

Abstract
Background
In global gene expression profiling experiments, variation in the expression of genes of interest can often be hidden by general noise. To determine how biologically significant variation can be distinguished under such conditions we have analyzed the differences in gene expression when Bacillus subtilis is grown either on methionine or on methylthioribose as sulfur source.
Results
An unexpected link between arginine metabolism and sulfur metabolism was discovered, enabling us to identify a high-affinity arginine transport system encoded by the yqiXYZ genes. In addition, we tentatively identified a methionine/methionine sulfoxide transport system which is encoded by the operon ytmIJKLMhisP and is presumably used in the degradation of methionine sulfoxide to methane sulfonate for sulfur recycling. Experimental parameters resulting in systematic biases in gene expression were also uncovered. In particular, we found that the late competence operons comE, comF and comG were associated with subtle variations in growth conditions.
Conclusions
Using variance analysis it is possible to distinguish between systematic biases and relevant gene-expression variation in transcriptome experiments. Co-variation of metabolic gene expression pathways was thus uncovered linking nitrogen and sulfur metabolism in B. subtilis.
Background
Genome programs have uncovered a vast number of genes of unknown function, prompting new types of functional investigation. In conjunction with expression-profiling experiments, knowledge of genome sequences provides an extremely powerful approach to interpreting global gene expression patterns. Paradoxically however, interpreting all the information potentially available from these experiments is hindered by the very accuracy of the physical measurements involved, so that the importance of the fluctuations inherent to biological experiments is often disregarded. This accounts for the emphasis placed by some on simple descriptive studies of gene expression, where patterns specific to a particular set of genetic or environmental conditions are displayed, usually for diagnostic purposes. Fortunately, transcription profiling can tell us much more, and can help to uncover new gene functions. Before jumping to conclusions, however, it is necessary, when observing variation in a large number of genes, to be able to distinguish specific biological effects from background noise and systematic variations due to the procedure itself.
In the present work, which uses Bacillus subtilis as a model, we have analyzed its transcriptome expression in conditions chosen to yield extremely similar transcription patterns: growth in the presence of either methionine or methylthioribose (MTR) as the sulfur source [1]. We discovered that, even with all the background constraints acting on the experiments and the similarity in the growth conditions, we could extract biologically significant observations, namely that there existed a strong link between sulfur metabolism and arginine metabolism. We also show that, in addition to finding biologically relevant patterns, the involuntary triggering of transitions in environmental conditions (with the regulation cascades they induce) plays an important role in creating systematic biases in the expression of some operons. The complex biochemistry of cDNA synthesis from mRNA is also a significant source of systematic errors, which makes it very difficult to compare experiments performed under different conditions [2]. Nevertheless, we were able to set up a simple and robust statistical protocol for transcriptome data analysis that enables one to distinguish fluctuations in expression of the genes of interest from fluctuations due to parasitic effects. This protocol does not require background noise subtraction nor normalization of the data, a notoriously difficult and controversial task [3].
We hope that, in addition to providing unexpected information about sulfur metabolism in Bacillus subtilis (a previously unrecorded relationship between the metabolism of arginine and that of methionine, as well as metabolism of methionine sulfoxide), this work will draw the attention of the molecular biology community to the risks of incorrect interpretation inherent in the large biological variability of transcriptome experiments.
Results
Effect of the sulfur source on gene expression
Variance analysis showed that the most important source of bias in comparing different experiments is a difference in the amount of RNA used to synthesize cDNA; in second place is the difficulty of exactly reproducing experimental conditions from one experiment to another (see Materials and methods). As expected [1], variance analysis showed that changing the sulfur source from methionine to MTR has little effect on the transcription level of most genes. Interestingly however, the list of the 20 most variable genes has several distinctive features. In this group, genes of known function are significantly more numerous than expected from their proportion in the genome (10/20; with p < 6%). Furthermore these genes correspond to correlated functions. Growth in methionine is characterized both by the higher expression of the arginine metabolic pathway and by that of operon yqiXYZ, which is thought to code for an amino-acid ABC permease function, whereas growth on MTR activates several genes involved in alleviating oxygen toxicity (catalase, hydroperoxide reductase, monooxygenase), as well as a long operon encoding a permease and known to be connected with sulfur metabolism (ytmIJKLMhisPytmOytnIJribRhipOytnM) [4, 5]. Thus, several genes group together in a physiologically consistent picture (oxido-reduction, arginine biosynthesis, ABC permeases, and so on). Remarkably, genes putatively expressed as an operon [6] appear together in the list: yqiXYZ, argGHytzD, argJDF, ytmIJLMPytmO. Finally, several genes belong to the preceding list and are also members of a putative operon (comGC, comGD, yqzE). Table 1 summarizes the information available on these genes.
Table 1.
Function of genes showing the largest differences as a function of the sulfur source
| Gene | Mean | Standard | Function |
| deviation | |||
| argD | 2.378 | 1.032 | Arginine biosynthesis |
| argF | 2.874 | 0.735 | Arginine biosynthesis |
| argG | 1.911 | 0.411 | Arginine biosynthesis |
| argH | 3.414 | 1.139 | Arginine biosynthesis |
| argJ | 3.252 | 0.873 | Arginine biosynthesis |
| comGC | 1.841 | 0.730 | Late competence operon required for DNA binding and uptake |
| comGD | 2.149 | 0.628 | Late competence operon required for DNA binding and uptake |
| levD | 2.653 | 0.757 | PTS system - fructose-specific IIA component |
| ycbD | 2.071 | 0.452 | Probable aldehyde dehydrogenase |
| yqiX | 3.052 | 0.297 | Probable amino-acid ABC permease (binding protein) |
| yqiY | 2.351 | 0.744 | Probable amino-acid ABC permease (permease) |
| yqiZ | 3.178 | 0.659 | Probable amino-acid ABC permease (ATP-binding protein) |
| yqzE | 2.000 | 0.946 | Hypothetical protein |
| ytzD | 3.378 | 0.839 | Hypothetical protein, downstream gene of operon argGH |
| ahpF | -3.013 | 0.601 | Alkyl hydroperoxide reductase (large subunit) - reduces organic hydroperoxides in their reduced dithiol form |
| hydroperoxides in their reduced dithiol form | |||
| katA | -1.964 | 0.914 | Vegetative catalase |
| ytmI | -2.418 | 0.489 | Possible methionine sulfoxide acetyl transferase (operon |
| ytmIJKLMhisPytmOytnIJribRhipOytnM) | |||
| ytmJ | -2.480 | 0.787 | Methionine - methionine sulfoxide ABC permease (binding protein) |
| (this paper) | |||
| ytmL | -1.852 | 0.979 | Methionine - methionine sulfoxide ABC permease (permease) (this |
| paper) | |||
| ytmO | -2.258 | 0.439 | Methionine sulfoxide monooxygenase (this paper) |
Arginine biosynthesis
In contrast to the situation in many other bacteria, the arginine biosynthesis genes in B. subtilis are split into two parts (argE, thought to code for acetylornithine deacetylase (EC 3.5.1.16), is part of a third operon, less clearly linked to arginine metabolism). This suggests some compartmentalization of the corresponding metabolites, as in eukaryotes where arginine biosynthesis is split between the mitochondria and the cytoplasm. Variance analysis shows that both sets of genes are stimulated when cells grow on methionine as the sulfur source (Table 1).
Using the absolute values for hybridization in the respective conditions, we found that the pathway including citrulline:ornithine acetyl transferase (EC 2.3.1.35), acetylornithine aminotransferase (EC 2.6.1.11) and ornithine carbamoyltransferase (EC 2.1.3.3) was stimulated 1.6-fold; and the pathway leading from citrulline and including arginine:argininosuccinate synthase (EC 6.3.4.5), argininosuccinate lyase (EC 4.3.2.1) and YtzD, which has an unknown (regulatory) function, was stimulated 1.7-fold (Table 2). YtzD is a small hydrophilic protein remarkable for its high content of methionine (and is therefore unusually rich in sulfur). The operon argGHytzD is found as a whole in the list in Table 1, whereas for the operon argCJBDcarABargF one finds only genes argJ, argD and argF in Table 1. This prompted us to look at the absolute values found for all genes of the operon: consistent with overexpression of the operon, we found that all other genes in this operon, except carA, were also overexpressed in methionine (Table 2). They do not show up among the first 20 overexpressed genes simply owing to the fact that there was more variability from experiment to experiment for these genes, resulting in a lower position in the variance analysis list. Taken together, however, these results show that both operons are indeed overexpressed, as a whole, in methionine growth conditions. As a control, we observed that there was no difference in expression of the argS gene, which is present in an entirely different operon (of unknown function).
Table 2.
Expression of arginine operons when cells were grown with methionine or with MTR as the sulfur source
| Gene and | |||
| Met | MTR | Ratio | |
| spot | |||
| argB a | 1286 | 907 | 1.43 |
| argB b | 1329 | 928 | |
| argC a | 1116 | 735 | 1.61 |
| argC b | 1145 | 666 | |
| argD a | 930 | 592 | 1.62 |
| argD b | 1014 | 605 | |
| argE a | 340 | 246 | 1.27 |
| argE b | 446 | 372 | |
| argF a | 699 | 406 | 1.77 |
| argF b | 717 | 392 | |
| argG a | 2304 | 1896 | 1.28 |
| argG b | 2827 | 2121 | |
| argH a | 2165 | 1120 | 1.94 |
| argH b | 2237 | 1147 | |
| argJ a | 1045 | 509 | 1.94 |
| argJ b | 1051 | 569 | |
| argS a | 991 | 838 | 1.16 |
| argS b | 984 | 858 | |
| carA a | 2329 | 2046 | 1.12 |
| carA b | 2326 | 2094 | |
| carB a | 932 | 635 | 1.47 |
| carB b | 912 | 618 | |
| yqzE a | 680 | 460 | 1.57 |
| yqzE b | 685 | 407 | |
| yqiX a | 2190 | 1401 | 1.58 |
| yqiX b | 2264 | 1412 | |
| yqiY a | 750 | 585 | 1.41 |
| yqiY b | 876 | 571 | |
| yqiZ a | 1527 | 926 | 1.70 |
| yqiZ b | 1595 | 907 |
cDNA hybridization after growth in methionine or MTR. The conditions are chosen to display roughly identical values for most genes in both conditions. The values obtained for the arginine operons described in the text are shown here. They indicate a higher expression in methionine as compared to MTR, all things being otherwise equal. Each spot in the experiment is referred to as a and b.
The yqiXYZ permease
It was expected that permeability to methionine would be affected in mutants of some of the genes involved in growth on methionine. As the yqiXYZ genes have significant similarity to amino-acid permease genes, we investigated permeability of mutant bacteria to methionine and arginine. We did not find any difference in methionine availability in mutants with disruptions in the putative permease genes ytmJKLMhisP or yqiXYZ (data not shown). In contrast, we found that growth of mutants in any of the yqiXYZ genes was strongly impaired in low concentrations of arginine as a sole nitrogen source. The observation that growth on methionine was related to arginine came as a surprise. We took advantage of the fact that arginine is a nitrogen source for B. subtilis to explore the affinity of this putative permease for arginine under in vivo conditions. Growth on arginine as the nitrogen source was impaired in all of the three mutants yqiX, yqiY or yqiZ (Figure 1, and data not shown). To evaluate the efficiency of the permease, we compared growth on increasing arginine concentrations (Figure 1). The permease allows cells to grow at half the maximum growth rate when arginine is at a concentration of 0.3 mM in these in vivo conditions. This operon therefore codes for a high-affinity arginine permease, similar to the artPIQMJ operon of Escherichia coli [7]. In contrast, the affinity of the residual permease(s) is much lower, of the order of 5 mM (Figure 1).
Figure 1.
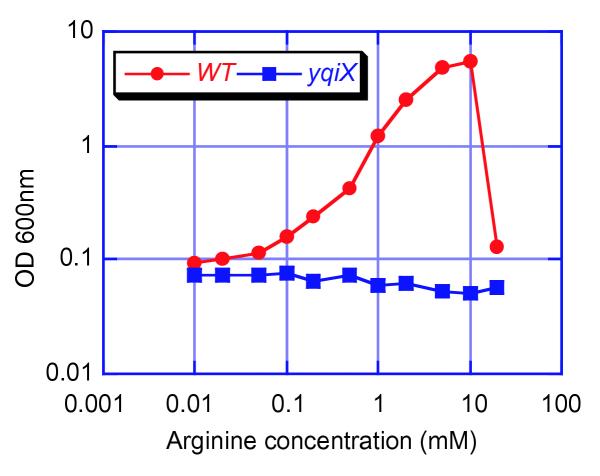
Comparison of the growth of B. subtilis wild type (WT) and yqiX mutant (BFS4740) in different concentrations of arginine as the nitrogen source in glucose minimal medium (from 10 μM to 20 mM; note that arginine is toxic at 20 mM). Both strains were grown at 37°C with agitation (180 rpm). The OD was taken for both strains after 8 h growth, when they entered stationary phase (due to nitrogen limitation, except for the highest arginine concentrations).
Methionine sulfoxide metabolism: the ytmIJKLMhisPytmOytnIJribR operon
The metabolism of methionine and MTR was further investigated. Because the permease for MTR is unknown, an obvious candidate was the permease encoded by genes ytmJKLMhisP. Growth of mutants in these genes on MTR was unaffected, however (data not shown). As in the case of yqiXYZ, comparison with known genes indicated that the genes code for a permease likely to transport amino acids or their analogs. The first gene in this operon, ytmI, is likely to code for an acetylase. One can expect, therefore, that the transported substrate is acetylated before being metabolized. This suggests that the substrate has to be diverted from a straightforward involvement in a process where it would be deleterious.
Sequence alignments of the products of genes that are adjacent to the permease gene in the operon using protein sequences in databases (data not shown), suggested that YtmO/YtnIJ/RibR is a FMN-dependent monooxygenase acting on a sulfur-containing substrate [4, 5]. This enzyme could be involved in some step of MTR degradation. Growth on MTR was, however, unaffected in the corresponding mutants (data not shown). We therefore attempted to see whether this operon was involved in the degradation of methionine and methionine sulfoxide, processes that scavenge sulfur and prevent methionine sulfoxide getting into protein synthesis. Growth of wild-type bacteria on methionine sulfoxide was better than on methionine (a 10% larger growth region from a disk impregnated with 10 μl of a 100 mM solution), suggesting that the former is indeed a common substrate in the environment. Growth was only slightly affected in the mutants ytmJ and ytmK when they were grown in the presence of a high concentration of methionine sulfoxide. Finally, no growth was observed with methionine sulfone, either in the wild type or the mutants, excluding this substance as an intermediate in degradation (data not shown). We therefore investigated the situation at low concentrations of either methionine or methionine sulfoxide, not only with the permease gene mutants but also with a mutant of gene ytnJ. As shown in Figure 2, mutations in these genes significantly impaired growth on both these substrates, showing that the permease is a transport system for these metabolites, and suggesting that methionine degradation proceeds, at least in part, through monooxygenation. This observation was further substantiated by the observation that the mutants were more resistant to selenomethionine than the wild type (data not shown).
Figure 2.
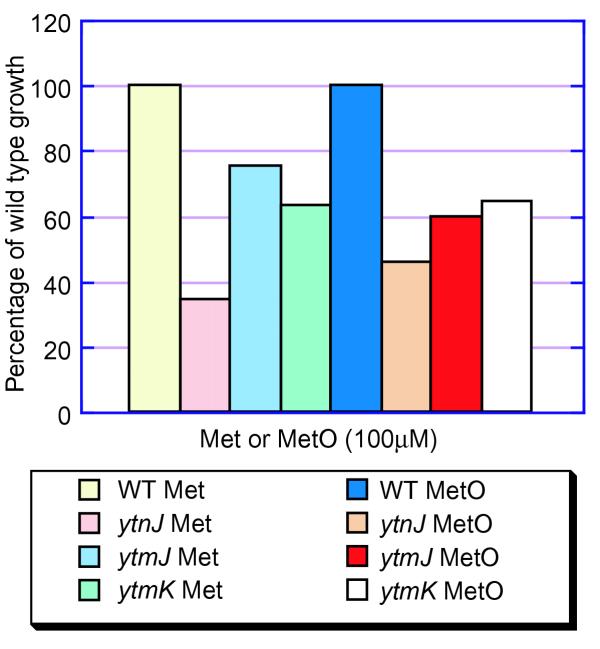
Comparison of the growth of B. subtilis wild type (WT) with ytnJ, ytmJ and ytmK mutants (BFS70, BFS85 and BFS86, respectively) on methionine (Met) and methionine sulfoxide (MetO) at 100 μM concentration. Growth was measured after 7 h and is represented as a percentage of wild-type growth.
Discussion
In most bacteria, sulfur metabolism has not been studied in as much depth as carbon, nitrogen or phosphorus metabolism. Our recent analysis of the distribution of sulfur metabolism genes in E. coli showed that they are clustered together into islands [8]. This indicates that selection pressure has forced the corresponding genes to group together (and/or that they have remained grouped together from very early times in the history of life). Sulfur recycling, in particular, was found to play an important role in cellular sulfur metabolism, with methylthioadenosine (MTA) being important; this is derived from many cell reactions and in particular the synthesis of polyamines [4]. The immediate degradation product of MTA, MTR, is not recycled in E. coli but is efficiently recycled in B. subtilis, presumably as methionine [1]. Our preliminary experiments showed that cells growing on MTR as the sulfur source behaved as did cells growing on methionine. This suggested that only a small set of genes was involved in the recycling process. Would it be possible to identify some of them?
We have used expression profiling to identify those genes that are differentially expressed when either MTR or methionine is used as the sulfur source, not to provide a global profile which would just be used for diagnostic purposes. In principle, expression profiling allows one to investigate the collective behavior of a group of genes of interest buried in the full genome. When there is much uncontrolled variation in the outcome of hybridization, however, it becomes difficult to distinguish random fluctuation from a specific effect. The work presented here was intended to identify at least some of the causes of variation in expression profiling in bacteria (simple fluctuations and systematic biases), trying to extract from general background noise those few genes that display differential expression. The fact that we came up with a sound biological explanation of the variations demonstrates that our experimental and statistical work plan is efficient, paving the way for further, more complex, research using transcriptome analysis.
Statistics
We have chosen to use variance analysis, a statistical approach that has long been used by economists and field scientists, who have to deal with a large number of pieces of data under conditions where the control of the environment is poor. These techniques have been validated through more than 60 years of hard work, and we think they should be more frequently considered for large-scale genomics experiments. Other techniques, such as rank analysis, are also relevant, but variance analysis is useful for analyzing the nature of systematic or random errors affecting data, when considering a large number of data points.
The basis of these techniques is that data treatment prior to analysis must be reduced to a minimum: for example, one should not have to subtract background values or use ratio measurements, because this can only be done without introducing specific bias under highly constrained statistical conditions (which are certainly not fulfilled in transcriptome experiments). Similarly, one should not introduce normalization conditions based on underlying biological hypotheses (for example, using the average of some housekeeping genes, which are supposed to be expressed at a constant level), because this introduces a systematic bias, with the assumption that, for two given genetic or environmental conditions, the expression level of such control genes would not vary. Many experiments [9] show that this is certainly not true in general.
The outcome of the variance analysis is to identify those genes that vary in a consistent manner, for a given experimental set-up. Once these genes have been identified, of course, it becomes necessary to make a quantitative evaluation of transcription and translation in the conditions of interest to be sure that they are indeed relevant.
Preparation of the cDNA
Bacterial expression profiling is particularly difficult to set up because, in contrast to eukaryotes, bacteria do not make stable polyadenylated mRNA. In fact, the average turnover of mRNA under usual growth conditions for model organisms such as E. coli and B. subtilis is of the order of 1-2 minutes. As shown in our analysis, experimental biases lie at the level of RNA recovery and synthesis of a labeled cDNA copy for hybridization to the DNA arrays. The amount of cDNA synthesized is not proportional to the amount of RNA and worse, the yield of cDNA synthesis appears to vary from gene to gene - average amounts of mRNA species giving the most variable cDNA outcome. In general, the reason given for using larger amounts of RNA is to avoid the situation where the lowest expression levels are hidden in the background noise. In contrast, it is the fear that the membrane would be saturated, and thus jeopardize the accuracy of the values corresponding to the highest expression levels, that guides the choice of a small amount of RNA in some experiments.
We evaluated the importance of the biases due to the background noise on the one hand and to the saturation problem on the other by studying the correlations in measurements performed using either 1 μg or 10 μg RNA from the same extract. A loss of sensitivity due to the background noise will manifest itself as a low correlation between the rank order of the genes in each experiment (genes having been ranked as a function of their signal intensity) for the genes giving low amounts of cDNA, and, conversely, saturation will lead to a poor correlation for genes giving larger amounts of cDNA. None of these biases had a significant influence in our experiments. In the absence of such bias, the ranking of the genes listed as a function of the signal intensity must be the same in each experiment. This is not what has been observed, except at the low and high ends of the scale.
An explanation for this lack of correlation has been proposed: in some genes, cDNA synthesis may be affected by the formation of secondary structures in the mRNA or by the presence of RNA segments of highly biased nucleotide composition [10]. These are conditions where the efficiency of cDNA synthesis might be affected for large RNA concentrations. A thorough study of the sequence of genes giving the most erratic hybridization values may allow one to estimate the value of this hypothesis: no straightforward reason could be found in the present experiments. As demonstrated by Hatfield and co-workers, the utilization of random hexanucleotide primers should alleviate some of this difficulty (namely, RNA folding near the 3'-end of the mRNA). Their work did not, however, explore in details the effect of RNA quantities on the outcome of the hybridization pattern, and it may well be that biases similar to those described here would be uncovered. Finally, the role of competition of a large number of primers of different sequences, as well as of the large quantity of rRNA present in the cDNA reaction mixture, has not yet been analyzed in depth. Further work is needed before we know the relative contributions of these parameters, and it can be expected that in the near future appropriate protocols will alleviate some of these hypothetical but plausible causes of bias.
Growth culture conditions
The experiments described here allowed us to prioritize the sources of biases and artifacts in the analysis of the bacterial transcriptome. In addition to controlling cDNA synthesis, we found that controlling culture conditions was more important than expected. Unappreciated differences in culture conditions seem to result in coordinated variations of gene expression.
Indeed, we observed differences in the expression of competence operons on different days. This indicates that subtle differences in growth conditions result in quite visible and self-consistent differences in gene expression. Competence as well as sporulation is monitored by the cell using subtle cues, most of which are still unknown. This experiment shows that these processes are extremely sensitive to growth conditions (experiments have been reproduced as exactly as possible, including control of agitation, monitoring growth and trying to keep everything as constant as possible). This warns against premature assignment of effects on competence or sporulation processes from experiments which have not been statistically controlled. It is clear that simply carrying out differential expression experiments would have led us to interpret an effect that is simply due to the extreme sensitivity of this process to subtle changes in environmental conditions as an effect of MTR on competence (see the common genes in Tables 1,3). Previous results on these interesting phenomena should be reconsidered in this light.
Table 3.
Function of genes showing the largest differences between days A and B
| Gene | Mean | Standard | Function |
| deviation | |||
| comEA | 2.588 | 0.930 | Late competence protein required for DNA binding and uptake |
| comFA | 3.213 | 0.809 | Late competence protein required for DNA uptake |
| comFC | 2.448 | 0.713 | Late competence protein required for DNA uptake |
| comGA | 4.794 | 0.956 | Late competence operon required for DNA binding and uptake |
| comGB | 3.649 | 0.604 | Late competence operon required for DNA binding and uptake |
| comGC | 2.644 | 0.724 | Late competence operon required for DNA binding and uptake |
| comGD | 3.512 | 0.828 | Late competence operon required for DNA binding and uptake |
| comGE | 4.067 | 1.038 | Late competence operon required for DNA binding and uptake |
| nucA | 3.134 | 0.954 | Competence-specific nuclease |
| rapH | 3.453 | 0.619 | Response regulator: aspartate phosphatase (sporulation) |
| ypzE | 2.261 | 0.143 | Hypothetical protein |
| yqzE | 3.856 | 0.810 | Hypothetical protein |
| yshA | 2.331 | 0.718 | Conserved hypothetical protein |
| yvrP | 2.697 | 0.966 | Conserved extracytoplasmic component of a permease |
| (hypothetical) | |||
| cotG | -2.547 | 0.784 | Spore coat protein |
| glgA | -2.335 | 1.053 | Bacterial glycogen synthase |
| rplV | -2.465 | 0.958 | Ribosomal protein L20 |
| ybfO | -2.884 | 0.880 | Similar to erythromycin esterase (detoxification) |
| yllA | -3.531 | 0.914 | Hypothetical protein |
| yojL | -2.631 | 0.710 | Similar to cell wall-binding protein |
Every biologist knows how difficult and time-consuming it is to reproduce exactly, in a highly quantitative manner, not only the experiments performed in another laboratory, but also those performed inside one's own laboratory. Indeed, it is rare that all experimental conditions can be controlled finely enough to allow for a rigorously exact repetition, despite impressions to the contrary. For example, in functional bacterial genomics experiments, one does not explicitly control what happens to the cells between preculture and growth in the final medium. Although these two steps occur usually at 37°C, most often bacteria are placed (for a variable time) in a room where temperature is significantly lower (and usually not controlled). Many epigenetic phenomena are triggered during this transition period, and this may lead to differences in gene expression. These differences will be non-random, but will correspond to uncontrolled differences between experiments thought to be totally identical. To give just one example: many experiments are performed at 'room temperature', which is a very poorly defined physical parameter! There are certainly other factors (see, for example, the role of glassware [11], carbon dioxide and the quality of the water) which are difficult to control and make the analysis of the experiments on gene expression difficult.
Methionine and methylthioribose (MTR)
The existence of large interactions of the type gene/condition limits the information that can be extracted from a transcriptome analysis. The statistical analysis, however, enabled us to identify genes that were almost certainly associated with given physiological conditions.
MTR is efficiently recycled in B. subtilis, using a yet unknown pathway. It seems reasonable to infer that this recycling is through methionine, as in Klebsiella pneumoniae [12]. No counterparts of the corresponding K. pneumoniae genes seem to be present in B. subtilis, however. The present experiment was therefore intended to identify the first element of the pathway (methylthioribose permease) and/or the genes responsible for its recycling, by comparing growth on MTR with that on methionine, its immediate derivative. The corresponding genes should indeed stand out if their expression is inducible by MTR. Naturally, because MTR is a normal product of the cell, the corresponding genes might be expressed constitutively; in which case they will not be identified by this procedure.
No significant induction was observed, but some repression of the arginine biosynthetic pathway was consistently found in growth on MTR. This interesting link between nitrogen and sulfur metabolism may be accounted for (and makes MTR, or one of its immediate derivatives, an interesting candidate for a co-repressor), if one notes that arginine is the precursor of polyamines in B. subtilis and that MTA, the immediate precursor of MTR, is produced in a stoichiometric fashion when the polyamine spermidine is synthesized [4]. It may be relevant to remark that the isolated argE gene is present in an operon of unknown function with several genes having significant homology with genes involved in sulfur metabolism. Moreover the ytzD gene in the argGHytzD operon encodes a sequence rich in sulfur-containing amino acids, which is unusual in B. subtilis, providing a further indication of a significant coupling between arginine and sulfur metabolism in this organism.
In addition, expression of two operons encoding permeases (yqiXYZ and ytmJKLMhisP) varies in opposite ways when cells are grown on methionine or MTR. This prompted us to measure the growth of mutants of each of these genes on various sulfur sources. No significant growth impairment was observed when concentration of the metabolites was high. The most plausible explanation is that MTR and methionine can be transported into the cell by several permeases (permeases transporting methionine are already known), so that inactivation of one of them does not alter growth. Further work showed that the yqiXYZ gene product was transporting arginine. To be consistent with the E. coli nomenclature supported by SwissProt, we propose to rename these genes artPQM (for arginine transport). The molecular nature of the link between sulfur and arginine remains to be investigated, however. A similar involvement in transport was witnessed with the ytmJKLMhisP genes. These genes appear to code for a permease for methionine and methionine sulfoxide, and the operon is meant to degrade methionine by a FMN-dependent monooxygenase, presumably after acetylation. So we have tentatively renamed the genes ytmI, ytmJKLMhisP, ytmOytnIJ and ribR respectively as mdgA, mdgOPQTS (mdg, methionine sulfoxide degradation, permease and transport), moxABC (mox, methionine sulfoxide oxidase) and rbfK (rbf, riboflavin kinase; thus avoiding 'rib', which usually stands for ribose). The existence of this operon is easy to account for, knowing that the usual biotope of B. subtilis is the phylloplane. Indeed, if methionine is exuded from plant leaves, then this occurs in the presence of a high level of dioxygen, which will efficiently metabolize the methionine to methionine sulfoxide. Modification, possibly acetylation, of the substrate will prevent it from getting into the protein synthesis pathway.
Finally, induction of genes involved in scavenging oxygen radicals further suggests that a process involving oxygen is carried out in the recycling of MTR. This is in line with the involvement of a dioxygenase in MTR recycling in K. pneumoniae [13].
Materials and methods
Bacterial strains and growth conditions
The following B. subtilis strains were used in this work: the wild-type strain 168 [14], BFS70 (ytnJ::lacZ), BFS85 (ytmJ::lacZ), BFS86 (ytmK::lacZ), BFS4740 (yqiX::lacZ), BFS4739 (yqiY::lacZ) and BFS4738 (yqiZ::lacZ). These strains have been constructed within the EC project for the functional characterization of the genome of B. subtilis in Europe (BFS70, BFS85, BFS86) [15], and in Japan (BFS4740, BFS4739, BFS4738) [16]. Bacteria were grown in ED minimal medium: K2HPO4, 8 mM; KH2PO4, 4.4 mM; glucose 27 mM; Na3-citrate, 0.3 mM; L-glutamine, 15 mM; L-tryptophan, 0.244 mM; ferric citrate, 33.5 μM; MgCl2, 2.61 mM; CaCl2, 49.5 μM; FeCl3, 49.9 μM; MnCl2, 5.05 μM; ZnCl2, 12.4 μM; CuCl2, 2.52 μM; CoCl2, 2.5 μM; Na2MoO4, 2.48 μM, with methionine or methylthioribose (MTR) (1 mM and 0.2 mM, respectively) in cultures for RNA preparation; or with methionine or methionine sulfoxide (concentrations indicated in the text) as sole sulfur source [17]. When arginine was tested as nitrogen source, glutamine was omitted from the medium and replaced with arginine (concentrations indicated in the text), MgSO4 at 2 mM was used as sulfur source. Cells were grown in 25 ml of ED medium in 250 ml Erlenmeyer flasks at 37°C with constant aeration. For growth on plates, minimal medium was supplemented with 17 g/l of agar Noble (Difco) and bacteria were grown at 37°C. All experiments were performed in accordance with the European regulation requirements concerning the use of genetically modified organisms (level 1 containment, agreement number 2735).
Isolation of total RNA
Total RNA was isolated from cells at an OD600 of 0.8-1.1. Samples of growing cells (5 ml) were centrifuged for 2 min at 8,000 rpm and 4°C. The pellets were immediately frozen in an ethanol/dry-ice bath. Before RNA preparation, pellets were briefly vortexed and 200 μl of lysozyme solution (1/2 vol glucose 20%, 1/2 vol Tris 25 mM EDTA 10 mM, lysozyme 40 mg/ml) was added. The suspensions were transferred to sterile 2 ml Eppendorf tubes and kept for 2 min at room temperature. The samples were subsequently mixed up with 37.5 μl of 0.5 M EDTA and then 375 μl of lysis buffer (2% SDS, 20 mM sodium acetate pH 4.8) was added and mixed. The samples were incubated for 3 min at 65°C in a dry-bath. After incubation, 700 μl of hot acid phenol was added, samples were vortexed for 15 sec and incubated for 3 min in a 65°C water-bath. Then, samples were cooled down on ice for 3 min and centrifuged for 5 min at 13,000 rpm. After centrifugation, the aqueous phase was transferred to a fresh Eppendorf tube and the hot acid phenol procedure was repeated twice. The third aqueous phase was extracted with 350 μl of acid phenol, 336 μl of chloroform and 14 μl of isoamyl alcohol by 15 sec vortexing followed by 5 min centrifugation at 13,000 rpm. The aqueous phase was transferred to a fresh Eppendorf tube and 672 μl of chloroform and 28 μl of isoamyl alcohol was added. The samples were vortexed for 15 sec and centrifuged for 5 min at 13,000 rpm. After transferring the aqueous phase to a fresh Eppendorf tube, 0.1 vol of sodium acetate pH 5.2 was added, mixed and followed by addition of 2.5 vol of absolute ethanol. Tubes were mixed by inversion and placed at -20°C for 1 h. Then, samples were centrifuged for 30 min at 13,000 rpm and 4°C, the supernatant was removed, the pellets were washed with 1 ml of cold 70% ethanol and centifugated as above. After removing the supernatant, the pellets were dried in speed-vac for 3 min. 300 μl of Tris 10 mM EDTA 1 mM (TE) was added and tubes were placed in a 37°C water-bath for 15 min.
To remove the genomic DNA, 120 μl of TE, 60 μl of 25 mM CaCl2 and 20 μl of DNaseI (DNaseI, RNase free, Roche) were added and tubes were incubated for 30 min in a 37°C water-bath. This treatment was followed by extraction with 700 μl acid phenol, 15 sec vortexing and 5 min centrifugation at 13,000 rpm. After centrifugation, the aqueous phase was transferred to a fresh Eppendorf tube and 350 μl acid phenol and 350 μl chloroform was added. Tubes were vortexed for 15 sec and centrifuged for 5 min at 13,000 rpm. After centrifugation, the aqueous phase was transferred to a fresh Eppendorf tube and 700 μl chloroform was added. Tubes were vortexed for 15 sec and centrifuged for 5 min at 13,000 rpm. The aqueous phase was transferred to a fresh Eppendorf tube and 50 μl sodium acetate pH 5.2 was added and mixed, followed by addition of 1 ml of absolute ethanol. Tubes were centrifuged for 30 min at 13,000 rpm and 4°C, the supernatant was removed, the pellets were washed with 1 ml of cold 70% ethanol and centifugated as above. After removing the supernatant, the pellets were dried in speed-vac for 3 min, resuspended in 100 μl of TE and placed for 15 min in a 37°C water bath for complete solubilization. The RNA concentration was determined by light absorption at 260 nm and 280 nm. RNA samples (2 μg) were loaded onto 1.2% agarose gel to check the RNA purity and integrity.
cDNA synthesis and labeling
For cDNA synthesis, CDS-specific primers (cDNA labeling primers optimized for B. subtilis, Sigma-GenoSys Biotechnologies) and two quantities of total RNA (1 and 10 μg) were used. Hybridization probes from 1 μg total RNA were generated in a final volume of 30 μl by mixing together RNA, 4 μl of B. subtilis labeling primers, 0.33 mM of each dATP, dTTP and dGTP. Tubes were heated at 90°C in a dry bath for 2 min and slow cooled to 42°C. After addition of 2 μl (50 U) of AMV reverse transcriptase (RNaseH-, Roche) and 2 μl (20 μCi) 33P-γ-dCTP (2,000-3,000 Ci/mmol; New England Nuclear), the reaction mixture was incubated at 42°C for 3 h in a water bath. Hybridization probes from 10 μg RNA were generated in the same way except that the reaction was performed in a final volume of 120 μl by mixing RNA, 4 μl of B. subtilis labeling primers and 0.25 mM unlabeled nucleotides. Tubes were heated up at 90°C for 2 min and slowly cooled down to 42°C. After reaching 42°C, 6 μl (150 U) of AMV reverse transcriptase and 6 μl (60 μCi) of 33P-γ-dCTP was added, and the reaction was allowed to proceed as described above. Unincorporated nucleotides were removed from labeled cDNA by gel filtration through a G-25 Sephadex column (Roche). Approximately 60-75% incorporation of labeled nucleotides was achieved in these conditions.
DNA array hybridization
Expression profiling measures global transcription either directly (RNA hybridization) or indirectly (hybridization of cDNA synthesized after total RNA has been extracted from the cells). The direct method cannot be used in general, notably because RNA is very unstable and is partially degraded during hybridization. Moreover, the RNA must be labeled before hybridization. This introduces a further source of variability as labeling after RNA extraction is not very sensitive and is prone to systematic bias. Furthermore, direct labeling during growth of the culture needs manipulation of very large quantities of radioactivity, generating an important background noise (to say nothing of safety problems). This led us, like most of our colleagues, to prefer the synthesis of a single cDNA copy for each RNA molecule, using appropriate primers and radioactive or fluorescent nucleotides. The primers were present in excess, and we carefully avoided reinitiation in reverse transcription, as PCR amplification at this stage would greatly enhance the risk of introducing large biases.
A commercial array on nylon membranes was chosen for the hybridization support, because this is the inexpensive solution chosen by laboratories which do not have access to large facilities for making glass arrays. Moreover, this allows one to compare results with those obtained in other laboratories. In addition, using nylon arrays allows one to reuse the same array several times, which provides internal consistency to the experiments. It must also be stressed that hybridization on nylon membranes is a three-dimensional process, where diffusion of the hybridizing molecule is relatively free, in contrast to the crowded situation on glass slides. Finally, the number of genes in a bacterial genome does not require extreme miniaturization.
Panorama nylon filters B. subtilis gene arrays (Sigma-GenoSys Biotechnologies) were soaked in 2x SSPE (10x SSPE: 1.8 M NaCl, 100 mM sodium phosphate pH 7.7, 10 mM EDTA) for 5 min and prehybridized in 15 ml hybridization solution (SSPE 5x, SDS 2%, Denhardt's reagent 1x and 100 μg/ml sonicated salmon sperm DNA) for 1 h at 65°C using roller bottles. The entire cDNA probe was heated for 10 min at 95°C, rapidly cooled down on ice, and added to 10 ml of hot hybridization solution. The prehybridization solution was removed and replaced with hybridization solution. Hybridization was carried out for 16-17 h at 65°C. Following hybridization, each filter was rinsed with 50 ml of washing solution (SSPE 0.5x, SDS 0.2%) for 3 min at room temperature. This operation was repeated twice and followed by three washes in 100 ml of washing solution at 65°C for 20 min each. Filters were then briefly dried in a Whatman paper, soaked with washing solution, and wrapped and sealed in a damp Saran film 25 μm (Bow). Filters were exposed to a PhosphorImager screen (Molecular Dynamics) for 22 h. Arrays were stripped with 500 ml of boiling dehybridization solution (Tris pH 7.6 10 mM, EDTA 1 mM, SDS 1 %) four times for 15 min, washed with 100 ml washing solution, and stored at room temperature in a sealed plastic bag with 20 ml of the same solution. Following this procedure, the same array could be used up to 10 times.
Each gene hybridization was performed in duplicate on pairs of spots deposited side by side on the nylon membrane. This provided an assay for repeatability in addition to a control for reproducibility, tested both by duplicating experiments and by comparing preparations with two different RNA concentrations. The linearity of the relation between the cDNA concentration and the corresponding RNA was analyzed by comparing syntheses performed using either 1 or 10 μg RNA, extracted from the same RNA preparation. This step in the protocol also allowed us to verify that the saturation threshold of the membranes is not reached. The variability introduced for the uncontrolled factors in the culture conditions and in the RNA preparations was estimated by reproducing the experiment on two different days. Overall assay of the reproducibility was further ensured by the fact that both environmental conditions were known to produce very similar behaviors of the cells.
Image acquisition and data analysis
Exposed PhosphorImager screens were scanned with a pixel size of 177 μm on a 445SI PhosphorImager (Molecular Dynamics). A commercial software, XdotsReader (COSE) was used to grid the phosphorimager image (TIFF image files) and to measure the intensity of each dot.
Statistical techniques
The study plan was meant to identify both the effect of the sulfur source on gene expression and the major sources of systematic errors in bacterial expression profiling.
Distribution of random fluctuations in gene expression
We used a standard analysis of variance with five factors on the logarithm of the gene expression. The logarithm transformation is used for normalizing and stabilizing the variance of the residuals (see [18, 19]), this can be justified by the multiplicative nature of the transcription signals. This statistical method has been used in a microarray data context [18] and possesses the following properties: It is well suited to this type of data with a factorial structure. It includes all the data in one pass and takes advantage of the size of the sample to improve the power of the statistical tests in contrast to the use of separate Student tests. There is a long experience of this method and a good knowledge of its limits and pitfalls. It is easy to (in)validate the basic hypothesis of the statistical model and to see whether it is an appropriate method to answer relevant questions about a specific data set.
Variance analysis showed a strong interaction between the amount of RNA used, the date of the experiment, the sulfur source and the genes. In other words, some genes are more sensitive than others to changing conditions. This is what is looked for when one changes the sulfur source, as ideally, only a few genes should have their expression modified in this case. This result is, however, unexpected when the amount of RNA is changed, as this means that the ratio between the quantity of cDNA produced and the quantity of mRNA varies from one gene to another.
The difficulties begin when we find interactions between two factors and genes (gene × date × sulfur source; gene × amount of RNA × sulfur source; and gene × date × amount of RNA) because this indicates that two factors act simultaneously on the values measured for some genes. In other words, one can compute the difference between two measurements on the same gene, but it is not possible to assign the value of this difference to the sole effect of changes in conditions between the two measurements. Tables listing results of this type must therefore be interpreted with extreme caution (it would be even worse, of course, if one used ratios, as is unfortunately often done). A prudent solution is to concentrate on the genes that do not show important variations, except for one of the factors. In practice, one computes for each gene the difference of measurements for a given factor (for example the sulfur source) in the four conditions (1 or 10 μg, day A or B), then one calculates the mean and the standard deviation of all four differences. The most interesting genes are those that combine the largest mean and the lowest standard deviation. The general solution to the problem, even when the factor of interest has more than two variables (for example the simultaneous study of three or more sulfur sources) is to perform for each gene a variance analysis with two factors (the factor of interest on the one hand, and all the other ones grouped together - for example, date, RNA amount - on the other), then to retain those genes that display both a weak interaction between factors and a large sum of the squares of the deviations from the mean, for the factor of interest.
Controlled factors
One can reasonably assume that the observed hybridization signal is proportional to the cDNA amount spotted by the robot, as the measurement of transcription expression is based on the hybridization of complex probes, at low Cot, under conditions where the target sequences are in large excess, that is, far from the saturation threshold [20]. We have taken five factors into account. First, the condition of the gene expression, denoted sulfur source, with two levels. Second, the culture day of RNA: experiments with same material and protocol were made at two different days. Third, the amount of RNA: 1 μg or 10 μg. Fourth, the robot lays two successive drops on the membrane, next to each other - this factor is denoted drop with two levels, 1 and 2. Experiments with level 1 and 2 are called replicates. Fifth, the gene: there are 4,107 genes.
We thus have 24 × 4,107 = 65,712 treatment units. Experiments have been made with two membranes. The membrane factor is superimposed on the interaction day × amount of RNA extraction, so we are not able to distinguish these two sources of variation.
Table 4 shows the characteristic values for the hybridization intensity distribution in the different experiments. The figures for each gene correspond to the average of the intensity of each one of the duplicate drops (in arbitrary units; the value given is that of the drop with the highest value in each quartile). The distribution is very asymmetrical. The spread of the distribution, as measured by the difference between the first and the third quartile, varies as a function of the amount of RNA used to synthesize cDNA (1 μg: 479 ± 81; 10 μg: 785 ± 52). The same holds for the average value, with a factor 1.6 between the two protocols, while the RNA amount used varied by a factor 10 (1 μg: 503 ± 88; 10 μg: 787 ± 36). There exists a clear difference between the measurements performed using 1 μg RNA on days A and B. This difference is not observed in the preparations performed using the very same extracts, with 10 μg RNA.
Table 4.
Characteristic values for the distribution of intensities (arbitrary units) in different experimental conditions
| Met A 1 | MTR A 1 | Met B 1 | MTR B 1 | Met A 10 | MTR A 10 | Met B 10 | MTR B 10 | |
| First | 406 | 381 | 300 | 293 | 523 | 548 | 532 | 488 |
| quartile | ||||||||
| Second | 597 | 559 | 434 | 422 | 762 | 833 | 797 | 756 |
| quartile | ||||||||
| Third | 970 | 913 | 719 | 693 | 1,230 | 1,371 | 1,335 | 1,294 |
| quartile |
The genes were ranked according to the intensity of the corresponding hybridization; the values were then arranged into four sets of equal numbers, with increasing hybridization values. The value for the highest hybridization in each quartile is given here. Met, methionine; MTR, methylthioribose; A and B refer to different days, and 1 and 10 refer to the amount of RNA (see text).
Variance analysis shows that the interactions involving the amount of RNA are more important than those due to all other factors. That is, variations from gene to gene, within a same experiment, of the ratio between the quantity of cDNA and that of mRNA, are more important than the variations resulting from the culture day or the sulfur source. This is, however, a factor often neglected in experimental protocols and therefore deserves special study.
To this end, the genes were ordered as a function of the average intensity of the signal, then distributed into five classes with the same number of elements. The first class (centiles from 0 to 20) contained the 823 genes having the lowest signal intensity, measured on the totality of the eight experiments. Class 20-40 contained the subsequent 823 genes, which have a signal with an intensity higher than that of class 0-20 and lower than that of class 40-60, and so on. The correlation between the values obtained using 1 μg and those performed using 10 μg of RNA was calculated for each class (Table 5). One observed a good correlation between the measurements performed with 1 μg and 10 μg of RNA, when the signal was either very low or very high. This showed first that the excess of sequence target DNA is sufficient for hybridization to be far from saturation, and second, perhaps unexpectedly, that the background noise (the residual radioactivity of the membrane, and so on) does not alter the measurement of hybridization of low cDNA amounts.
Table 5.
Variation of the correlation in the measurements performed with 1 μg and 10 μg RNA
| Centile | Met A 1 × Met A 10 | Met B 1 × Met B 10 | MTR A 1 × MTR A 10 | MTR B 1 × MTR B 10 |
| 0-20 | 0.589 | 0.589 | 0.563 | 0.668 |
| 20-40 | 0.132 | 0.248 | 0.103 | 0.422 |
| 40-60 | 0.018 | 0.087 | -0.022 | 0.332 |
| 60-80 | 0.191 | 0.179 | 0.120 | 0.350 |
| 80-100 | 0.698 | 0.763 | 0.706 | 0.807 |
The results for experiments with 1 or 10 μg RNA were compared after ordering the genes with the corresponding hybridization values in increasing order. A high correlation indicates that the results are very similar for both conditions. The correlation is very good for highly expressed or poorly expressed genes whereas it is low for those with medium expression. Met, methionine; MTR, methylthioribose; A and B refer to different days, and 1 and 10 refer to the amount of RNA (see text).
In contrast, the correlation was very poor for the medium hybridization values. To verify that this was not an artifact due to the lower variance in the central classes, we calculated the correlation between the values obtained using the same amount of RNA extracted from a culture grown with methionine and another with methylthioribose (Table 6). The correlations were much higher than those found in Table 5, despite the fact that the classes contained exactly the same genes, and that measurements varied very little, for the vast majority of the genes, from one sulfur source to the other.
Table 6.
Variation of the correlation in the measurements performed with two sources of sulfur
| Centile | Met A 1 × MTR A 1 | Met A 10 × MTR A 10 | Met B 1 × MTR B 1 | Met B 10 × MTR B 10 |
| 0-20 | 0.751 | 0.767 | 0.636 | 0.777 |
| 20-40 | 0.685 | 0.680 | 0.514 | 0.681 |
| 40-60 | 0.730 | 0.663 | 0.608 | 0.749 |
| 60-80 | 0.821 | 0.736 | 0.738 | 0.843 |
| 80-100 | 0.945 | 0.912 | 0.946 | 0.969 |
The data is arranged as in Table 5. Correlation is good, indicating that there is not a large difference between growth on methionine and growth on MTR (as expected).
The absence of a good correlation between the measurements performed using 1 μg or 10 μg RNA for medium hybridization values is thus real. In other terms, the estimation of the expression of a gene depends considerably on the amount of RNA used to synthesize its cDNA complement, despite the fact that we used two aliquots of a unique RNA preparation. This phenomenon is particularly important for the genes that give a medium hybridization (and therefore transcription) signal. For this reason we can assert that the observations made in the present study draw the minimal conclusions that can be extracted from the data, and that it is likely that there are further effects buried in the data, in particular involving genes with medium expression.
A rapid browse through the list of the 20 most variable genes did not reveal any special feature. Namely, there is a perfectly normal representation of unknown ('y') genes (14/20 as compared to 69.4% in the genome) and those were not the same genes as in the two preceding lists.
The repeatability of the robot spotting is good, and this source of artifact can therefore be disregarded (95% of the genes display a difference between both spots lower than 20%). It is not possible to know, when a spot is systematically more intense than its counterpart, whether the bias is due to some specific feature of the gene or due to the membrane (for example, irregularity of the robot, heterogeneity of the background) because each gene keeps the same position in the membrane in all experiments.
Uncontrolled factors
The difficulty of finely controlling the experimental conditions from one day to the other is the second source of systematic errors. The list of the 20 most variable genes according to this parameter has several interesting features. Genes of known function were abnormally abundant (13/20; the difference from the proportion in the genome is highly significant, p < 0.001) indicating a non-random gene distribution. Furthermore, day A is characterized by the expression of an ensemble of genes involved in competence and day B by the expression of genes involved in sporulation.
Unidentified factors
These controlled and uncontrolled factors (such as contribution of the quantity of residual rRNA in the initial RNA preparation, fluctuation in the state of the culture) manifest themselves in the model. In theory, there remains a residual term in the statistical model. As the residue is very small in the present study, this indicates that unidentified factors do not play an important role in the experiment presented.
Acknowledgments
Acknowledgements
This work benefited from the support of the BACELL European Union program. Some of the mutants used for confirmatory studies came from the collection of the Japanese-European consortium for functional analysis of the Bacillus subtilis genome: we are grateful to N. Ogasawara and S.D. Ehrlich for providing access to the strains. We thank I. Martin-Verstraete for her interest and her help in supplying the strains.
References
- Sekowska A, Danchin A. Identification of yrrU as the methylthioadenosine nucleosidase gene in Bacillus subtilis. DNA Res. 1999;6:255–264. doi: 10.1093/dnares/6.5.255. [DOI] [PubMed] [Google Scholar]
- Danchin A, Sekowska A. Expression profiling in reference bacteria: dreams and reality. GenomeBiology. 2000;1:1024.1–1024.5. doi: 10.1186/gb-2000-1-4-reviews1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SA. Primer of Biostatistics 4th edition New York: McGraw Hill, 1997.
- Sekowska A, Kung HF, Danchin A. Sulfur metabolism in Escherichia coli and related bacteria: facts and fiction. J Mol Microbiol Biotechnol. 2000;2:145–177. [PubMed] [Google Scholar]
- Coppée J-Y, Auger S, Turlin E, Sekowska A, Caer J-PL, Labas V, Vagner V, Danchin A, Martin-Verstraete I. Sulfur-limitation-regulated proteins in Bacillus subtilis : a two-dimensional gel electrophoresis study. Microbiology. 2001;147:1631–1640. doi: 10.1099/00221287-147-6-1631. [DOI] [PubMed] [Google Scholar]
- Bacillus subtilis Genome Sequencing Project http://genolist.pasteur.fr/SubtiList
- Wissenbach U, Six S, Bongaerts J, Ternes D, Steinwachs S, Unden G. A third periplasmic transport system for L-arginine in Escherichia coli : molecular characterization of the artPIQMJ genes, arginine binding and transport. Mol Microbiol. 1995;17:675–686. doi: 10.1111/j.1365-2958.1995.mmi_17040675.x. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Sekowska A, Danchin A. Sulphur islands in the Escherichia coli genome: markers of the cell's architecture? FEBS Lett. 2000;476:8–11. doi: 10.1016/s0014-5793(00)01660-4. [DOI] [PubMed] [Google Scholar]
- Gourse RL, Gaal T, Bartlett MS, Appleman JA, Ross W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu Rev Microbiol. 1996;50:645–677. doi: 10.1146/annurev.micro.50.1.645. [DOI] [PubMed] [Google Scholar]
- Arfin SM, Long AD, Ito ET, Tolleri L, Riehle MM, Paegle ES, Hatfield GW. Global gene expression profiling in Escherichia coli K12. The effects of intergration host factor. J Biol Chem. 2000;275:29672–29684. doi: 10.1074/jbc.M002247200. [DOI] [PubMed] [Google Scholar]
- Bigay J, Deterre P, Pfister C, Chabre M. Fluoroaluminates activate transducin-GDP by mimicking the gamma-phosphate of GTP in its binding site. FEBS Lett. 1985;191:181–185. doi: 10.1016/0014-5793(85)80004-1. [DOI] [PubMed] [Google Scholar]
- Myers RW, Abeles RH. Conversion of 5-S-methyl-5-thio-D-ribose to methionine in Klebsiella pneumoniae. Stable isotope incorporation studies of the terminal enzymatic reactions in the pathway. J Biol Chem. 1990;265:16913–16921. [PubMed] [Google Scholar]
- Wray JW, Abeles RH. The methionine salvage pathway in Klebsiella pneumoniae and rat liver. Identification and characterization of two novel dioxygenases. J Biol Chem. 1995;270:3147–3153. doi: 10.1074/jbc.270.7.3147. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:741–746. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micado Release 6.1 http://locus.jouy.inra.fr/cgi-bin/genmic/madbase/progs/madbase.operl
- JAFAN: Japan Functional Analysis Network for Bacillus subtilis http://bacillus.genome.ad.jp/
- Spizizen J. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci USA. 1958;44:1072–1078. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. 2001. Preprint available online at http://www.jax.org/research/churchill/research/expression/kerr-synteni.pdf. [DOI] [PubMed]
- Efron B, Tibshirani R, Goss V, Chu G. Microarrays and their use in a comparative experiment. 2001. Preprint available online at http://www.biostat.wisc.edu/geda/seminars.html.
- Bertucci F, Bernard K, Loriod B, Chang YC, Granjeaud S, Birnbaum D, Nguyen C, Peck K, Jordan BR. Sensitivity issues in DNA array-based expression measurements and performance of nylon microarrays for small samples. Hum Mol Genet. 1999;8:1715–1722. doi: 10.1093/hmg/8.9.1715. [DOI] [PubMed] [Google Scholar]