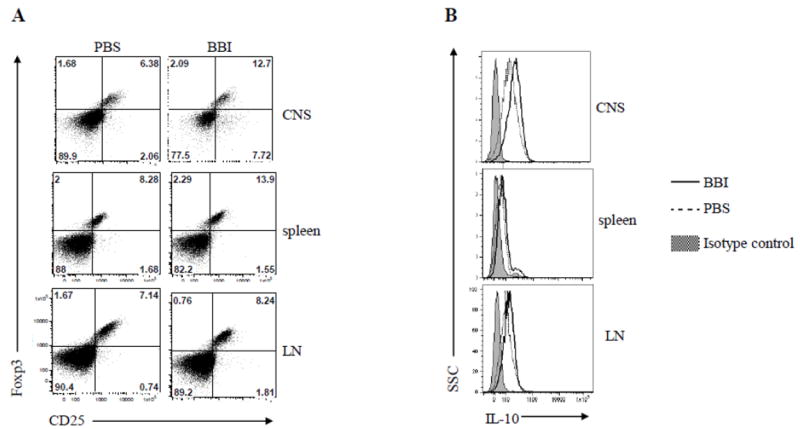
C57BL/6 mice (n=6 per group) were immunized with MOG35-55 and treated with BBI (1mg/day) or PBS by oral gavage from day 0 p.i.; mice were sacrificed on day 14 p.i. (A) CD4+ T cells from spinal cord, spleen and LN were gated and their CD25 and Foxp3 expression analyzed by flow cytometry. (B) Flow cytometric analysis of IL-10 intracellular levels in CD4+CD25+ T cells from spinal cord, spleen and LN of mice treated with BBI for 14 days p.i. Data are representative of three experiments.
DISCUSSION
Our previous studies on BBI have shown that this protease inhibitor suppresses EAE development, and that treatment with BBI enhances IL-10 production by immune cells (Touil et al., 2008). However, it has not been determined whether increased IL-10 production plays a role in observed anti-inflammatory effects of BBI. The goal of the current study was to extend our previous findings and in particular to determine the role of IL-10 in EAE suppression caused by BBI treatment. The most important novel finding presented here is that the beneficial effects of BBI in EAE indeed depend on IL-10 production. We also demonstrate that BBI enhances Treg development, and that the major source of BBI-induced IL-10 is CD4+ T cells.
We show here, and in our recent publication (Dai et al., 2011), that BBI treatment delayed the onset of EAE and slowed the pace of clinical deficit accumulation, but ultimately did not stop disease development, with incidence and severity reaching levels similar to control mice between three and four weeks p.i. A possible reason for prolonged EAE development in BBI-treated mice was initially slower development of MOG35-55-specific responses in the periphery, which was only temporary, as later on they became either equivalent to those in control mice, or even stronger (Dai et al., 2011). We interpreted these findings as an indication that BBI delays EAE development primarily by retarding CNS infiltration by inflammatory cells, and not by suppression of peripheral encephalitogenic responses. Given that post initial priming phase (day 14, 21 p.i.) BBI-treated mice had stronger MOG35-55-specific responses in the periphery, but fewer inflammatory cells in their CNS than the control mice, it seems likely that BBI slowed egress of MOG35-55-specific CD4+ T cells from peripheral lymphoid organs. Thus, in this scenario BBI alters the distribution of encephalitogenic T cells between the periphery and CNS, a mechanism similar to that of fingolimod, a recently approved oral therapy for MS (Cohen et al., 2011). The mechanism by which BBI delays exit of CD4+ T cells from peripheral lymphoid organs is not known, but it likely involves enhanced IL-10 production, as, in the absence of IL-10 BBI, has no effect. Consistent with the delay in clinical disease development, infiltration of inflammatory cells into the CNS was also delayed and reduced, although without dramatic perturbation in cellular composition.
Given that recruitment of myelin-reactive encephalitogenic T cells into the CNS and myelin destruction are hallmarks of EAE (Anderton, 2010), one potential mechanism of BBI-mediated suppression of CNS inflammation might be inhibition of BBB breakdown by immune cells, resulting in reduced infiltration into the CNS (Touil et al., 2008). We consistently observed that mice treated with BBI had markedly lower numbers of CNS infiltrating cells. Further, BBI administration reduced the percentage of Th17 cells in the CNS. As a result, both the absolute numbers and percentage of Th17 cells were lower in the CNS of BBI-treated mice compared to PBS-treated mice. Given the crucial role of Th17 cells in the development of EAE (EI-Behi et al., 2011; Komiyama et al., 2006), suppression of their infiltration into the CNS is likely an important mechanism underlying the beneficial effects of BBI on this disease. Although the importance of Th1 cells in the development of EAE has become uncertain, recent studies support their pathogenic role. For example, Kroenke et al. (Kroenke et al., 2008) showed that adoptive transfer of either IL-12p70-polarized Th1 or IL-23-polarized Th17 cells into naive syngeneic mice resulted in clinically indistinguishable EAE between the two groups. Our results show that BBI treatment suppressed both Th1 and Th17 cells in the CNS, thus reducing CNS autoimmune inflammation.
BBI treatment did not have a beneficial effect in IL-10-/- mice, as evidenced by normal development of clinical EAE, unaffected CNS inflammation and similar extent of BBB damage. These findings clearly demonstrate that IL-10 is required for BBI-induced beneficial effects in EAE. The anti-inflammatory properties of IL-10 have been well documented. This cytokine targets the induction of various pro-inflammatory mediators as well as antigen presentation and thereby indirectly inhibits T-cell responses (Mekala et al., 2005; Dai et al., 2009; Zhu et al., 2008). It has been shown that IL-10 confers protection against disease in the C57BL/6 model of EAE as well (Anderson et al., 2004; Mendel and Shevach, 2002; Zhang et al., 2004). In the present study, in line with our previous observation (Touil et al., 2008), we observe that BBI treatment induces increased IL-10 production not only in the peripheral immune system, but also in CNS-infiltrating cells. Further, we have shown that CD4+ T cells, but not other cells (including APCs), are the main contributors to IL-10 production brought about by BBI treatment. Most important, the protection from EAE mediated by BBI was completely lacking in IL-10-/- mice. These results provide direct evidence for the essential role of IL-10 in BBI-mediated suppression in EAE.
There is increasing evidence indicating that CD4+CD25+Foxp3+ Treg accumulate and function within the inflamed CNS during EAE (Ephrem et al., 2008). In agreement with those observations, our data show that the protective effects of BBI may also result from an increase in Treg numbers. Although the molecular mechanisms by which BBI shifts the balance between Th1/Th17 and Treg cells are currently unknown, the elevated numbers of Tregs in the CNS might be the result of direct expansion of CD4+CD25+Foxp3+ T cells or their recruitment and accumulation into the CNS due to BBI treatment. We have also observed that Tregs from BBI-treated mice produce somewhat greater levels of IL-10 than those from control mice, which, in combination with their higher numbers, likely leads to dramatically increased IL-10 production in the CNS. This increase in IL-10 production may be responsible for reduced CNS inflammation through mechanisms such as reduced production of pro-inflammatory cytokines and chemokines, which is a well established anti-inflammatory mechanism of IL-10 (Peron et al., 2010).
In summary, the suppressive effects of BBI in EAE are fully dependent on IL-10 production. Questions that remain to be answered are whether BBI induces IL-10 production by CD4+ T cells through its direct action on them, or indirectly, by acting on other cells, such as APCs. Furthermore, it is not clear whether increased production of IL-10 mediates anti-inflammatory effects of BBI in EAE; or if normal IL-10 production, in combination with some other effect(s) of BBI, unrelated to the increase in IL-10 expression, would have similar anti-inflammatory effect. In addition, molecular mechanisms by which BBI induces IL-10 production remain to be elucidated.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (to AR). We would like to thank Katherine Regan for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akirav EM, Bergman CM, Hill M, Ruddle NH. Depletion of CD4(+)CD25(+) T cells exacerbates experimental autoimmune encephalomyelitis induced by mouse, but not rat, antigens. J Neurosci Res. 2009;87:3511–3519. doi: 10.1002/jnr.21981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Reddy J, Nazareno R, Sobel RA, Nicholson LB, Kuchroo VK. IL-10 plays an important role in the homeostatic regulation of the autoreactive repertoire in naive mice. J Immunol. 2004;173:828–834. doi: 10.4049/jimmunol.173.2.828. [DOI] [PubMed] [Google Scholar]
- Anderton SM. Treg and T-effector cells in autoimmune CNS inflammation: a delicate balance, easily disturbed. Eur J Immunol. 2010;40:3321–3324. doi: 10.1002/eji.201041100. [DOI] [PubMed] [Google Scholar]
- Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- Birk Y. The Bowman-Birk inhibitor trypsin- and chymotrypsin-inhibitor from soybeans. Int J Pept Protein Res. 1985;25:113–31. doi: 10.1111/j.1399-3011.1985.tb02155.x. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69:759–777. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]
- Croxford JL, Feldmann M, Chernajovsky Y, Baker D. Different therapeutic outcomes in experimental allergic encephalomyelitis dependent upon the mode of delivery of IL-10: a comparison of the effects of protein, adenoviral or retroviral IL-10 delivery into the central nervous system. J Immunol. 2001;166:4124–4130. doi: 10.4049/jimmunol.166.6.4124. [DOI] [PubMed] [Google Scholar]
- Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, McCandless EE, Patel JR, Luker GD, Littman DR, Russell JH, Klein RS. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208:327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Groux H, Hinton DR, Stohlman SA, Coffman RL. Transgenic interleukin 10 prevents induction of experimental autoimmune encephalomyelitis. J Exp Med. 1999;189:1005–1010. doi: 10.1084/jem.189.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Zhu H, Lei P, Yagita H, Liu J, Wen X, Zhou W, Gong F, Shen G, Fang M. Programmed death-1 signaling is essential for the skin allograft protection by alternatively activated dendritic cell infusion in mice. Transplantation. 2009;88:864–73. doi: 10.1097/TP.0b013e3181b6ea74. [DOI] [PubMed] [Google Scholar]
- Dai H, Ciric B, Zhang G-X, Rostami A. Bowman–Birk inhibitor attenuates experimental autoimmune encephalomyelitis by delaying infiltration of inflammatory cells into the CNS. Immunol Res. 2011 doi: 10.1007/s12026-011-8254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly NL, Chen YK, Foley FM, Bansal PS, Bharathi R, Clark RJ, Sommerhoff CP, Craik DJ. The absolute structural requirement for a proline in the P3’-position of Bowman-Birk protease inhibitors is surmounted in the minimized SFTI-1 scaffold. J Biol Chem. 2006;281:23668–23675. doi: 10.1074/jbc.M601426200. [DOI] [PubMed] [Google Scholar]
- de Waal Malefyt R, Haanen J, Spits H, Roncarolo M, te Velde A, Figdor C, Johnson K, Kastelein R, Hans Yssel H, de Vries JE. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–924. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-behi M, Ciric B, Yu S, Zhang GX, Fitzgerald DC, Rostami A. Differential effect of IL-27 on developing versus committed Th17 cells. J Immunol. 2009;183:4957–67. doi: 10.4049/jimmunol.0900735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-behi M, Rostami A, Ciric B. Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–75. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, Delignat S, Elluru S, Bayry J, Lacroix-Desmazes S, Cohen JL, Salomon BL, Kazatchkine MD, Kaveri SV, Misra N. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O’Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan BM, Noseworthy JH. Multiple sclerosis. Annu Rev Med. 2002;53:285–302. doi: 10.1146/annurev.med.53.082901.103909. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larionova NI, Gladysheva IP, Tikhonova TV, Kazanskaia NF. Inhibition of cathepsin G and elastase from human granulocytes by multiple forms of the Bowman-Birk type of soy inhibiton. Biokhimiia. 1993;58:1437–44. [PubMed] [Google Scholar]
- Losso JN. The biochemical and functional food properties of the bowman-birk inhibitor. Crit Rev Food Sci Nutr. 2008;48:94–118. doi: 10.1080/10408390601177589. [DOI] [PubMed] [Google Scholar]
- Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+regulatory T cells by targeting of antigens to immature dendritic cells. Blood. 2003;101:4862–4869. doi: 10.1182/blood-2002-10-3229. [DOI] [PubMed] [Google Scholar]
- Marin-Manzano MC, Ruiz R, Jimenez E, Rubio LA, Clemente A. Anti-carcinogenic soyabean Bowman-Birk inhibitors survive faecal fermentation in their active form and do not affect the microbiota composition in vitro. Br J Nutr. 2009;101:967–971. doi: 10.1017/s0007114508057590. [DOI] [PubMed] [Google Scholar]
- Matejuk A, Buenafe AC, Dwyer J, Ito A, Silverman M, Zamora A, Subramanian S, Vandenbark AA, Offner H. Endogenous CD4+BV8S2- T cells from TG BV8S2+ donors confer complete protection against spontaneous experimental encephalomyelitis (Sp-EAE) in TCR transgenic, RAG-/- mice. J Neurosci Res. 2003;71:89–103. doi: 10.1002/jnr.10450. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Anderton SM. Cytokines in the induction and resolution of experimental autoimmune encephalomyelitis. Cytokine. 2005;32:81–4. doi: 10.1016/j.cyto.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Mekala DJ, Alli RS, Geiger TL. IL-10-dependent suppression of experimental allergic encephalomyelitis by Th2-differentiated, anti-TCR redirected T lymphocytes. J Immunol. 2005;174:3789–3797. doi: 10.4049/jimmunol.174.6.3789. [DOI] [PubMed] [Google Scholar]
- Mendel I, Shevach EM. The IL-10-producing competence of Th2 cells generated in vitro is IL-4 dependent. Eur J Immunol. 2002;32:3216–3224. doi: 10.1002/1521-4141(200211)32:11<3216::AID-IMMU3216>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Park JH, Jeong HJ, Lumen BO. In vitro digestibility of the cancer-preventive soy peptides lunasin and BBI. J Agric Food Chem. 2007;55:10703–10706. doi: 10.1021/jf072107c. [DOI] [PubMed] [Google Scholar]
- Peron Jean Pierre S, Yang Kayong, Chen Mei-Ling, Brandao Wesley Nogueira, Basso Alexandre S, Commodaro Alessandra G, Weiner Howard L, Rizzo Luiz V. Oral tolerance reduces Th17 cells as well as the overall inflammation in the central nervous system of EAE mice. J Neuroimmunol. 2010;227:10–17. doi: 10.1016/j.jneuroim.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Qi RF, Song ZW, Chi CW. Structural features and molecular evolution of Bowman-Birk protease inhibitors and their potential application. Acta Biochim Biophys Sin (Shanghai) 2005;37:283–292. doi: 10.1111/j.1745-7270.2005.00048.x. [DOI] [PubMed] [Google Scholar]
- Rynda-Apple A, Huarte E, Maddaloni M, Callis G, Skyberg JA, Pascual DW. Active immunization using a single dose immunotherapeutic abates established EAE via IL-10 and regulatory T cells. Eur J Immunol. 41:313–323. doi: 10.1002/eji.201041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Regulatory T cells in autoimmmunity. Annu Rev Immunol. 2000;18:423–44918. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol. 2009;39:1108–1117. doi: 10.1002/eji.200839073. [DOI] [PubMed] [Google Scholar]
- Touil T, Ciric B, Ventura E, Shindler KS, Gran B, Rostami A. Bowman-Birk inhibitor suppresses autoimmune inflammation and neuronal loss in a mouse model of multiple sclerosis. J Neurol Sci. 2008;271:191–202. doi: 10.1016/j.jns.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark AA, Finn T, Barnes D, Culbertson N, Chou YK, Hicks K, Bakke A, Mass M, Whitham R, Offner H, Bourdette D. Diminished frequency of interleukin-10-secreting, T-cell receptor peptide-reactive T cells in multiple sclerosis patients might allow expansion of activated memory T cells bearing the cognate BV gene. J Neurosci Res. 2001;66:171–176. doi: 10.1002/jnr.1209. [DOI] [PubMed] [Google Scholar]
- Ware JH, Wan XS, Rubin H, Schechter NM, Kennedy AR. Soybean Bowman-Birk protease inhibitor is a highly effective inhibitor of human mast cell chymase. Arch Biochem Biophys. 1997;344:133–8. doi: 10.1006/abbi.1997.0182. [DOI] [PubMed] [Google Scholar]
- Zhu H, Qiu W, Lei P, Zhou W, Wen X, He F, Li L, Dai H, Shen G, Gong F. IL-10 gene modified dendritic cells inhibit T helper type 1-mediated alloimmune responses and promote immunological tolerance in diabetes. Cell Mol Immunol. 2008;5:41–6. doi: 10.1038/cmi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]