

Abstract
Viruses are intracellular pathogens responsible for a vast number of human diseases. Due to their small genome size, viruses rely primarily on the biosynthetic apparatus of the host for their replication. Recent work has shown that the molecular chaperone Hsp90 is nearly universally required for viral protein homeostasis. As observed for many endogenous cellular proteins, numerous different viral proteins have been shown to require Hsp90 for their folding, assembly, and maturation. Importantly, the unique characteristics of viral replication cause viruses to be hypersensitive to Hsp90 inhibition, thus providing a novel therapeutic avenue for the development of broad-spectrum antiviral drugs. The major developments in this emerging field are hereby discussed.
Keywords: Hsp90, Antivirals, Chaperones, Drug resistance, Hsp90 inhibitors, Virus, protein folding
1. Introduction
The capacity of all proteins to carry out their function is dependent on their ability to fold correctly and folding of many cellular proteins critically relies on the assistance of molecular chaperones. Molecular chaperones are highly conserved and often essential for viability and for the ability to survive cellular stress. Chaperones reside in all cellular compartments where they promote the folding, maturation, complex assembly, and trafficking of proteins in an ATP dependent manner [1-3]. In addition, chaperones provide a conduit to the protein degradation machinery and regulate many cellular pathways [3].
Viruses are intracellular obligate parasites that hijack the biosynthetic machinery of the host for their replication. Given the structural and functional complexity of many viral proteins, it is not surprising that they, like cellular proteins, are also dependent on chaperones for their folding and function. In this review we discuss the role of Hsp90 in viral replication and the potential of targeting Hsp90 as an antiviral strategy.
2. Hsp90: Overview of cellular functions
Hsp90s are a family of highly abundant, essential, and evolutionary conserved molecular chaperones. In mammals, there are two cytoplasmic Hsp90 isoforms, the stress induced Hsp90α and the constitutively expressed Hsp90β [4], as well as an ER resident homologue Grp94 (also called gp96) [5], and a mitochondrial variant, TRAP1 [6]. Hsp90s are ATPases that mostly form homodimeric complexes, with each monomer sharing a common domain organization comprised of a C terminal dimerization domain, a middle domain, and a structurally unique N terminal ATPase domain (see review [7, 8]). In the cytoplasm, Hsp90 regulates the activity, maturation, localization, and turnover of a large yet select set of substrates or “clients” [9, 10]. Hsp90 function is regulated by a cohort of co-chaperones that modulate its ATPase cycle, enable client acquisition and selection, and provide a link to other chaperone systems as well as the ubiquitin-proteasome system [11-13]. Hsp90 is believed to recognize a metastable structural element in its clients rather than a primary amino acid sequence. Unlike other chaperone systems in the cell, Hsp90 does not appear to interact with newly synthesized proteins. Instead, Hsp90 receives its client proteins from other chaperone systems, such as Cdc37 or the co-translationally acting Hsp70 chaperone system, likely following the acquisition of a partially folded state recognized by Hsp90. Thus, some of the client specificity of Hsp90 is determined by these collaborating chaperones. For protein kinases, which comprise the most intensely studied client group, binding to Hsp90 requires the Cdc37 co-chaperone, which binds to both Hsp90 and protein kinase clients [14]. For other clients, such as steroid hormone receptors, Hsp70 binds first, and the co-chaperone Hop bridges transfer to Hsp90. Hop physically interacts with both chaperone systems by virtue of specific tetratricopeptide (TPR) interaction domains that recognize the C-terminal tails of both Hsp70 and Hsp90 [15, 16]. In addition to its role in assisting the maturation of proteins, Hsp90 was shown to be specifically required for the degradation of certain proteins [17]. In this regard, it is noteworthy that CHIP, another TPR containing protein that binds to Hsp90 also contains a U-box domain with E3 ubiquitin ligase activity. CHIP has been shown to direct Hsp90 and Hsp70 bound proteins to proteasomal degradation [18].
The cellular functions and targets of Hsp90 are highly diverse. In addition to its originally described function assisting the maturation of protein kinases and steroid hormone receptors [19], Hsp90 is required for the function of a large number of proteins and protein complexes, both under normal growth conditions and under conditions of stress, such as elevated temperatures. Hsp90 clients include proteins involved in transcription, translation, mitochondrial function, kinetochore assembly, centrosome function and cell cycle [20-22]. Hsp90 is also central to secretory pathway function and associates with many complexes involved in membrane trafficking [3, 22]. Many of the Hsp90 clients are part of multisubunit complexes. The current understanding of Hsp90 function as a chaperone that stabilizes or remodels polypeptides that are substantially folded suggests that its action promotes a conformational maturation step that enables clients to bind ligands, interact with cofactors, or carry out their biological function [23]. For instance, many clients of Hsp90 undergo ordered assembly and disassembly processes, and Hsp90 may stabilize subunits of these complexes prior to assembly or facilitate their conformational transitions [24]. During cellular stress, e.g. at elevated temperatures, Hsp90 may also become important to stabilize the labile conformations of many proteins, most notably cell cycle components. In this regard, it is noteworthy that aberrant proteins with a few amino acid changes exhibit radically higher dependence on Hsp90 than their native counterparts [25].
Among the Hsp90 clientele identified to date are many proteins involved in signal transduction and cell division, including numerous proteins related to tumorigenesis [26, 27]. The dependence of multiple oncogenes on Hsp90 renders cancer cells hypersensitive to Hsp90 inhibition and forms the basis for using Hsp90 inhibitors in cancer treatment. Numerous specific pharmacological inhibitors of Hsp90 have been identified. The majority of these, including radicicol, Geldanamycin (GA), and the GA derivatives 17-allyl-17-demethoxygeldanamycin (17-AAG) and 17-desmethoxy-17-N,N-dimethylaminoethylaminogeldanamycin (17-DMAG), inhibit Hsp90 activity by competing with ATP for binding to the N-terminal ATP/ADP-binding domain of Hsp90 (see [28] for review). Inhibitors that bind to the C-terminal domain, such as novobiocin and coumermycin A1, have also been identified. Due to the fact that many Hsp90 clients are involved in cancer progression, these inhibitors possess broad anticancer activity and several are currently undergoing advanced stage clinical evaluation [26, 29].
3. Hsp90 in viral replication
Viral proteins, like cellular proteins, are dependent on chaperone function for folding and assembly [30]. Hsp90 has been shown to play a role in the replication of many different viruses including DNA viruses, RNA viruses of both positive and negative sense genomes, and double-stranded RNA viruses (see Table 1). In fact, the dependence of viruses on Hsp90 appears to be nearly universal. Strikingly, for viruses tested to date, replication appears to be sensitive to Hsp90 inhibitors at concentrations not affecting cellular viability. The hypersensitivity of viral replication to Hsp90 inhibition may stem from several unique characteristics of viral protein homeostasis, which present a distinct set of challenges to the protein folding machinery, and thus render it more sensitive to fluctuations in cellular folding capacity. First, the limited size of most viral genomes often determines that viral proteins be multifunctional, likely resulting in structurally complex proteins that are dependent on chaperone function. In addition, many viruses, and in particular cytopathic viruses, must produce large quantities of a limited number of viral proteins within a short period of time. The need to rapidly produce a limited array of structurally complex proteins in high abundance is likely to tax the capacity of the chaperone systems required to fold them. Moreover, the unique complexity and structural requirements of viral capsids make their precursors particularly vulnerable to aggregation and misfolding [31-34]. Thus, capsid proteins must fold to a precursor conformation that is soluble yet poised to assemble in the presence of the viral genome to form a capsid, which may contain over 1000 identical subunits and retain infectivity despite extreme thermal and chemical conditions [35]. The high mutation rates of RNA viruses, which are the highest in nature, inevitably lead to the production of a swarm of mutant viral proteins during infection. Such viral population diversity has been shown to be critical for the ability of viruses to adapt to adverse conditions [36-38]. However, the mutant proteins thus generated can have dominant negative effects on viral protein complex assemblies [39]. As mutant proteins are known to be hyperdependent on chaperone function for activity [25], chaperones are likely to be instrumental in buffering the deleterious effects of mutant viral proteins and thus facilitate viral adaptation. Finally, since chaperones regulate many cellular functions including signaling networks, cell cycle progression, and apoptosis, viruses are likely to manipulate chaperones to render cells conducive to viral replication.
Table. 1. Summary of the viruses and viral proteins currently known to require the Hsp90 chaperone.
| Viral family | virus | viral protein | Hsp90 dependent process | Additional factor | References |
|---|---|---|---|---|---|
| Herpesviridae | HSV1/HSV2 | UL30 | Polymerase localization | Hsp70, BAG3 | [51, 52, 57] |
| VZV | ORF29F | Localization of orf29 | Hsp70, BAG3 | [54] | |
| HCMV | - | Expression of immediate early protein IE2 | PI3K | [53] | |
| EBV | KH | Apoptosis prevention | - | [96, 111] | |
| EBNA | Cell proliferation | - | [112] | ||
| KSHV | KI | Apoptosis prevention | Hsp40 | [94] | |
| v-FLIP | Apoptosis prevention | IKK, Cdc37 | [93] | ||
| Polyomaviridae | SV40 | LT | Stabilization of LT protein | - | [63] |
| Poxviridae | Vaccinia virus | 4a core protein | Capsid assembly/ Virus gene expression | Hsp70 | [34, 113] |
| Reoviridae | Reovirus | σ1 | C' trimerization of σ1 | Hsp70, p23 | [76-79] |
| Rotavirus | NSP3 | Dimerization of NSP3 | - | [66] | |
| - | - | PI3K | [70] | ||
| Birnaviridae | IBDV | VP2 (Capsid) | Virus Internalization | - | [88] |
| Picornaviridae | Poliovirus | P1 capsid protein | Cleavage of P1 into VP1, VP2 & VP3 | p23 | [33] |
| Rhinovirus | P1 capsid protein | Cleavage of P1 into VP1, VP2 & VP3 | p23 | [33] | |
| Coxsackievirus | P1 capsid protein | Cleavage of P1 into VP1, VP2 & VP3 | p23 | [33] | |
| Flaviviridae | HCV | NS3 Protease | Cleavage at NS2/3 junction, NS3 function | - | [71, 72, 99] |
| NS5A | Replication complex formation/Genome replication | FKBP8/hB-ind1 | [73, 74, 114, 115] | ||
| DENV | Viral receptor | Virus Internalization | Hsp70/GRP78 | [85, 86] | |
| Arenaviridae | LCMV | NP | Antigen cross presentation | - | [116] |
| Nodaviridae | FHV | Protein A | Replication complex formation | - | [58, 59, 117] |
| Hepeviridae | HEV | Capsid | Intracellular transfer | - | [82] |
| Rhabdoviridae | VSV | L protein | Protein stabilization | - | [60] |
| Paramyxoviridae | HPIV2/3 | L protein | Protein stabilization | - | [60] |
| SV5/41 | L protein | Protein stabilization | - | [60] | |
| Measles virus | - | Enhanced oncolytic activity | - | [118] | |
| Sendai virus | - | Innate immunity activation | TBK1 | [110] | |
| Bunyaviridae | La Crosse virus | L protein | Protein stabilization | - | [60] |
| Orthomyxoviridae | Influenza A Virus | PB1, PB2 | Nuclear localization | - | [47] |
| vRNP complex formation | - | [48] | |||
| RNA synthesis | - | [49] | |||
| Filoviridae | Ebola virus | - | Virus propagation | - | [119] |
| Retrovirus | HTLV1 | tat | Transcription | - | [95] |
| HIV1 | tat | Transcription/Cell survival | - | [89, 90, 120] | |
| Hepadnaviridae | DHBV | P protein | Reverse transcriptase priming | p23, Cdc37, Hsp40, Hsc70 | [41, 43, 44, 121] |
| HBV | P protein | Reverse transcriptase priming | Hsp70, Hsp40 Hop, P23 | [43, 45, 46] |
Abbreviations: HSV, herpes simplex virus; VZV, varicella zoster virus; HCMV, human cytomegalovirus; EBV, Epstein-Bar virus; KSHV, Kaposi sarcoma-associated herpesvirus; SV, simian virus; IBDV, infectious bursal disease virus; HCV, hepatitis C virus; DENV, dengue virus; LCMV, lymphocytic choriomeningitis virus; FHV, flock house virus; HEV, Hepatitis E virus; VSV, vesicular stomatitis virus; HPIV, human parainfluenza virus; vRNP, viral ribonucleoprotein complex; HTLV, human adult T cell leukemia virus; HIV, human immunodeficiency virus; DHBV, duck hepatitis B virus; HBV, hepatitis B virus.
While the antiviral effects of Hsp90 inhibition have been broadly demonstrated, detailed understanding of the role of Hsp90 in viral replication is only available for a few viruses. In most cases, the antiviral activity of Hsp90 inhibitors is accompanied by the degradation of a single viral protein, implicating such viral proteins as dependent on Hsp90 for their maturation or stability. The major categories of viral Hsp90 clients are outlined below.
4. Viral Non-Structural Proteins as Hsp90 clients
4.1. Viral Polymerases
Viral polymerases constitute the largest class of identified Hsp90 viral client proteins. The role of Hsp90 in the function of viral polymerases appears to be variable, although detailed knowledge is only available in a few cases. The best-elucidated example of Hsp90's role in polymerase function comes from the reverse transcriptases (RT) of two viruses belonging to the Hepadnaviridae family: duck hepatitis B virus (DHBV) and human hepatitis B virus (HBV). These enzymes mediate both the incorporation of viral pregenomic RNA (pgRNA) into nucleocapsids and the reverse transcription of pgRNA into DNA [40]. For DHBV, Hsp90 stimulates the ability of the RT to initiate and maintain stable reverse transcription. This function of Hsp90 requires the presence of other chaperones, such as Hsp70 and Hsp40, and co-chaperones Hop and p23, which together function as substrate release factors and facilitated incorporation of the pgRNA into nucleocapsids [41-43]. The Hsp90 cofactor Cdc37, normally required for the folding of Hsp90 dependent cellular kinases, was also shown to interact with the DHBV RT independently of Hsp90 [44]. Overexpression of Cdc37 resulted in increased reverse transcription and pgRNA packaging into nucleocapsids, while overexpression of a dominant negative Cdc37 mutant had opposite effects. For HBV, Hsp90 inhibition with GA has been shown to reduce both reverse transcription and pgRNA incorporation into nucleocapsids. Furthermore, the production of active reverse transcriptase in vitro requires the addition of recombinant Hsp90, Hsp70, Hsp40, Hop and p23, similar to DHBV [45, 46].
The polymerase of influenza virus A also requires Hsp90 for genome replication (Fig. 1A). The RNA dependent RNA polymerase of influenza virus is comprised of three subunits, PB1, PB2 and PA. Hsp90 associates with both PB1 and PB2 in infected cells and re-localizes to the nucleus where it facilitates viral RNA synthesis [47, 48]. Hsp90 inhibition by treatment with GA or 17-AAG was shown to enhance the degradation of both PB1 and PB2 as well as to reduce levels of the fully assembled polymerase complex, thus reducing the levels of viral derived RNAs [49].
Figure 1. Distinct functions of Hsp90 during viral replication.
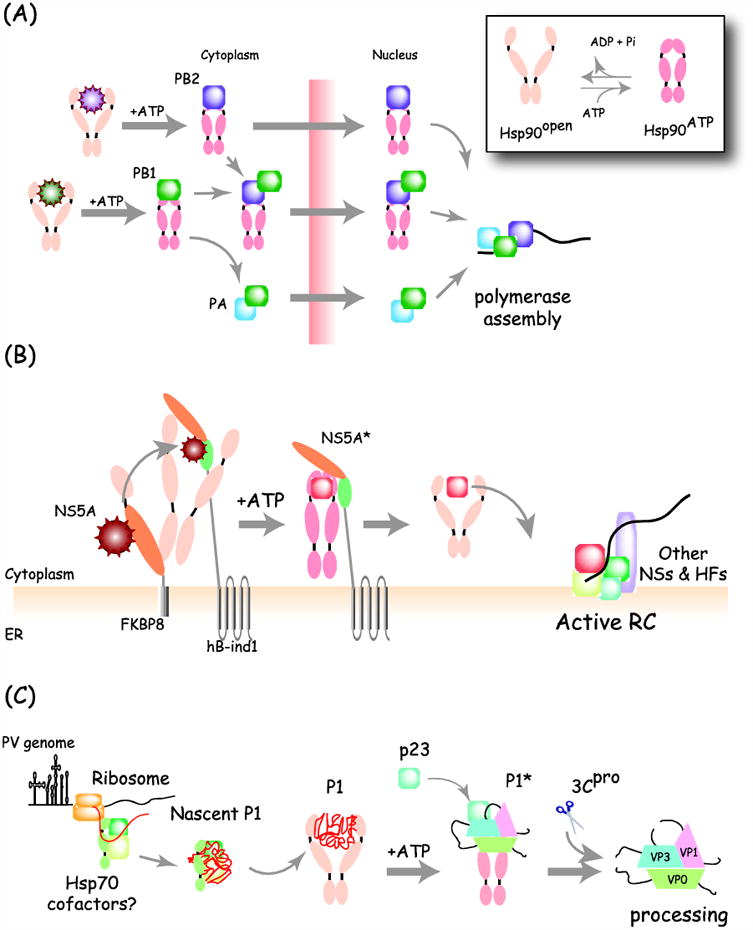
(A) Hsp90-facilitates assembly of Influenza polymerase. Newly made influenza PB2 interacts with Hsp90 in the cytoplasm and the complex translocates into the nucleus. Another polymerase subunit PB1, which itself interacts with Hsp90, is also transported into the nucleus together with the PB2/Hsp90 complex. Once in the nucleus, Hsp90 dissociates as PB2 binds to PB1/PA to form the trimeric active polymerase. Inset: the two conformations of Hsp90 are regulated by its ATPase cycle. (B) Formation of a multi-chaperone complex with non-structural protein NS5A during HCV replication. HCV NS5A assembles into a complex containing Hsp90, FKBP8 and hB-ind1. Interaction with FKBP8 is required for NS5A entry into the Hsp90 cycle whereas hB-ind1 action stimulates dissociation from Hsp90 and correct NS5A folding (NS5A*), leading incorporation into an active replication complex (RC) containing other NS proteins and host factors (HFs). (C) Role of Hsp90 in picornavirus capsid maturation. Picornavirus capsid precursor poly-protein P1 binds associates with Hsp90, likely following an upstream interaction with Hsp70. Hsp90 is required to fold P1 to a mature conformation (P1*) that is competent for proteolytic cleavage by the viral protease (3CPro) into the mature capsid protein. The processed capsid proteins no longer interact with Hsp90.
Hsp90 is important for the polymerase functions of Herpesviridae family. The alpha herpesvirus, herpes simplex virus type 1 (HSV-1) induces formation of specific chaperone-rich nuclear structures called “Virus-induce Chaperone enriched” (VICE) compartments, which appear to facilitate viral replication in the nucleus and nuclear protein quality control [50]. During HSV infection, a subpopulation of Hsp90 localizes to VICE compartments and its ATPase activity is required for the proper nuclear localization of viral DNA-dependent DNA polymerase, UL30. Inhibition of Hsp90 with GA results in mislocalization of the viral polymerase to the cytoplasm instead of the nucleus, proteasomal degradation of the polymerase, and consequent reduction of viral DNA replication and production of progeny viral particles [51, 52]. Hsp90 is also required for replication of beta-herpesviruses. Hsp90 inhibition with GA led to degradation of the viral polymerase as well as reduced viral gene expression via the disruption of PI3-kinase pathway during human cytomegalovirus (HCMV) infection [53]. Whether Hsp90 is required for the nuclear localization of the HCMV polymerase, as in the case of HSV1, remains to be tested. On the other hand, during the infection with another alpha-herpesvirus, varicella zoster virus (VZV), the viral single-stranded DNA binding protein ORF29p was shown to colocalize with Hsp90 and Hsp70 in the nucleus through a specific interaction with BAG3 [54]. BAG3 is one of a cohort of Hsp70 interacting Bag-family proteins that modulate Hsp70 function [55, 56]. Hsp90 inhibition by treatment with GA or 17-DMAG resulted in diffuse cytosolic localization of ORF29p and BAG3, suggesting that herpesvirus DNA binding proteins may generally require Hsp90 for nuclear import. BAG3 co-immunoprecipitates with ICP0, the HSV E3 ubiquitin ligase and major pathogenicity factor, but whereas knockdown of BAG3 reduced the replication of VZV it did not affect wild type HSV replication [57]. This raises the possibility that viruses utilize different chaperone complexes for the same purpose. It will be of interest to establish whether the polymerases of herpesviruses require Hsp90 for folding and/or nuclear localization directly or whether Hsp90 is required for the folding or activity of cellular factors required for polymerase stability or nuclear import.
Protein A, the RNA dependent RNA polymerase protein of the Nodaviridae flock house virus (FHV), is also dependent on Hsp90. Interestingly in the case of FHV, Hsp90 appears to be required for translation of protein A but not for substantial polymerase activity [58]. Inhibition of Hsp90 with GA during FHV infection results in reduced levels of polysomes translating protein A, while no effect is observed on other viral proteins or cellular RNA associated with polysomes [59]. Whether the role of Hsp90 in protein A synthesis is due to a direct effect on protein A or via the chaperoning of a cellular factor is unknown. In addition to the above examples, other viral families also require Hsp90 for polymerase activity since pharmacological inhibition of Hsp90 by GA during infection results in the degradation of the RNA-dependent RNA polymerases of rhabdoviruses (vesicular stomatitis virus), paramyxoviruses (human parainfluenza 2 and 3, simian virus 5 and 41), and bunyaviruses (La Cross virus), and the concomitant suppression of viral propagation [60].
The dependence of viral polymerases on Hsp90 is very striking as it extends to the RNA-dependent RNA polymerases of both positive and negative-strand RNA viruses, DNA-dependent DNA polymerases, as well as to the reverse transcriptase of hepadnaviruses, despite the evolutionary, structural and functional divergence of these proteins. In this regard, viral polymerases may share features of their cellular counterparts, as telomerase and DNA polymerase α are known Hsp90 client proteins [61, 62]. The general dependence of many polymerases on Hsp90 suggests that this chaperone may recognize a conserved feature of these diverse enzymes, or may facilitate a common step in their maturation and/or mechanism, such as the interaction with nucleic acid. However, not all viral polymerases require Hsp90 for their function [33]. Nonetheless, it will be of interest to elucidate the reason underlying the wide dependence of polymerases function on hsp90.
4.2. Other Non-structural proteins
Hsp90 is also required for the activity and folding of non-structural proteins other than polymerases. In the case of the DNA tumor virus SV40, Hsp90 associates with the large T antigen (LT) [63]. LT protein is a multifunctional protein that interacts with several cellular proteins, including the chaperones Hsp70 and Hsp90, as well as regulatory proteins, such as p53 and DNA polymerase α [64, 65]. Hsp90 physically interacts with LT and Hsp90 inhibition with GA or radicicol leads to its degradation in cells. Similarly, for rotavirus, nonstructural protein 3 (NSP3) also requires Hsp90 for folding and stabilization [66]. NSP3 shuts off cellular translation by relocalizing the cytoplasmic poly(A) binding protein (PABP-C1) to the nucleus, thereby evicting it from translation initiation complexes in infected cells [67-69]. Inhibition of Hsp90 by 17-AAG or 17-DMAG resulted in reduced NSP3 translation and abolishes nuclear translocation of PABP-C1, thus suppressing viral replication [66, 70].
In the case of hepatitis C virus (HCV), Hsp90 is required for the activity and stability of two non-structural proteins, the protease and helicase NS3 and the multifunctional protein NS5A (Fig. 1B). Inhibition of Hsp90 by GA, 17-AAG, or radicicol reduced the protease activity of NS3 required to liberate it from the NS2/3 protein precursor, and caused NS3 proteasomal degradation [71, 72]. Interestingly, even though Hsp90 is required for the proteolytic activity of NS3, Hsp90 binds the helicase domain and not the protease domain. Accordingly, deletion of the helicase domain prevents the degradation of NS3 induced by Hsp90 inhibitors. NS5A also associates with Hsp90 and Hsp90 inhibition by GA results in a modest level of NS5A degradation [73]. As discussed above for the polymerase-Hsp90 interactions, the study of HCV encoded clients of Hsp90 also implicates specific Hsp90 co-chaperones in viral replications. Interestingly, the analysis of the NS5A-Hsp90 interactions revealed a novel type of membrane-anchored Hsp90 complex, where Hsp90 and NS5A are both associated with the proline-isomerase FKBP8 and a novel membrane-bound Hsp90 cofactor called human butyrate-induced transcript 1 (hB-ind1), which harbors a p23 homology domain [74]. These findings illustrate how the large collection of Hsp90 cofactors can direct individual viral proteins to specific cellular locales and functions. It is also noteworthy that replication of all positive-strand RNA viruses, such as HCV, occurs on intracellular membranes, raising the possibility that membrane-anchored Hsp90 cofactors, such as hB-ind1, target the chaperone to sites of viral replication and help assemble large replication complexes. Investigating the role of hB-ind1 and other Hsp90 cofactors in the replication of additional RNA viruses may lead to more specific therapeutic avenues to inhibit viral replication, since inhibition of specific Hsp90-cochaperone interactions could reduce pleiotropic adverse effects on the protein homeostasis of uninfected cells.
5. Viral structural proteins as Hsp90 clients
Viral structural proteins assemble to form the viral capsid, a complex protein structure that encapsulates the viral genome, protecting it from degradation and releassing it upon re-infection of a new cell. Viral capsids can be comprised of one or more proteins and can contain over 1000 subunits. These structures must be sufficiently stable both within the host and outside to withstand harsh environmental conditions, such as low pH and high temperature, and yet they must also disassemble readily upon entry into cells to deliver the viral genome. Therefore, the complexity of viral capsids is likely to be especially demanding on the cellular protein folding and assembly machinery.
Hsp90 is known to facilitate folding of several viral structural proteins. Hsp90 is required for replication of Picornaviridae family members poliovirus, rhinovirus, and coxsackievirus [33]. Mechanistic analyses of poliovirus and rhinovirus replication traced the Hsp90 requirement to a role in the maturation of the viral capsid proteins [33] (Fig. 1C). The picornavirus capsid consists of 60 copies of each of four subunits, which are generated by the cleavage of the P1 precursor protein by a viral encoded protease. Hsp90, in combination with the p23 cochaperone, was found to interact with P1 and to be required for the cleavage of P1 into the capsid subunits by the viral protease. Interestingly, Hsp90 did not interact with any other viral proteins and its inhibition did not affect the cleavage of virus encoded precursor proteins that are similarly cleaved by the same viral protease. Hence, as protease activity is not diminished with other viral proteins, it is likely that Hsp90 is required for inducing a P1 confirmation that enables recognition and/or cleavage by the protease [33]. Moreover, following cleavage, Hsp90 is no longer associated with the mature, cleaved capsid subunits, suggesting Hsp90 recognizes a state of P1 that is lost following cleavage. Since the Picornaviridae family is the largest viral family and both capsid structure and maturation pathway are conserved within this family, the wealth of sequence and structural information make P1 an interesting model for studying Hsp90-substrate interactions. It is interesting to note, however, that hepatitis A replication is not sensitive to Hsp90 inhibition; this hepatovirus is distinguished from other Picornaviridae in its extremely inefficient translation and replication kinetics, and may accordingly employ alternative strategies for P1 maturation [75].
The reovirus attachment protein σ1 also requires Hsp90 for folding. The σ1 protein forms a homotrimeric complex that decorates the viral capsid and mediates binding to host cell receptors [31, 32]. The folding of the σ1 protein occurs in a stepwise manner: upon synthesis on polysomes the emerging nascent chains begin to trimerize co-translationally, forming a folded, trimeric N-terminal domain; subsequently to completion of translation the C-terminal domain folds and trimerizes post-translationally [76, 77]. Notably, Hsp90 is essential for the post-translational folding step of the C-termini [78]. Accordingly, inhibition of Hsp90 blocks folding of the C-termini but does not affect trimerization of the N-terminal domain. Hsp90 recognizes a conformation that is lost upon folding and does interact with fully assembled σ1 trimers. Strikingly, translation of σ1 leads to Hsp90 phosphorylation, which in turn blocks the Hsp90-σ1 interaction, suggesting a possible novel feedback mechanism to release Hsp90 upon σ1 folding [79].
During vaccinia virus infection, inhibition of Hsp90 by GA or novobiocin reduces viral DNA replication by specifically inhibiting intermediate and late gene expression but not early gene expression [80, 81]. Hsp90 interacts with the 4a core protein of vaccinia virus, implicating Hsp90 in the conformational maturation of the vaccinia capsid. Furthermore, Hsp90 only colocalizes with the viral core protein 4a early during infection but not at later stages, suggesting a transient role for Hsp90 in virion morphogenesis [34]. Since vaccinia virus encodes over 250 proteins, it remains likely that Hsp90 and other chaperones play additional roles in the replication of this virus.
While all the above findings illustrate to a role of Hsp90 in viral capsid folding and assembly, it is also possible that Hsp90 associates with viral capsids at an early entry step such as penetration or uncoating, in which capsids must be transported in the cell and disassembled. This has in fact been suggested for Hepatitis E virus (HEV), where Hsp90 appears important for intracellular transport of the incoming viral particle [82].
6. Cellular factors associated with viral propagation, pathogenesis and the immune system
In addition to a direct interaction with viral proteins that mediates their folding, assembly and activity Hsp90 can also facilitate or modulate infection through regulation of host processes and cellular Hsp90 client proteins. The examples below illustrate the plurality of levels at which Hsp90 regulates viral infection. First, Hsp90 was reported to be present on the surface of some cells [83]. While the actual mechanism by which a cytosolic chaperone reaches the cell surface is not fully clear, plasma membrane associated Hsp90 and Hsp70 have been observed in both stressed and antigen-presenting cells [84]. Such surface exposed Hsp90 is proposed to play a role in the internalization of both dengue virus (DENV) and infectious bursal disease virus (IBDV) [85, 86]. In the case of dengue virus, the cell-surface Hsp90 is proposed to interact with an unknown viral receptor complex [86]. Addition of anti-Hsp90α or Hsp70 antibodies to the extracellular medium inhibits virus infection in monocytes and macrophages but does not effect dengue virus propagation in liver cells, suggesting a cell type specific requirement for plasma membrane localized Hsp90 [85]. Moreover, transient heat shock was shown to increase cell surface expression of Hsp90 and Hsp70 and also increase dengue virus infectivity [87]. Similarly to dengue virus, antibodies against Hsp90α were shown to inhibit the ability of IBDV to infect a cell line of chicken origin [88].
Hsp90 was implicated in supporting the efficient production of human immunodeficiency virus (HIV) proviral DNA by promoting the formation of a Cdk9/cyclin T1 complex, the components of the human positive transcription elongation factor P-TEFβ [89]. This complex phosphorylates cellular RNA polymerase II, resulting in its increased processivity. The HIV accessory protein Tat can then recruit P-TEFβ to the 5′ of HIV RNA and stimulate transcription of HIV proviral DNA. Hsp90, together with the co-chaperone Cdc37, were shown to bind Cdk9 and render it competent for binding cyclin T1. Inhibition of Hsp90 impaired formation of Cdk9/cyclin T1 complexes. While the relevance of this finding to HIV replication is unknown, Hsp90 inhibition by GA and coumermycin A1 was reported to reduce the replication of HIV [89, 90].
Viruses causing chronic infection might take advantage of chaperone machinery to modulate host cell viability to ensure persistence of the viral infection. Since Hsp90 modulates the activity of many signal transduction pathways and immune modulators, Hsp90 inhibitors may help clear such chronic viral infections. For instance, both the glycoprotein K1 and the viral FLIP protein (v-FLIP) of the gamma herpesvirus Kaposi sarcoma-associated herpesvirus (KSHV) activate the NF-kB pathway and block apoptosis to ensure survival of infected host cells [91, 92]. v-FLIP forms an IKK kinase complex with assistance of Hsp90 and Cdc37 [93]. Hsp90, together with other chaperones such as Hsp40, also interacts with K1 and prevents its proteasomal degradation [94]. Accordingly, inhibitors of Hsp90 block the activity of K1 and v-FLIP. As a result, NF-kB activation is impaired and apoptosis of infected cells is increased, helping clear the viral infection. Similar to KSHV, Epstein Bar virus (EBV) and human T-cell leukemia virus (HTLV)-1 also exploit Hsp90 function to promote survival of infected host cells thus prolonging the viral infection [95, 96].
7. Potential of Hsp90 inhibitors as antivirals
Viral infections are amongst the principal causes of human morbidity and mortality worldwide and impose severe economic burdens on society. Despite their importance, few effective antivirals currently exist and many medically and economically important viral infections lack any treatment (see [97] for review). One of the main hurdles to the development of effective antivirals is the rapid acquisition of drug resistance by viruses, which can limit the utility of even the most effective drugs and preclude their long-term use [98]. The high mutation rates, short replication times, large population sizes, and compact genomes enable viruses to rapidly acquire mutations conferring drug resistance, thus circumventing inhibition by antiviral drugs. The ability of viruses to develop drug resistance to antiviral compounds targeting viral proteins is enormous, since the drug target is under the replicative control of the virus and therefore escape mutations are easily generated. In the case of antivirals targeting cellular factors required for viral replication, acquisition of drug resistance has also been observed, although it is more difficult as the virus must either dispense with the affected function or evolve to employ an alternative cellular pathway. The lack of successful antiviral therapies for most viruses create a pressing need for identification of novel antivirals that do not elicit drug resistance.
Inhibition of Hsp90 constitutes a promising antiviral approach. First, as can be inferred from the multiplicity of different viruses requiring Hsp90 for replication, Hsp90 inhibitors are among the broadest-spectrum antivirals identified to date. Currently, Hsp90 inhibitors have been demonstrated to possess antiviral activity in tissue culture against picornaviruses (poliovirus, coxsackievirus, rhinovirus), influenza virus, paramyxoviruses (HPIV2, HPIV3, SV5, SV41), HCV, Ebola virus, vesicular stomatitis virus, La crosse virus, severe acquired respiratory syndrome (SARS), FHV, HIV, vaccinia virus, and herpes viruses (HSV1/2, HCMV, VZV). This property makes Hsp90 inhibitors particularly attractive antivirals for existing viral diseases lacking therapies and for rapid response to newly emerging viral diseases. Secondly, administration of Hsp90 inhibitors to infected animals was shown to reduce the replication of two different viruses, poliovirus and HCV, with little toxicity to the infected host [33, 99]. These experiments highlight the feasibility of using these inhibitors therapeutically in humans. Thirdly, Hsp90 inhibitors were demonstrated to be refractory to development of drug resistance. This was clearly highlighted by poliovirus experiments, as this virus has been demonstrated to gain drug resistance to all antivirals tested to date whether they target viral or host factors. However, when poliovirus was repeatedly treated with Hsp90 inhibitors, no drug resistance was observed despite extensive passaging of the virus in the presence of Hsp90 inhibitors in cultured cells. Similarly, no drug resistance was observed in viruses recovered from Hsp90 inhibitor treated mice. The lack of viral drug resistance to Hsp90 inhibitors suggests such an antiviral approach may be particularly useful for treatment of chronic viral infections and treatment of RNA virus infections for which drug resistance is most frequently observed.
The feasibility of transitioning Hsp90 inhibitors for antiviral use in humans is further supported by the fact that Hsp90 constitutes a highly druggable target. To date, numerous structurally diverse pharmacological inhibitors have been identified. The majority of these inhibit Hsp90 by blocking ATP binding, although inhibitors that block alternate sites on Hsp90 have also been identified. Moreover, Hsp90 activity is inhibited by acetylation [100] and histone deacetylase (HDAC) inhibitors have been reported to block Hsp90 activity [101-103]. The fact that Hsp90 inhibitors possess anti-cancer activity and cancer cells display ∼100 fold hypersensitivity to Hsp90 inhibition relative to normal cells has led to the synthesis and human evaluation of many Hsp90 inhibitors, the most advanced of which are currently undergoing phase III clinical trials [104]. From data available to date, Hsp90 inhibitors appear to be reasonably well tolerated at concentrations sufficient for anticancer activity and show good tissue distribution. Importantly, for an acute viral infection, the course of inhibitor administration would be much shorter than those currently used for cancer, thus potentially lessening side effects observed in the much longer cancer therapies. Similarly, HDAC inhibitors are used in the clinic for cancer treatment. Together, these results show that Hsp90 inhibitors are safe for use in humans and could provide a viable antiviral therapeutic strategy.
Additional important consideration regarding the use of Hsp90 inhibitors as antivirals comes from the role of Hsp90 in inflammation, immunology, cellular antiviral defense pathways, and apoptosis [84, 105]. Hsp90 inhibitors have been shown to reduce inflammation [106-108]. This property may be useful during viral infections where pathology is in part due to inflammatory processes. Additional roles of Hsp90 in folding and activity of intracellular antiviral and inflammatory responses, such as interferon regulatory factor 3 (IRF3), TBK1, and the double-stranded RNA activated protein kinase (PKR) [109, 110], may play important roles in modulating the course of viral infection during treatment with Hsp90 inhibitors.
8. Future Directions and Perspectives
Proper folding is the essential ritual undergone by all proteins to achieve their correct structure and activity. Although it is clear that all viruses depend on the host chaperone machinery to fold their proteins, most of the mechanisms and cellular factors underlying viral protein homeostasis are still to be unveiled. Understanding this fascinating process is likely to reveal many important principles and aspects of chaperone function that are fundamental to not only viral biology but likely relevant to our understanding of normal protein homeostasis. With regards to broad-spectrum therapeutic avenues to combat viral infection, Hsp90 inhibitors hold considerable promise, particularly as new compounds with low toxicity and high specificity emerge. Characterization of specific co-chaperones involved in viral replication may provide additional targets to develop antivirals with fewer pleiotropic side effects. Nonetheless, the success of Hsp90 inhibitors for anticancer therapies, and the fact that many Hsp90 inhibitors are relatively well tolerated, bode well for harnessing them to be used as antivirals. To further support the development of Hsp90 inhibitors as antiviral therapeutics, future efforts should be aimed at careful evaluation of both the effectiveness and toxicity of Hsp90 inhibitors in animal models of infection for those viral infection where good animal models exist (e.g. influenza virus, coxsackievirus, HSV).
Finally, it is important to note that Hsp90 does not work alone, and other chaperones, such as Hsp70, are also conscripted to facilitate viral protein folding and propagation. Hence, comprehensive understanding of chaperone use during viral infection will provide new insight into viral replication mechanisms and potential therapeutics.
Highlights.
Viruses are intracellular pathogens responsible for a vast number of human diseases.
The molecular chaperone Hsp90 is nearly universally required for viral protein replication.
The unique characteristics of viral replication cause viruses to be hypersensitive to pharmacological inhibitors of Hsp90
The near-universal requirement for Hsp90 provides a novel therapeutic avenue for development of broad-spectrum antiviral drugs.
Acknowledgments
We thank Dr. Raul Andino for useful discussions and Dr. Christine Livingston for comments on the manuscript. Work in the Frydman lab is supported by NIH grant GM56433. S.T. acknowledges support of grants-in-aid from the Naito Foundation and the Uehara Memorial Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 2.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 3.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 4.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–637. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. The Journal of biological chemistry. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 6.Schneider C, Sepp-Lorenzino L, Nimmesgern E, Ouerfelli O, Danishefsky S, Rosen N, Hartl FU. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci U S A. 1996;93:14536–14541. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 8.Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Quarterly reviews of biophysics. 2011;44:229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chadli A, Bouhouche I, Sullivan W, Stensgard B, McMahon N, Catelli MG, Toft DO. Dimerization and N-terminal domain proximity underlie the function of the molecular chaperone heat shock protein 90. Proc Natl Acad Sci U S A. 2000;97:12524–12529. doi: 10.1073/pnas.220430297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prodromou C, Panaretou B, Chohan S, Siligardi G, O'Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. The ATPase cycle of Hsp90 drives a molecular ‘;clamp’ via transient dimerization of the N-terminal domains. Embo J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandal AK, Gibney PA, Nillegoda NB, Theodoraki MA, Caplan AJ, Morano KA. Hsp110 chaperones control client fate determination in the hsp70-Hsp90 chaperone system. Mol Biol Cell. 2010;21:1439–1448. doi: 10.1091/mbc.E09-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riggs DL, Cox MB, Cheung-Flynn J, Prapapanich V, Carrigan PE, Smith DF. Functional specificity of co-chaperone interactions with Hsp90 client proteins. Crit Rev Biochem Mol Biol. 2004;39:279–295. doi: 10.1080/10409230490892513. [DOI] [PubMed] [Google Scholar]
- 13.Theodoraki MA, Caplan AJ. Quality control and fate determination of Hsp90 client proteins. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbamcr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearl LH. Hsp90 and Cdc37 -- a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Kimmins S, MacRae TH. Maturation of steroid receptors: an example of functional cooperation among molecular chaperones and their associated proteins. Cell Stress Chaperones. 2000;5:76–86. doi: 10.1379/1466-1268(2000)005<0076:mosrae>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 17.Dickey CA, Koren J, Zhang YJ, Xu YF, Jinwal UK, Birnbaum MJ, Monks B, Sun M, Cheng JQ, Patterson C, Bailey RM, Dunmore J, Soresh S, Leon C, Morgan D, Petrucelli L. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci U S A. 2008;105:3622–3627. doi: 10.1073/pnas.0709180105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol. 2003;35:572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- 19.Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 21.Stemmann O, Neidig A, Kocher T, Wilm M, Lechner J. Hsp90 enables Ctf13p/Skp1p to nucleate the budding yeast kinetochore. Proc Natl Acad Sci U S A. 2002;99:8585–8590. doi: 10.1073/pnas.082223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 23.Peterson LB, Blagg BS. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med Chem. 2009;1:267–283. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–2235. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- 25.Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A. 1996;93:8379–8383. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 27.Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206:149–157. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 28.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–495. [PubMed] [Google Scholar]
- 30.Nagy PD, Wang RY, Pogany J, Hafren A, Makinen K. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology. 2011;411:374–382. doi: 10.1016/j.virol.2010.12.061. [DOI] [PubMed] [Google Scholar]
- 31.Mah DC, Leone G, Jankowski JM, Lee PW. The N-terminal quarter of reovirus cell attachment protein sigma 1 possesses intrinsic virion-anchoring function. Virology. 1990;179:95–103. doi: 10.1016/0042-6822(90)90278-y. [DOI] [PubMed] [Google Scholar]
- 32.Nagata L, Masri SA, Pon RT, Lee PW. Analysis of functional domains on reovirus cell attachment protein sigma 1 using cloned S1 gene deletion mutants. Virology. 1987;160:162–168. doi: 10.1016/0042-6822(87)90056-0. [DOI] [PubMed] [Google Scholar]
- 33.Geller R, Vignuzzi M, Andino R, Frydman J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007;21:195–205. doi: 10.1101/gad.1505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hung JJ, Chung CS, Chang W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J Virol. 2002;76:1379–1390. doi: 10.1128/JVI.76.3.1379-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossmann MG. Constraints on the assembly of spherical virus particles. Virology. 1984;134:1–11. doi: 10.1016/0042-6822(84)90267-8. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS pathogens. 2005;1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vignuzzi M, Stone JK, Andino R. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance and biological implications. Virus research. 2005;107:173–181. doi: 10.1016/j.virusres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowder S, Kirkegaard K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet. 2005;37:701–709. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- 40.Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as wel as for reverse transcription. Nature. 1990;344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- 41.Hu J, Anselmo D. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J Virol. 2000;74:11447–11455. doi: 10.1128/jvi.74.24.11447-11455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J, Toft D, Anselmo D, Wang X. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J Virol. 2002;76:269–279. doi: 10.1128/JVI.76.1.269-279.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu J, Toft DO, Seeger C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. Embo J. 1997;16:59–68. doi: 10.1093/emboj/16.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Grammatikakis N, Hu J. Role of p50/CDC37 in hepadnavirus assembly and replication. The Journal of biological chemistry. 2002;277:24361–24367. doi: 10.1074/jbc.M202198200. [DOI] [PubMed] [Google Scholar]
- 45.Hu J, Flores D, Toft D, Wang X, Nguyen D. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol. 2004;78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu J, Seeger C. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A. 1996;93:1060–1064. doi: 10.1073/pnas.93.3.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. The Journal of biological chemistry. 2002;277:45306–45314. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 48.Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol. 2007;81:1339–1349. doi: 10.1128/JVI.01917-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology. 2008;377:431–439. doi: 10.1016/j.virol.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 50.Burch AD, Weller SK. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol. 2004;78:7175–7185. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burch AD, Weller SK. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol. 2005;79:10740–10749. doi: 10.1128/JVI.79.16.10740-10749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li YH, Tao PZ, Liu YZ, Jiang JD. Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother. 2004;48:867–872. doi: 10.1128/AAC.48.3.867-872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basha W, Kitagawa R, Uhara M, Imazu H, Uechi K, Tanaka J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir Chem Chemother. 2005;16:135–146. doi: 10.1177/095632020501600206. [DOI] [PubMed] [Google Scholar]
- 54.Kyratsous CA, Silverstein SJ. BAG3, a host cochaperone, facilitates varicella-zoster virus replication. J Virol. 2007;81:7491–7503. doi: 10.1128/JVI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nollen EA, Kabakov AE, Brunsting JF, Kanon B, Hohfeld J, Kampinga HH. Modulation of in vivo HSP70 chaperone activity by Hip and Bag-1. The Journal of biological chemistry. 2001;276:4677–4682. doi: 10.1074/jbc.M009745200. [DOI] [PubMed] [Google Scholar]
- 56.Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci. 2008;65:1390–1402. doi: 10.1007/s00018-008-7535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kyratsous CA, Silverstein SJ. The co-chaperone BAG3 regulates Herpes Simplex Virus replication. Proc Natl Acad Sci U S A. 2008;105:20912–20917. doi: 10.1073/pnas.0810656105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kampmueller KM, Miller DJ. The cellular chaperone heat shock protein 90 facilitates Flock House virus RNA replication in Drosophila cells. J Virol. 2005;79:6827–6837. doi: 10.1128/JVI.79.11.6827-6837.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castorena KM, Weeks SA, Stapleford KA, Cadwallader AM, Miller DJ. A functional heat shock protein 90 chaperone is essential for efficient flock house virus RNA polymerase synthesis in Drosophila cells. J Virol. 2007;81:8412–8420. doi: 10.1128/JVI.00189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Connor JH, McKenzie MO, Parks GD, Lyles DS. Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology. 2007;362:109–119. doi: 10.1016/j.virol.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crevel G, Bates H, Huikeshoven H, Cotterill S. The Drosophila Dpit47 protein is a nuclear Hsp90 co-chaperone that interacts with DNA polymerase alpha. J Cell Sci. 2001;114:2015–2025. doi: 10.1242/jcs.114.11.2015. [DOI] [PubMed] [Google Scholar]
- 62.Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M, Trager JB, Morin GB, Toft DO, Shay JW, Wright WE, White MA. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13:817–826. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyata Y, Yahara I. p53-independent association between SV40 large T antigen and the major cytosolic heat shock protein, HSP90. Oncogene. 2000;19:1477–1484. doi: 10.1038/sj.onc.1203475. [DOI] [PubMed] [Google Scholar]
- 64.Sawai ET, Butel JS. Association of a cellular heat shock protein with simian virus 40 large T antigen in transformed cells. J Virol. 1989;63:3961–3973. doi: 10.1128/jvi.63.9.3961-3973.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gannon JV, Lane DP. p53 and DNA polymerase alpha compete for binding to SV40 T antigen. Nature. 1987;329:456–458. doi: 10.1038/329456a0. [DOI] [PubMed] [Google Scholar]
- 66.Dutta D, Chattopadhyay S, Bagchi P, Halder UC, Nandi S, Mukherjee A, Kobayashi N, Taniguchi K, Chawla-Sarkar M. Active Participation of Cellular Chaperone Hsp90 in Regulating the Function of Rotavirus Nonstructural Protein 3 (NSP3) The Journal of biological chemistry. 2011;286:20065–20077. doi: 10.1074/jbc.M111.231878. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Harb M, Becker MM, Vitour D, Baron CH, Vende P, Brown SC, Bolte S, Arold ST, Poncet D. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J Virol. 2008;82:11283–11293. doi: 10.1128/JVI.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michel YM, Poncet D, Piron M, Kean KM, Borman AM. Cap-Poly(A) synergy in mammalian cell-free extracts. Investigation of the requirements for poly(A)-mediated stimulation of translation initiation. The Journal of biological chemistry. 2000;275:32268–32276. doi: 10.1074/jbc.M004304200. [DOI] [PubMed] [Google Scholar]
- 69.Vende P, Piron M, Castagne N, Poncet D. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J Virol. 2000;74:7064–7071. doi: 10.1128/jvi.74.15.7064-7071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutta D, Bagchi P, Chatterjee A, Nayak MK, Mukherjee A, Chattopadhyay S, Nagashima S, Kobayashi N, Komoto S, Taniguchi K, Chawla-Sarkar M. The molecular chaperone heat shock protein-90 positively regulates rotavirus infection. Virology. 2009;391:325–333. doi: 10.1016/j.virol.2009.06.044. [DOI] [PubMed] [Google Scholar]
- 71.Ujino S, Yamaguchi S, Shimotohno K, Takaku H. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus nonstructural protein NS3. The Journal of biological chemistry. 2009;284:6841–6846. doi: 10.1074/jbc.M806452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci U S A. 2001;98:13931–13935. doi: 10.1073/pnas.241510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. Embo J. 2006;25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taguwa S, Kambara H, Omori H, Tani H, Abe T, Mori Y, Suzuki T, Yoshimori T, Moriishi K, Matsuura Y. Cochaperone activity of human butyrate-induced transcript 1 facilitates hepatitis C virus replication through an Hsp90-dependent pathway. J Virol. 2009;83:10427–10436. doi: 10.1128/JVI.01035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aragones L, Guix S, Ribes E, Bosch A, Pinto RM. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS pathogens. 2010;6:e1000797. doi: 10.1371/journal.ppat.1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leone G, Coffey MC, Gilmore R, Duncan R, Maybaum L, Lee PW. C-terminal trimerization, but not N-terminal trimerization, of the reovirus cell attachment protein Is a posttranslational and Hsp70/ATP-dependent process. The Journal of biological chemistry. 1996;271:8466–8471. doi: 10.1074/jbc.271.14.8466. [DOI] [PubMed] [Google Scholar]
- 77.Leone G, Maybaum L, Lee PW. The reovirus cell attachment protein possesses two independently active trimerization domains: basis of dominant negative effects. Cell. 1992;71:479–488. doi: 10.1016/0092-8674(92)90516-f. [DOI] [PubMed] [Google Scholar]
- 78.Gilmore R, Coffey MC, Lee PW. Active participation of Hsp90 in the biogenesis of the trimeric reovirus cell attachment protein sigma1. The Journal of biological chemistry. 1998;273:15227–15233. doi: 10.1074/jbc.273.24.15227. [DOI] [PubMed] [Google Scholar]
- 79.Zhao YG, Gilmore R, Leone G, Coffey MC, Weber B, Lee PW. Hsp90 phosphorylation is linked to its chaperoning function. Assembly of the reovirus cell attachment protein. The Journal of biological chemistry. 2001;276:32822–32827. doi: 10.1074/jbc.M105562200. [DOI] [PubMed] [Google Scholar]
- 80.Sekiguchi J, Shuman S. Novobiocin inhibits vaccinia virus replication by blocking virus assembly. Virology. 1997;235:129–137. doi: 10.1006/viro.1997.8684. [DOI] [PubMed] [Google Scholar]
- 81.Sekiguchi J, Stivers JT, Mildvan AS, Shuman S. Mechanism of inhibition of vaccinia DNA topoisomerase by novobiocin and coumermycin. The Journal of biological chemistry. 1996;271:2313–2322. doi: 10.1074/jbc.271.4.2313. [DOI] [PubMed] [Google Scholar]
- 82.Zheng ZZ, Miao J, Zhao M, Tang M, Yeo AE, Yu H, Zhang J, Xia NS. Role of heat-shock protein 90 in hepatitis E virus capsid trafficking. J Gen Virol. 2010;91:1728–1736. doi: 10.1099/vir.0.019323-0. [DOI] [PubMed] [Google Scholar]
- 83.Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007;98:1536–1539. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009;85:905–910. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 85.Cabrera-Hernandez A, Thepparit C, Suksanpaisan L, Smith DR. Dengue virus entry into liver (HepG2) cells is independent of hsp90 and hsp70. J Med Virol. 2007;79:386–392. doi: 10.1002/jmv.20786. [DOI] [PubMed] [Google Scholar]
- 86.Reyes-Del Valle J, Chavez-Salinas S, Medina F, Del Angel RM. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. 2005;79:4557–4567. doi: 10.1128/JVI.79.8.4557-4567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chavez-Salinas S, Ceballos-Olvera I, Reyes-Del Valle J, Medina F, Del Angel RM. Heat shock effect upon dengue virus replication into U937 cells. Virus research. 2008;138:111–118. doi: 10.1016/j.virusres.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 88.Lin TW, Lo CW, Lai SY, Fan RJ, Lo CJ, Chou YM, Thiruvengadam R, Wang AH, Wang MY. Chicken heat shock protein 90 is a component of the putative cellular receptor complex of infectious bursal disease virus. J Virol. 2007;81:8730–8741. doi: 10.1128/JVI.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O'Keeffe B, Fong Y, Chen D, Zhou S, Zhou Q. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. The Journal of biological chemistry. 2000;275:279–287. doi: 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- 90.Vozzolo L, Loh B, Gane PJ, Tribak M, Zhou L, Anderson I, Nyakatura E, Jenner RG, Selwood D, Fassati A. Gyrase B inhibitor impairs HIV-1 replication by targeting Hsp90 and the capsid protein. The Journal of biological chemistry. 2010;285:39314–39328. doi: 10.1074/jbc.M110.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 92.Tomlinson CC, Damania B. The K1 protein of Kaposi's sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–1927. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;116:3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 94.Wen KW, Damania B. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene. 2010;29:3532–3544. doi: 10.1038/onc.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kawakami H, Tomita M, Okudaira T, Ishikawa C, Matsuda T, Tanaka Y, Nakazato T, Taira N, Ohshiro K, Mori N. Inhibition of heat shock protein-90 modulates multiple functions required for survival of human T-cell leukemia virus type I-infected T-cell lines and adult T-cell leukemia cells. Int J Cancer. 2007;120:1811–1820. doi: 10.1002/ijc.22403. [DOI] [PubMed] [Google Scholar]
- 96.Jeon YK, Park CH, Kim KY, Li YC, Kim J, Kim YA, Paik JH, Park BK, Kim CW, Kim YN. The heat-shock protein 90 inhibitor, geldanamycin, induces apoptotic cell death in Epstein-Barr virus-positive NK/T-cell lymphoma by Akt down-regulation. J Pathol. 2007;213:170–179. doi: 10.1002/path.2219. [DOI] [PubMed] [Google Scholar]
- 97.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.zur Wiesch PA, Kouyos R, Engelstadter J, Regoes RR, Bonhoeffer S. Population biological principles of drug-resistance evolution in infectious diseases. Lancet Infect Dis. 2011;11:236–247. doi: 10.1016/S1473-3099(10)70264-4. [DOI] [PubMed] [Google Scholar]
- 99.Nakagawa S, Umehara T, Matsuda C, Kuge S, Sudoh M, Kohara M. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun. 2007;353:882–888. doi: 10.1016/j.bbrc.2006.12.117. [DOI] [PubMed] [Google Scholar]
- 100.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 101.Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, Wang HG, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther. 2003;2:971–984. [PubMed] [Google Scholar]
- 102.Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–5135. [PubMed] [Google Scholar]
- 103.Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 104.Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem. 2009;9:1479–1492. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Joly AL, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun. 2010;2:238–247. doi: 10.1159/000296508. [DOI] [PubMed] [Google Scholar]
- 106.Dello Russo C, Polak PE, Mercado PR, Spagnolo A, Sharp A, Murphy P, Kamal A, Burrows FJ, Fritz LC, Feinstein DL. The heat-shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin suppresses glial inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J Neurochem. 2006;99:1351–1362. doi: 10.1111/j.1471-4159.2006.04221.x. [DOI] [PubMed] [Google Scholar]
- 107.Kim JM, Lee DH, Kim JS, Lee JY, Park HG, Kim YJ, Oh YK, Jung HC, Kim SI. 5,7-dihydroxy-3,4,6-trimethoxyflavone inhibits the inflammatory effects induced by Bacteroides fragilis enterotoxin via dissociating the complex of heat shock protein 90 and I kappaB alpha and I kappaB kinase-gamma in intestinal epithelial cell culture. Clin Exp Immunol. 2009;155:541–551. doi: 10.1111/j.1365-2249.2008.03849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rice JW, Veal JM, Fadden RP, Barabasz AF, Partridge JM, Barta TE, Dubois LG, Huang KH, Mabbett SR, Silinski MA, Steed PM, Hall SE. Small molecule inhibitors of Hsp90 potently affect inflammatory disease pathways and exhibit activity in models of rheumatoid arthritis. Arthritis Rheum. 2008;58:3765–3775. doi: 10.1002/art.24047. [DOI] [PubMed] [Google Scholar]
- 109.Donze O, Abbas-Terki T, Picard D. The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. Embo J. 2001;20:3771–3780. doi: 10.1093/emboj/20.14.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang K, Shi H, Qi R, Sun S, Tang Y, Zhang B, Wang C. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol Biol Cell. 2006;17:1461–1471. doi: 10.1091/mbc.E05-09-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kotsiopriftis M, Tanner JE, Alfieri C. Heat shock protein 90 expression in Epstein-Barr virus-infected B cells promotes gammadelta T-cell proliferation in vitro. J Virol. 2005;79:7255–7261. doi: 10.1128/JVI.79.11.7255-7261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun X, Barlow EA, Ma S, Hagemeier SR, Duellman SJ, Burgess RR, Tellam J, Khanna R, Kenney SC. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:3146–3151. doi: 10.1073/pnas.0910717107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jindal S, Young RA. Vaccinia virus infection induces a stress response that leads to association of Hsp70 with viral proteins. J Virol. 1992;66:5357–5362. doi: 10.1128/jvi.66.9.5357-5362.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Okamoto T, Omori H, Kaname Y, Abe T, Nishimura Y, Suzuki T, Miyamura T, Yoshimori T, Moriishi K, Matsuura Y. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J Virol. 2008;82:3480–3489. doi: 10.1128/JVI.02253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taguwa S, Okamoto T, Abe T, Mori Y, Suzuki T, Moriishi K, Matsuura Y. Human butyrate-induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J Virol. 2008;82:2631–2641. doi: 10.1128/JVI.02153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Basta S, Stoessel R, Basler M, van den Broek M, Groettrup M. Cross-presentation of the long-lived lymphocytic choriomeningitis virus nucleoprotein does not require neosynthesis and is enhanced via heat shock proteins. J Immunol. 2005;175:796–805. doi: 10.4049/jimmunol.175.2.796. [DOI] [PubMed] [Google Scholar]
- 117.Weeks SA, Miller DJ. The heat shock protein 70 cochaperone YDJ1 is required for efficient membrane-specific flock house virus RNA replication complex assembly and function in Saccharomyces cerevisiae. J Virol. 2008;82:2004–2012. doi: 10.1128/JVI.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu C, Erlichman C, McDonald CJ, Ingle JN, Zollman P, Iankov I, Russell SJ, Galanis E. Heat shock protein inhibitors increase the efficacy of measles virotherapy. Gene Ther. 2008;15:1024–1034. doi: 10.1038/gt.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Smith DR, McCarthy S, Chrovian A, Olinger G, Stossel A, Geisbert TW, Hensley LE, Connor JH. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res. 2010;87:187–194. doi: 10.1016/j.antiviral.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell. 2004;15:2347–2360. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beck J, Nassal M. Efficient Hsp90-independent in vitro activation by Hsc70 and Hsp40 of duck hepatitis B virus reverse transcriptase, an assumed Hsp90 client protein. The Journal of biological chemistry. 2003;278:36128–36138. doi: 10.1074/jbc.M301069200. [DOI] [PubMed] [Google Scholar]