

Abstract
Soil, water and sediment samples collected from in and around Jaduguda, Bagjata and Turamdih mines were analyzed for physicochemical parameters and cultured, and yet to be cultured microbial diversity. Culturable fraction of microbial community measured as Colony Forming Unit (CFU) on R2A medium revealed microbes between 104 and 109 CFU/g sample. Community DNA was extracted from all the samples; 16S rRNA gene amplified, cloned and subject to Amplified Ribosomal DNA Restriction Analysis. Clones representing each OTU were selected and sequenced. Sequence analyses revealed that non-contaminated samples were mostly represented by Acidobacteria, Bacteroidetes, Firmicutes and Proteobacteria (β-, γ-, and/or δ-subdivisions) along with less frequent phyla Nitrospira, Deferribacteres, Chloroflexi. In contrast, samples obtained from highly contaminated samples showed distinct abundance of β-,γ- and α-Proteobacteria along with Acidobacteria,Bacteroidetes and members of Firmicutes, Chloroflexi, Candidate division, Planctomycete, Cyanobacteria and Actinobacteria as minor groups. Our data represented the baseline information on bacterial community composition within non-contaminated samples which could potentially be useful for assessing the impact of metal and radionuclides contamination due to uranium mine activities.
Keywords: Uranium mines, 16S rRNA gene, Bacterial community, ARDRA
Introduction
Dispersion and migration of uranium (U) and other toxic metals and radionuclides from uranium mines due to mining operations and waste piling is a serious environmental concern to all uranium producing states (Foster et al. 2008; Islam et al. 2011). Once released into the environment, fate and toxicity of these metallic contaminants are strongly regulated by abiotic and biotic components such as minerals and bacteria (Lloyd and Renshaw 2005). On the other hand, the toxic and bioavailable forms of these metals and radionuclides often affect adversely the diversity and function of autochthonous microorganisms of neighboring habitats that bequeath ecological sustains by maintaining biogeochemical cycles (Torsvik et al. 2002; Islam et al. 2011). Diversity and distribution of microbes is often site-specific, influenced by composition of geochemical matrix of microhabitats. Perturbation to this due to environmental contamination could cause change in inhabitant microbial community structure, diversity and function (Herrera et al. 2007; Desai et al. 2009). In order to gauge the impact of environmental contamination, microbial community composition and diversity are increasingly being considered as highly sensitive ecological parameters (Wang et al. 2007; Desai et al. 2009; Islam and Sar 2011b). Therefore, studies on diversity and composition of indigenous microbial communities within sites having high risk of contamination may serve as baseline information to assess the subsequent impact of contamination.
In the recent years, advances in culture-based and -independent molecular approaches have elucidated microbial diversity and function in sites contaminated with radioactive wastes or wastes generated from uranium, copper and zinc mines. These studies have revealed the presence of diverse (phylogenetically and metabolically) populations of viable and active microorganisms organized in complex communities (Radeva and Selenska-Pobell 2005; Akob et al. 2007; Rastogi et al. 2010). Particularly, the abundance of Acidithiobacillus, Pseudomonas, Acinetobacter, Nitrosomonas was observed in various U mine waste sites in Germany (Radeva and Selenska-Pobell 2005), while high abundance of Proteobacteria (Sphingomonas, Acidovorax, Acinetobacter and Ralstonia) was reported from U-contaminated radioactive waste (Akob et al. 2007). Recently, Rastogi et al. (2010) has reported abundance of Proteobacteria, Acidobacteria and Bacteriodetes in a U mine impacted site in USA. Studies conducted in our laboratory have revealed microbial communities within two U mines (Jaduguda and Banduhurang) along with potential of inhabitant bacteria in U and other metal resistance and sequestration and impact of U ore contamination on soil microbial diversity (Sar et al. 2007; Choudhary and Sar 2011; Islam et al. 2011; Islam and Sar 2011a, b). With vast genetic and metabolic diversity, these microorganisms were found to interact with metals and radionuclides directly or indirectly by redox transfer, biosorption, bioaccumulation or bioprecipitation affecting their environmental mobility and toxicity (Suzuki and Banfield 2000; Tabak et al. 2005; Nedelkova et al. 2007). Considering the significance of geomicrobiology of contaminated sites, it is therefore imperative to decipher phylogenetic diversity of indigenous microbial populations in sites having high risk of contamination or already contaminated to illuminate community resilience, their potential role in affecting metal biogeochemistry and in designing appropriate bioremediation strategies (Tabak et al. 2005; Akob et al. 2007).
Uranium mines at Jaduguda, Bagjata, and Turamdih are all located in highly mineral-rich areas of East Singhbhum district, Jharkhand, India. Jaduguda mine is the oldest U mine in India operating since 1968, while the other two mines are commissioned relatively recently (2002–2003) (Gupta and Sarangi 2005). Present work was undertaken to ascertain diversity and structure of bacterial communities within sites in and around these U mines as a means to obtain the baseline data on microbial diversity. Sites contaminated with U-ores/-mine wastes as well as sites located away from such contaminants (ores and wastes; especially, agriculture fields, streams, etc. located outside the mine areas) were considered. Samples collected from all these sites were analyzed for their physicochemical properties (pH, conductivity, total organic carbon (TOC), total nitrogen (TN), total phosphorus (TP) and heavy metal content). Culturable bacterial counts were recorded and finally the compositions of microbial communities were determined at molecular level with the determination of diversity indices and identification of dominant ribotypes.
Materials and methods
Collection of samples
Soil, water and sediment samples were collected from various locations of proposed and existing uranium mines of at Jaduguda (N 22°39′, E 86°20′), Bagjata (N 22°28′, E 86°29′), and Turamdih (N 22°43′, E 86°11′). Samples B209, B210, B211, B214, B218 and T112, T219, T221, T222 were collected from agriculture field; pond and small streams located outside the boundary of Bagjata and Turamdih mines, respectively, and are designated as non-contaminated samples. Samples CW1, CW2 and CW3 were obtained from sites within the mines and contaminated with mine wastes and ores and are designated as contaminated samples. All samples were collected aseptically and stored immediately in ice till further analysis.
Geomicrobiological analysis
Physiochemical parameters including pH, conductivity/salinity of the samples were measured by an Orion star™ series meter (Thermo Electron Corporation) following the procedure as described elsewhere (Islam and Sar 2011a). Heavy metals and actinide elements were estimated by ICP-MS (Varian) and/or AAS (Perkin-Elmer). Culturable microbial populations were enumerated by CFU counts using R2A medium. Measured quantities of samples were dispersed in sterile saline (0.9%), shake (1 h), diluted and plated in triplicate on R2A medium. The composition of R2A medium was (g/lit): yeast extract, 0.5; peptone, 0.5; casamino acid, 0.5; dextrose, 0.5; soluble starch, 0.5; sodium pyruvate, 0.3; dipotassium phosphate, 0.3; magnesium sulphate, 0.05; (Fredrickson et al. 2004). Plates were incubated at 30 °C in dark and colonies were counted after 1 week.
Extraction of community metagenomes, PCR amplification of 16S rRNA genes, cloning and clone library analysis
Community metagenome from each sample was extracted using Power soil™ DNA kit (MO BIO laboratories, Inc.). 16S rRNA genes from each extracted metagenomes were PCR amplified using universal and bacteria-specific primers (27F and 1492R, respectively) (Islam et al. 2011). Amplified products were purified using gel-purification kit (Qiagen), cloned into pGEM-T easy vector (Promega) and transformed into E.coli JM109 following manufacturer’s instructions. For each sample, positive clones were selected for clone library construction. Detailed description of methodology was same as described previously (Islam and Sar 2011a).
Amplified ribosomal DNA restriction analysis (ARDRA)
From each library 50–100 clones were used for reamplification of cloned 16S rRNA gene by colony PCR with vector-specific primers. Colony PCR products were purified and digested with restriction enzymes in separate reactions. Portion (10 μl) of the amplification products were digested at 37 °C for 3 h with the restriction endonuclease RsaI and HaeIII. Digestion products (10 μl) were run on 2.5% agarose gel for 3 h and band patterns were compared visually.
Phylogenetic analysis 16S rRNA gene
All positive 16S rRNA gene clones were grouped according to their ARDRA pattern and each group was referred as an operational taxonomic unit (OTU). About first 600–700 nucleotides of representative clones from each dominant OTU were sequenced. All sequences were examined as potential chimeras using the CHIMERA_CHECK program at the Ribosomal Database Project II (RDP-II) with default settings. Both BLAST program of NCBI database and Hierarchy Browser from Ribosomal Database Project II (RDP-II) were used to find out nearly identical sequences for the 16S rRNA gene sequences determined. Phylogenetic trees were constructed using the Neighbor-joining method with Jukes-Cantor distance correction in MEGA software version 4 using selected sequences (>500 nucleotides and positive polarity) (Tamura et al. 2007).
Statistical analysis
Based on ARDRA profiles, Shannon’s equitability and diversity indices were calculated for each sample (Shannon 1948).
Nucleotide sequence accession numbers
Nucleotide sequences obtained in the present study were deposited in GenBank database under the accession numbers HM469536-HM469615 and HQ909283-HQ909337.
Results and discussion
Physicochemical and microbiological analyses
All samples were analyzed for their physicochemical and microbiological properties (Figs. 1, 2). As evident from the Fig. 1, samples were mostly having near neutral pH (except a few, e.g., B209, B210, T112, T222 with relatively lower pH and CW2 having an alkaline pH). Conductivity of the contaminated samples showed considerably higher values (955–4,340 μs/cm) than that of non-contaminated samples (10–244) (data not shown). It was observed that overall nutrient (TOC, TN and TP) content in contaminated samples, particularly in CW1 and CW3, was relatively higher than other contaminated and even non-contaminated samples. Microbiological counts as obtained by CFU counts were higher in few samples like B218, T221, CW1 and CW3. Heavy metal content as analyzed in non-contaminated samples indicated that for the elements tested the concentrations were within the limits for background concentrations of trace elements estimated in non-anthropogenic soils (Burt et al. 2003). Level of Th was found to be within a relatively narrow range of 3–17 mg/kg across all the samples tested. Concentrations of metals like Co, Ni and Zn were considerably higher in contaminated samples (over the non-contaminated counterparts) exceeding the values reported for non-anthropogenic soils or matches well with those reported for anthropogenically contaminated soils (Burt et al. 2003). Uranium, though present in several non-contaminated samples as well, was present at elevated level in two contaminated samples (CW1 and CW2) (Fig. 2). Noticeably, in all three non-contaminated samples simultaneous presence of three or more metals (including U) at higher concentrations was observed. Presence of U and other metals in non-contaminated samples could be explained considering the fact that this whole region is highly mineral-rich and natural weathering may easily enrich the soil and other parts with metals (Sarangi and Singh 2006).
Fig. 1.

Geomicrobial properties of the samples
Fig. 2.

Analysis of heavy metals and radionuclides (U and Th) content within the samples
16S rRNA gene clone library analysis and bacterial diversity indices
For analyzing bacterial diversity nearly 50 or 100 clones were used to construct individual clone library for each sample. Based on ARDRA profiles, statistical analysis was followed to determine the bacterial diversity within these samples. Interestingly, all three contaminated samples showed relatively higher Shannon diversity indices (H) but the equitability value (EH) varied considerably among the samples [B209 (2.88, 0.71), B210 (3.0, 0.93), B211 (2.77, 0.89), B214 (2.48, 0.63), B218 (2.99, 0.94), T112 (3.22, 0.95), T219 (3.52, 0.97), T221 (2.6, 0.70), T222 (2.9, 0.95), CW1 (3.65, 0.92), CW2 (3.51, 0.89) and CW3 (3.69, 0.92)]. Based on diversity result, we inferred that high nutrient content of the samples possibly make the heavy metal contamination as less stressful event as the inhabitant bacterial members might be nutritionally well supported to withstand metal toxicity. Nevertheless, near neutral pH may allow formation of relatively insoluble metal complexes for most cations thereby reducing their availability as well.
Sequence analysis and affiliation of major ribotypes
Compositions of bacterial communities within the samples were ascertained by analyzing 16S rRNA gene sequences of major ribotypes from each library (Figs. 3a, b and 4). Our analysis revealed that samples from non-contaminated sites were mostly represented by Acidobacteria, Bacteroidetes, Firmicutes and Proteobacteria (β-, γ-, and/or δ-subdivisions) along with less frequent Nitrospira, Deferribacteres, Chloroflexi in one or few samples. Among the samples collected from Bagjata, bacterial communities in B209 (agriculture field) and B211 (river sediment) showed representatives of Bacteroidetes, Acidobacteria and Firmicutes. Presence of β-, γ- and δ-Proteobacteria in B211 was noticeable. Sample B210 (pond sediment) comprised Bacteroidetes, Gemmmatimonadetes and Candidate division along with γ-Proteobacteria. Although members of β- and γ-Proteobacteria were found in B 214 (garden soil) and B218 (river sediment), the absence of all three major phyla like Acidobacteria, Bacteroidetes and Firmicutes was notable. Additionally, phyla Chloroflexi and Deferribacteres were present in high percentage in B214, while Cyanobacteria and Chloroflexi were present in B218. All the samples from Turamdih showed the presence of Acidobacteria as a major group along with Bacteroidetes/Firmicutes/Cyanobacteria or Gemmmatimonadetes in one or more samples. Members of Proteobacteria were not so abundant in these samples. Particularly, γ-Proteobacteria was not found, although the presence of either β- or δ-Proteobacteria was detected in few samples.
Fig. 3.

Distribution of different bacterial groups within (a) non-contaminated and (b) contaminated samples
Fig. 4.

Relative abundance of bacterial phyla detected within the samples
Samples obtained from highly contaminated locations of Jaduguda mine showed distinct abundance of β-Proteobacteria, along with γ- and α-Proteobacteria, Acidobacteria,Bacteroidetes and Firmicutes. Members of Actinobacteria, Chloroflexi, Planctomycetes, Cyanobacteria and Gemmmatimonadetes were detected as relatively minor groups. Sample CW1 showed diverse assemblage of bacterial populations well represented by Proteobacteria (β- > γ- > δ- > α-subdivisions) > Acidobacteria > Gemmmatimodadetes > unclassified bacteria > Bacteroidetes. The observed community structure within this sample corroborates well with its relatively higher organic carbon, nitrogen and phosphorous content and CFU counts suggesting that in spite of higher metal contamination bacterial flora can flourish very well possibly by developing appropriate homeostatic mechanisms (Akob et al. 2008).
Phylogenetic analysis of microbial groups
Sequences representing ARDRA OTUs were analyzed to ascertain their phylogenetic lineage with similar taxa/groups from diverse ecological habitats and with environmentally relevant metabolic functions (Table 1; Figs. 5, 6, and 7).
Table 1.
Details of 16S rRNA gene sequences retrieved in this study and used for phylogenetic analysis
| Sl no. | Accession number | Clone id | Phylum | Present in |
|---|---|---|---|---|
| 1 | HM469555 | T112-61C1 | Acidobacterium | Fig. 5 |
| 2 | HM469561 | B209-3B2 | Bacteroidetes | Fig. 7 |
| 3 | HM469565 | B209-18B5 | Nitrospira | Fig. 7 |
| 4 | HM469566 | B209-3B6 | Bacteroidetes | Fig. 7 |
| 5 | HM469560 | B209-8B1 | Firmicutes | Fig. 7 |
| 6 | HM469567 | B209-33B8 | Acidobacteria | Fig. 5 |
| 7 | HM469568 | B209-35B9 | Acidobacteria | Fig. 5 |
| 8 | HM469570 | B210-6S4 | Acidobacteria | Fig. 5 |
| 9 | HM469571 | B210-7S5 | Acidobacteria | Fig. 5 |
| 10 | HM469573 | B210-14S1 | γ-Proteobacteria | Fig. 6 |
| 11 | HM469574 | B210-44S6 | Acidobacteria | Fig. 5 |
| 12 | HM469575 | B210-1S8 | Acidobacteria | Fig. 5 |
| 13 | HM469578 | B211-9R5 | δ-Proteobacteria | Fig. 6 |
| 14 | HM469579 | B211-39R2 | Bacteroidetes | Fig. 7 |
| 15 | HM469580 | B211-27R7 | Firmicutes | Fig. 7 |
| 16 | HM469581 | B211-10R1 | β-Proteobacteria | Fig. 6 |
| 17 | HM469584 | B214-22N1 | Chloroflexi | Fig. 7 |
| 18 | HM469585 | B214-3N2 | β-Proteobacteria | Fig. 6 |
| 19 | HM469586 | B214-5N3 | Chloroflexi | Fig. 7 |
| 20 | HM469591 | B218-31E1 | Cyanobacteria | Fig. 7 |
| 21 | HM469594 | B218-6E4 | Chloroflexi | Fig. 7 |
| 22 | HM469597 | B218-10 | δ-Proteobacteria | Fig. 6 |
| 23 | HM469592 | B218-15E2 | γ-Proteobacteria | Fig. 6 |
| 24 | HM469593 | B218-2E3 | β-Proteobacteria | Fig. 6 |
| 25 | HM469596 | B218-35E7 | β-Proteobacteria | Fig. 6 |
| 26 | HM469598 | T219-15D1 | Cyanobacteria | Fig. 7 |
| 27 | HM469600 | T219-36D3 | Bacteroidetes | Fig. 7 |
| 28 | HM469601 | T219-04D8 | Bacteroidetes | Fig. 7 |
| 29 | HM469602 | T219-27D5 | Acidobacteria | Fig. 5 |
| 30 | HM469603 | T219-31D4 | Cyanobacteria | Fig. 7 |
| 31 | HM469604 | T219-03D7 | Cyanobacteria | Fig. 7 |
| 32 | HM469610 | T221-14M5 | Acidobacteria | Fig. 5 |
| 33 | HM469612 | T222-8G2 | Acidobacteria | Fig. 5 |
| 34 | HM469613 | T222-5G3 | Acidobacteria | Fig. 5 |
| 35 | HM469614 | T222-18G5 | Acidobacteria | Fig. 5 |
| 36 | HM469538 | CW1-8A4 | β-Proteobacteria | Fig. 6 |
| 37 | HM469540 | CW1-60A8 | Acidobacteria | Fig. 5 |
| 38 | HM469536 | CW1-14A2 | Gemmatimonadetes | Fig. 7 |
| 39 | HM469543 | CW1-3A11 | Acidobacteria | Fig. 5 |
| 40 | HM469550 | CW1-77A19 | Acidobacteria | Fig. 5 |
| 41 | HM469541 | CW1-36A9 | α-Proteobacteria | Fig. 6 |
| 42 | HM469545 | CW1-22A13 | Acidobacteria | Fig. 5 |
| 43 | HM469548 | CW1-56A17 | γ-Proteobacteria | Fig. 6 |
| 44 | HM469551 | CW1-78A20 | β-Proteobacteria | Fig. 6 |
| 45 | HM469549 | CW1-57A18 | Bacteroidetes | Fig. 7 |
| 46 | HQ909289 | CW2-36 | Cyanobacteria | Fig. 7 |
| 47 | HQ909290 | CW2-44 | β-Proteobacteria | Fig. 6 |
| 48 | HQ909293 | CW2-77 | α-Proteobacteria | Fig. 6 |
| 49 | HQ909288 | CW2-35 | Cyanobacteria | Fig. 7 |
| 50 | HQ909294 | CW2-96 | Cyanobacteria | Fig. 7 |
| 51 | HQ909295 | CW2-97 | Actinobacteria | Fig. 7 |
| 52 | HQ909296 | CW2-128 | γ-Proteobacteria | Fig. 6 |
| 53 | HQ909324 | CW3-166 | β-Proteobacteria | Fig. 6 |
| 54 | HQ909306 | CW3-12 | β-Proteobacteria | Fig. 6 |
| 55 | HQ909307 | CW3-16 | β-Proteobacteria | Fig. 6 |
| 56 | HQ909309 | CW3-25 | β-Proteobacteria | Fig. 6 |
| 57 | HQ909326 | CW3-145 | Acidobacteria | Fig. 5 |
| 58 | HQ909316 | CW3-130 | α-Proteobacteria | Fig. 6 |
| 59 | HQ909319 | CW3-144 | Firmicutes | Fig. 7 |
| 60 | HQ909317 | CW3-133 | α-Proteobacteria | Fig. 6 |
| 61 | HQ909318 | CW3-136 | β-Proteobacteria | Fig. 6 |
| 62 | HQ909327 | CW3-20 | α-Proteobacteria | Fig. 6 |
| 63 | HQ909330 | CW3-167 | β-Proteobacteria | Fig. 6 |
| 64 | HQ909311 | CW3-138 | Firmicutes | Fig. 7 |
| 65 | HQ909308 | CW3-88 | β-Proteobacteria | Fig. 6 |
| 66 | HQ909312 | CW3-33 | α-Proteobacteria | Fig. 6 |
| 67 | HQ909314 | CW3-52 | β-Proteobacteria | Fig. 6 |
| 68 | HQ909320 | CW3-147 | α-Proteobacteria | Fig. 6 |
| 69 | HQ909322 | CW3-161 | β-Proteobacteria | Fig. 6 |
| 70 | HQ909332 | CW3-142 | Acidobacterium | Fig. 5 |
| 71 | HQ909337 | CW3-45 | γ-Proteobacteria | Fig. 6 |
| 72 | HQ909335 | CW3-11 | α-Proteobacteria | Fig. 6 |
Fig. 5.
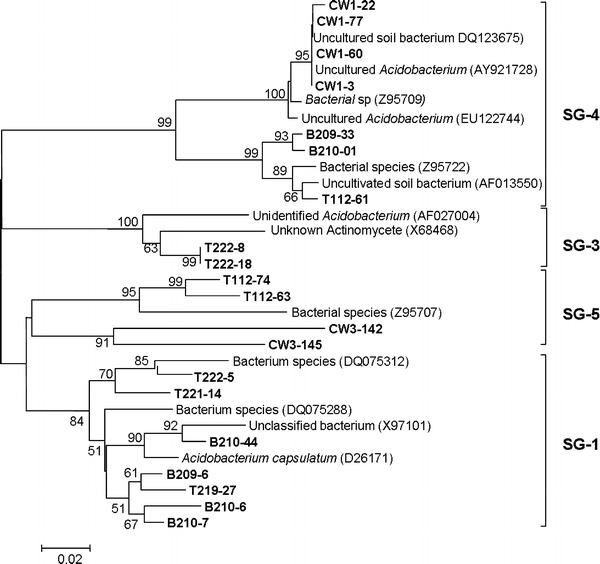
Phylogenetic dendrogram of the Acidobacterium division based on Neighbor-joining analysis. Subdivisions (see the text) are indicated in brackets at the right of the tree
Fig. 6.
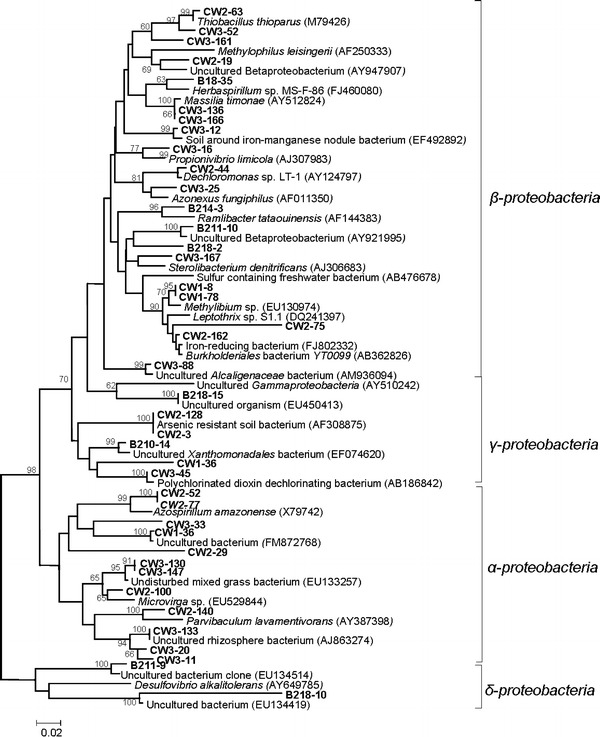
Neighbor-joining tree constructed based on proteobacterial 16S rRNA gene sequences detected in the present study along with similar sequences retrieved from NCBI and RDP databases. Numbers at nodes indicate percent bootstrap values above 80 supported by 1,000 replicates. Bar indicates Jukes-Cantor evolutionary distance
Fig. 7.
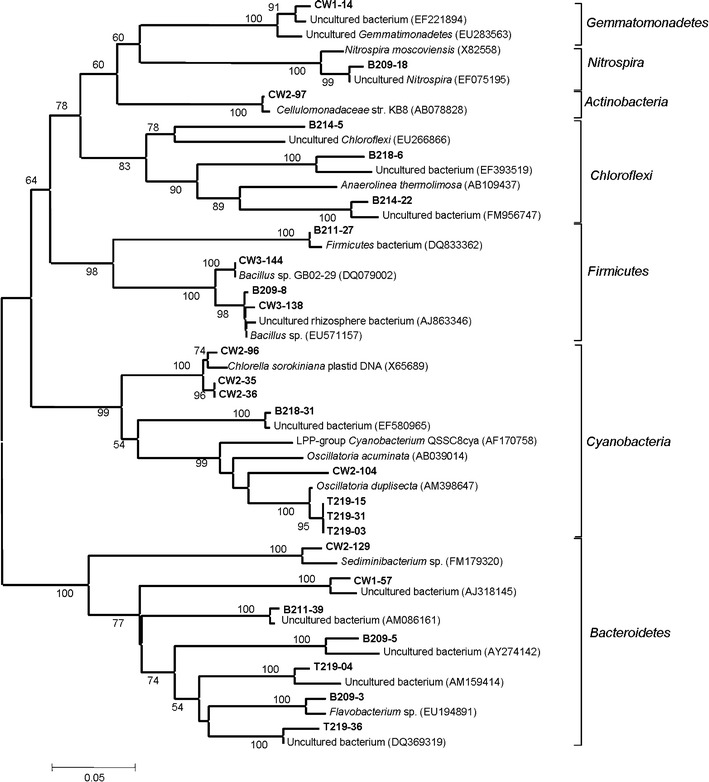
Neighbor-joining tree constructed based on 16S rRNA gene sequences of phyla Bacteroidetes, Firmicutes, Actinobacteria, Gemmatimonadetes and Cyanobacteria detected in the present study along with similar sequences retrieved from NCBI and RDP databases. Numbers at nodes indicate percent bootstrap values above 80 supported by 1,000 replicates. Bar indicates Jukes-Cantor evolutionary distance
Acidobacteria
Similarity search in NCBI and RDP databases indicated that all the Acidobacterial sequences had strong identity with uncultured members of this phylum. Particularly, sequences from non-contaminated samples showed a strong similarity with uncultured Acidobacteria from undisturbed mixed grass prairie and rice field soils. In contrast, Acidobacteria sequences retrieved from contaminated samples showed high similarity with uncultured clones mostly obtained from hydrocarbon-contaminated soil and sediment, soil adjacent to silage storage bunker and Altamira cave, etc. In order to determine the distribution of Acidobacteria sequences retrieved from ten clone libraries into different subgroups, a Neighbor-joining tree was constructed using representative Acidobacteria sequences of different subgroups along with our sequences (Fig. 5). Representative sequences for various subgroups were obtained from previously published works of Sait et al. 2006 (subgroup 1) and Hugenholtz et al. 1998 (subgroup 2, 3, 4, 5, 6, 7 and 8). Hugenholtz et al. (1998) mentioned that Acidobacteria subdivisions 1, 3, 4, and 6 are well represented by environmental clone sequences with only five cultured representatives. In this regard, our analysis indicated that members of subgroup 1, 3, 4 and 5 were present within all eight libraries. Members that belong to other subgroups (2, 6, 7 and 8) were not detected in the present libraries. Phylogenetic tree further revealed that the most dominant Acidobacteria clones from libraries B209, B210, T219, T221 and TD222 were present within subgroup 1. Within this subgroup, seven sequences representing the major ribotypes from the above libraries were included. Sequence of the clone B210-44 although shared identity with an uncultured bacterium, showed a close phylogenetic lineage with culturable representative Acidobacterium capsulatumi with 90% bootstrap value. The next cluster that accommodated maximum numbers of ribotypes showed their affiliation to subgroup 4. In this subgroup, four Acidobacteria clones from the contaminated site CW1, and three from non-contaminated sites (T112, B209 and B210) were included. Subgroup 5 was composed of four dominant Acidobacteria clones; two retrieved from contaminated library CW3 and other two from non-contaminated library T112. Subgroup 3 was represented by members from the library T222. Abundance of Acidobacteria in uranium-contaminated samples was reported by Barns et al. 2007. The ubiquity and abundance of Acidobacteria in soils and their ability to withstand polluted and extreme environments suggest that they serve functions important in the environment and are potentially quite varied (Ward et al. 2009).
Proteobacteria
Predominance of phylum Proteobacteria was observed mainly in contaminated samples, although their presence was detected within non-contaminated samples as well. Among the Proteobacteria, members of the class β-Proteobacteria was found to represent the abundant ribotypes in contaminated as well as non-contaminated samples. As evident from Fig. 5, within the contaminated samples nine ribotypes of CW3 library (42% coverage), five ribotypes (29% coverage) from CW2 library and two ribotypes (11% coverage) of library CW1 were affiliated to β-Proteobacteria. Several major ribotypes from non-contaminated samples (B18, B211 and B214) were belonging to β-Proteobacteria. As evident from the tree topology, three clones (CW3-63, CW3-161 and CW3-52) from CW3 formed a separate monophyletic clade showing their relatedness with thiosulfate oxidizing autotrophic bacterium Thiobacillus thioparus (ATCC 8158). In another clade clone B218-35 (covering 9% of the library) showed affiliation with N2-fixing bacterium Herbaspirillum sp. MS-F-86 (FJ460080) isolated from Ninghai harbor, China. In the same clade clone, CW3-136 and CW3-166 of library CW3 showed strong phylogenetic affiliation and lineage (100% bootstrap support) with nickel-resistant Massilia timonae (AY512824). Clone CW3-12 showed a strong affiliation with uncultured β-Proteobacteria (EF492892) isolated from soil around iron-manganese nodule from China. Other clones in the same library formed a single clade showing their affiliation with hydrocarbon degrader Propionivibrio limicola (AJ307983). Clone CW2-44 and CW3-25 showed affiliation with perchlorate-reducing bacterium Dechloromonas sp. LT-1 (AY124797) and denitrifying Azonexus fungiphilus (AF011350), respectively. Clone B214-3, the most abundant ribotype (covering 22%) of the library B218 formed a monophyletic node showing close relatedness with ‘nanometric’ bacterium Ramlibacter tataouinensis (AF144383) isolated from meteorite-impacted subdesert soil in Tunisia, France. The other node was comprised six clones. Clones B218-167 and B211-10 of this cluster showed a strong affiliation with uncultured β-Proteobacteria (AY921995). Clone CW3-167 showed affiliation with cholesterol-oxidizing, denitrifying bacterium Sterolibactrium denitrificans (AJ306683). In another clade, two clones CW1-8 and CW1-78 together covering 11% of the CW1 library showed affiliation with Methylibium sp. (EU130974) isolated from granular activated carbon filters. Along with clones CW2-75 and CW2-162 having relatedness with Burkholderiales bacterium YT0099, the later clones showed a strong lineage with members of Burkholderiaceae capable of redox transformation of metals to satisfy metabolic requirements. Sequence of clone CW3-88 formed a separate clade showing its distinctness from other members but strong phylogenetic lineage (99% bootstrap value) with uncultured Alcaligenaceae (AM936094) isolated from hydrocarbon contaminated soil. Members of the class γ-Proteobacteria were relatively more abundant in contaminated samples. Sequence of clone B218-15 was highly related to an uncultured member of γ-Proteobacteria. Two clones (CW2-3 and CW2-128 covering 21% of CW2 library), showed a strong phylogenetic affiliation with arsenic-resistant soil bacterium (AF308875). Sequence of clone B210-14 marked its affiliation with uncultured Xanthomonadace bacterium, while two clones CW1-36 and CW3-45 showed their lineage to polychlorinated dioxin dechlorinating bacterium. Members of the class α-Proteobacteria were represented by ribotypes from contaminated samples CW2 and CW3, covering 25 or 27% in respective libraries. Within this class, sequences of clones CW2-52 and CW2-77 showed a strong relatedness with N2-fixing bacteria Azospirillum amazonense. Sequences of clones CW3-33 and CW1-36 showed phylogenetic affiliation with uncultured bacterium isolated from floor dust. Together with sequence of CW2-29, both these two clades were found to be related sharing the same node, thus indicating a common phylogenetic origin. The remaining clade within this class was composed of five clones. Clone CW3-130 and CW3-147 (covering 7% of the CW3 library) showed affiliation with mixed grass bacterium where as clone CW2-100 showed affiliation with Microvirga sp. (EU529844) isolated from soil of cucumber farming greenhouse. Sequence of clone CW2-140 showed a strong affiliation with Parvibaculum lavamentivorans (AY387398) bacterium capable of alkylbenzenesulfonate surfactant (LAS) biodegradation. Sequences of three other clones CW3-133, CW3-20 and CW3-11, although strongly related to each other showed phylogenetic affiliation with uncultured bacterium isolated from a poplar tree microcosm. Class δ-Proteobacteria was represented by two clones (B218-10 and B211-9). Sequence of clone B218-10 was the most abundant ribotype in library B218 that showed affiliation with Desulfovibrio alkalitolerans (AY649785) isolated from a heating water of Denmark. Dominancy of Proteobacteria in contaminated sites is well reported (Rastogi et al. 2010; Ellis et al. 2003). Lineages belonging to Proteobacteria are well known to survive in oligotrophic environments, capable of metal reduction and resistance and play very important roles in uranium immobilization in contaminated sediments or groundwater.
Bacteroidetes, Firmicutes, Actinobacteria, Gemmatimonadetes and Cyanobacteria
Members of these phyla were more abundant in non-contaminated samples (Fig. 6). Phylum Bacteroidetes was represented by five ribotypes. Sequence of clone B209-3 showed a strong phylogenetic lineage (100% bootstrap value) with Flavobacterium sp. (EU194891) while clone (B211-39) showed a strong affiliation with uncultured bacterium isolated from gold mine tailings of Nederland. Clone CW2-129 showed affiliation with Sediminibacterium sp. (FM179320) where as clone CW1-57 showed affiliation with uncultured bacterium isolated from a waste-gas biofilter. Presence of the phylum Bacteroidetes in heavy metal and uranium-contaminated soils and sediments were reported previously by Ellis et al. 2003; Reardon et al. 2004; Brodie et al. 2006 and Akob et al. 2008.
Phylum Cyanobacteria was represented by eight clones. Among the four clones from non-contaminated sites, three from library T219 showed lineage with Oscillatoria duplisecta (AM398647) isolated from a thermal mud of Euganean thermal springs (Italy). Clone CW2-104 from sample CW2 also showed affiliation with Oscillatoria acuminata (AB039014). Other three clones (CW2-96, CW2-34 and CW2-35) from contaminated sample CW2, covering 11% of the clone library, and showed affiliation with uncultured Chlorophyta chloroplast 16S rRNA gene (AB374385). Cyanobacteria were the ubiquitous group present in most soil habitats and possessing high tolerance to environmental stress (Chong et al. 2010). Our results indicated that it may be present both in contaminated and non-contaminated samples. It also can act as a biosorbent of heavy metals in sewage water (El-Enany and Issa 2000). Phylum Firmicutes was represented by four clones. Clone B211-27 showed a strong phylogenetic affiliation (100% bootstrap value) with Firmicutes bacterium isolated from Pere Marquette River sediment. Clone B219-8, CW3-144 and CW3-138 showed a strong phylogenetic affiliation (100% bootstrap value) with Manganese (II)-oxidizing Bacillus sp. Akob et al. (2008) showed the presence of Firmicutes in radionuclide-contaminated subsurface sediments during the early part of the ethanol microcosm incubation suggesting that these groups may have actively involved in nitrate reduction and subsequent uranium reduction both important for the immobilization of U(VI) in contaminated sediments. Merroun et al. (2005) showed that Bacillus sp., can be recovered from uranium mining waste pile and can make complexion with uranium by either whole cells or S-Layer sheets. Phylum Chloroflexi was mainly represented by ribotypes from sample B214. Sequences of clone B214-22 and B214-6 formed a distinct monophyletic clade along with Anaerolinea thermolimosa (AB109437) isolated from thermophilic granular sludge. Phylum Actinobacteria, Nitrospira and Gemmatimonadetes were all represented by single ribotype. In the phylum Actinobacteria, clone CW2-97 was found to be phylogenetically affiliated (with 100% bootstrap value) with Cellulomonadaceae str. KB8 isolated from paddy rice. Clone B209-18 showed a strong phylogenetic affiliation with nitrite oxidizing Nitrospira moscoviensis. In the phylum Gemmatimonadetes, clone CW1-14 showed affiliation with uncultured bacterium isolated from oil palm empty fruit bunches compost heap.
Acknowledgments
Authors acknowledge the financial support from Board of Research in Nuclear Sciences (BRNS), Department of Atomic Energy (DAE), Govt. of India. Ekramul Islam acknowledges support from Council of Scientific and Industrial Research (CSIR), Govt. of India for his fellowship. Generous support from Uranium Corporation of India Limited, Jaduguda is gratefully acknowledged. Authors greatly acknowledge anonymous reviewers for their critical comments in improving the manuscript.
References
- Akob DM, Mills HJ, Kostka JE. Metabolically active microbial communities in uranium-contaminated subsurface sediments. FEMS Microbiol Ecol. 2007;59:95–107. doi: 10.1111/j.1574-6941.2006.00203.x. [DOI] [PubMed] [Google Scholar]
- Akob DM, et al. Functional diversity and electron donor dependence of microbial populations capable of U(VI) reduction in radionuclide-contaminated subsurface sediments. Appl Environ Microbiol. 2008;74:3159–3170. doi: 10.1128/AEM.02881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barns SM, Cain EC, Sommerville L, Kuske CR. Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol. 2007;73:3113–3116. doi: 10.1128/AEM.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie EL, et al. Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl Environ Microbiol. 2006;72:6288–6298. doi: 10.1128/AEM.00246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt R, et al. Major and trace elements of selected pedons in the USA. J Environ Qual. 2003;32:2109–2121. doi: 10.2134/jeq2003.2109. [DOI] [PubMed] [Google Scholar]
- Chong CW, et al. High levels of spatial heterogeneity in the biodiversity of soil prokaryotes on Signy Island, Antarctica. Soil Biol Biochem. 2010;42:601–610. doi: 10.1016/j.soilbio.2009.12.009. [DOI] [Google Scholar]
- Choudhary S, Sar P. Uranium biomineralization by a metal resistant Pseudomonas aeruginosa strain isolated from contaminated mine waste. J Hazard Mater. 2011;186:336–343. doi: 10.1016/j.jhazmat.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Desai C, Parikh RY, Vaishnav T, Shouche YS, Madamwar D. Tracking the influence of long-term chromium pollution on soil bacterial community structures by comparative analyses of 16S rRNA gene phylotypes. Res Microbiol. 2009;160:1–9. doi: 10.1016/j.resmic.2008.10.003. [DOI] [PubMed] [Google Scholar]
- El-Enany AE, Issa AA. Cyanobacteria as a biosorbent of heavy metals in sewage water. Environ Toxicol Pharmacol. 2000;8:95–101. doi: 10.1016/S1382-6689(99)00037-X. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Morgan P, Weightman AJ, Fry JC. Cultivation-dependent and -independent approaches for determining bacterial diversity in heavy-metal-contaminated soil. Appl Environ Microbiol. 2003;69:3223–3230. doi: 10.1128/AEM.69.6.3223-3230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AL, et al. Relationships between microbial communities and environmental parameters at sites impacted by mining of volcanogenic massive sulfide deposits, Prince William Sound, Alaska. Appl Geochem. 2008;23:279–307. doi: 10.1016/j.apgeochem.2007.10.012. [DOI] [Google Scholar]
- Fredrickson JK, et al. Geomicrobiology of high-level nuclear waste-contaminated vadose sediments at the Hanford site, Washington state. Appl Environ Microbiol. 2004;70:4230–4241. doi: 10.1128/AEM.70.7.4230-4241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Sarangi AK (2005) Emerging trend of uranium mining: the Indian scenario presented in the IAEA symposium on “Uranium production and raw materials for the nuclear fuel cycle—Supply and demand, economics, the environment and energy security”, June 20–24, Vienna
- Herrera A, et al. Species richness and phylogenetic diversity comparisons of soil microbial communities affected by nickel-mining and revegetation efforts in New Caledonia. Eur J Soil Biol. 2007;43:130–139. doi: 10.1016/j.ejsobi.2006.12.001. [DOI] [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam E, Sar P. Culture-dependent and -independent molecular analysis of the bacterial community within uranium ore. J Basic Microbiol. 2011;4:372–384. doi: 10.1002/jobm.201000327. [DOI] [PubMed] [Google Scholar]
- Islam E, Sar P (2011b) Molecular assessment on impact of uranium ore contamination in soil bacterial diversity. Int Biodet Biodegrad 65:1043–1051
- Islam E, Dhal PK, Sufia KK, Sar P. Molecular analysis of bacterial communities in uranium ores and surrounding soils from Banduhurang open cast uranium mine, India: A comparative study. J Environ Sci Health A. 2011;46:271–280. doi: 10.1080/10934529.2011.535433. [DOI] [PubMed] [Google Scholar]
- Lloyd JR, Renshaw JC. Microbial transformations of radionuclides: fundamental mechanisms and biogeochemical implications. Met Ions Biol Syst. 2005;44:205–240. [PubMed] [Google Scholar]
- Merroun ML, Raff J, Rossberg A, Hennig C, Reich T, Selenska-Pobell S. Complexation of uranium by cells and S-layer sheets of Bacillus sphaericus JG-A12. Appl Environ Microbiol. 2005;71:5532–5543. doi: 10.1128/AEM.71.9.5532-5543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelkova M, Merroun ML, Rossberg A, Hennig C, Selenska-Pobell S. Microbacterium isolates from the vicinity of a radioactive waste depository and their interactions with uranium. FEMS Microbiol Ecol. 2007;59:694–705. doi: 10.1111/j.1574-6941.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Radeva G, Selenska-Pobell S. Bacterial diversity in water samples from uranium wastes as demonstrated by 16S rDNA and ribosomal intergenic spacer amplification retrievals. Can J Microbiol. 2005;51:910–923. doi: 10.1139/w05-082. [DOI] [PubMed] [Google Scholar]
- Rastogi G, et al. Microbial diversity in uranium mining-impacted soils as revealed by high-density 16S microarray and clone library. Microbiol Ecol. 2010;59:94–108. doi: 10.1007/s00248-009-9598-5. [DOI] [PubMed] [Google Scholar]
- Reardon CL, et al. Composition and diversity of microbial communities recovered from surrogate minerals incubated in an acidic uranium-contaminated aquifer. Appl Environ Microbiol. 2004;70:6037–6046. doi: 10.1128/AEM.70.10.6037-6046.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sait M, Davis KE, Janssen PH. Effect of pH on isolation and distribution of members of subdivision 1 of the phylum Acidobacteria occurring in soil. Appl Environ Microbiol. 2006;72:1852–1857. doi: 10.1128/AEM.72.3.1852-1857.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sar P, et al. Molecular assessment of microbial diversity and community structure at uranium mines of Jaduguda, India. Adv Mater Res. 2007;20–21:413–416. doi: 10.4028/www.scientific.net/AMR.20-21.413. [DOI] [Google Scholar]
- Sarangi AK, Singh AS (2006) Vein type uranium mineralisation in Jaduguda, uranium deposits, Singhbhum, India. In: Proceedings of the International Symposium on understanding the genesis of ore deposits to meet the demands of 21st Century, association on the genesis of ore deposits, Moscow, Russia, vol 12, pp 54–61
- Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- Suzuki Y, Banfield JF. Geomicrobiology of uranium, in mineralogy, geochemistry and the environment. Mineral Soc Am. 2000;38:21–27. [Google Scholar]
- Tabak HH, et al. Developments in bioremediation of soils and sediments polluted with metals and radionuclides: 1. Microbial processes and mechanisms affecting bioremediation of metal contamination and influencing metal toxicity and transport. Rev Environ Sci Biol/Technol. 2005;4:115–156. doi: 10.1007/s11157-005-2169-4. [DOI] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Ovreas L, Thingstad TF. Prokaryotic diversity—magnitude, dynamics, and controlling factors. Science. 2002;296:1064–1066. doi: 10.1126/science.1071698. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shi J, Wang H, Lin Q, Chen X, Chen Y. The influence of soil heavy metals pollution on soil microbial biomass, enzyme activity, and community composition near a copper smelter. Ecotoxicol Environ Saf. 2007;67:75–81. doi: 10.1016/j.ecoenv.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Ward NL, et al. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol. 2009;75:2046–2056. doi: 10.1128/AEM.02294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]