

Abstract
Background
As more complete genomes are sequenced, conservation of gene order between different organisms is emerging as an informative property of the genomes. Conservation of gene order has been used for predicting function and functional interactions of proteins, as well as for studying the evolutionary relationships between genomes. The reasons for the maintenance of gene order are still not well understood, as the organization of the prokaryote genome into operons and lateral gene transfer cannot possibly account for all the instances of conservation found. Comprehensive studies of gene order are one way of elucidating the nature of these maintaining forces.
Results
Gene order is extensively conserved between closely related species, but rapidly becomes less conserved among more distantly related organisms, probably in a cooperative fashion. This trend could be universal in prokaryotic genomes, as archaeal genomes are likely to behave similarly to bacterial genomes. Gene order conservation could therefore be used as a valid phylogenetic measure to study relationships between species. Even between very distant species, remnants of gene order conservation exist in the form of highly conserved clusters of genes. This suggests the existence of selective processes that maintain the organization of these regions. Because the clusters often span more than one operon, common regulation probably cannot be invoked as the cause of the maintenance of gene order.
Conclusions
Gene order conservation is a genomic measure that can be useful for studying relationships between prokaryotes and the evolutionary forces shaping their genomes. Gene organization is extensively conserved in some genomic regions, and further studies are needed to elucidate the reason for this conservation.
Background
Completely sequenced genomes enable the study of relations between organisms in terms of the complete set of genes they possess. Genomic properties have been proposed as the most convenient tool for studying these relationships, as they are global properties that may circumvent many of the difficulties of classical molecular phytogenies [1]. Common gene content [2, 3] or conservation of families of proteins [4] are examples of this kind of genomic information. From this genomic perspective, conservation of gene order is a very informative measure that may provide information both about the function and interactions of the proteins these genes encode [5, 6], and about the evolution of the genomes and the organisms themselves.
Gene order is generally well preserved at close phylogenetic distances [7]. When the species are not closely related, the degree of gene order conservation is usually low, and consequently it was proposed that conservation of gene order is easily lost during evolution [8]. This loss also extends to the disruption of operons, in some cases wiping them out completely [9].
Nevertheless, some instances of especially well-preserved clusters of genes are known, even in divergent species. The best examples are the genes for ribosomal proteins [10] and the dcw cluster [11]. Lathe and co-workers [12] recently identified genomic regions in which gene order is especially highly conserved. Even if some rearrangement does occur in these regions, the general trend is to keep the genes closer together than in other regions. This shows that selection for gene location and ordering could exist in some cases. The operon structure and common regulation cannot easily account for the conservation, as these conserved regions extend for more than a single operon; hence the proposed nomenclature of uber-operons [12].
Conservation of gene order can be due to any one of the following three reasons. First, the species have diverged only recently and gene order has not yet been destroyed; second, there has been lateral gene transfer of a block of genes; and third, the integrity of the cluster is important to the fitness of the cell. Only in this latter case is gene order conservation selectable.
Proposed explanations for selection for gene ordering include helping the interaction of proteins encoded by the genes of the cluster [13], favoring lateral gene transfer [14], or co-localization of the mRNAs in the same region of the cell [15]. These explanations are not mutually exclusive. Recent studies of the structure of the dcw cluster suggest that, in this particular case, conservation of gene order within the cluster may be linked to cellular morphology, thus connecting gene order with a selectable phenotype [16].
The importance of gene order in the study of evolution is starting to be recognized. Even if the loss of gene order conservation is faster than the loss of sequence similarity, a large amount of conservation remains at medium phylogenetic distances, such as that between Escherichia coli and Bacillus subtilis [8]. Conservation is a valuable clue to the relationships between organisms and the influence of events such as lateral gene transfer on the evolution of genomes.
I present here an analysis of the extent and characteristics of gene order conservation in prokaryotes and attempt to answer two questions. Does conservation of gene order occur similarly throughout the prokaryotes? Are the conserved regions distributed uniformly within the genomes?
Results and discussion
Conservation of gene order in evolution
General trends in gene order conservation
To address the issue of how gene order is conserved during evolution, I measured gene order conservation in prokaryotes in relation to evolutionary distance in terms of small subunit rRNA (SSU rRNA) substitutions. The results are shown in Figure 1.
Figure 1.
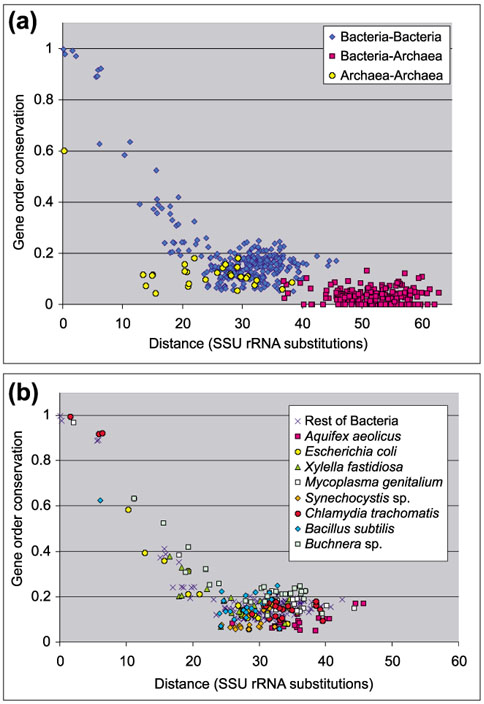
Conservation of gene order in prokaryotic genomes in relation to phylogenetic distance, measured as the number of substitutions in SSU rRNA. Each point represents a pair of species. (a) Results for all species. (b) Same plot as in (a), but with Archaea removed and values for some bacterial species highlighted.
In the Bacteria, conservation of gene order apparently follows a common trend for all species. The loss of gene order conservation when phylogenetic distance increases is clear, but even at long distances some conservation is maintained. This is mainly because of clusters of genes that remain well conserved during bacterial evolution [12]. Gene order is extensively conserved at small phylogenetic distances, mostly because rearrangement has not yet had time to occur.
The distribution in Figure 1a fits to a sigmoid curve, revealing the existence of a cooperative process in the loss of gene ordering. This might be related to the existence of operons, in which the displacement of a single gene can facilitate the rearrangement of the rest of the operon. Previous studies proposed an exponential shape for the distribution [8, 17]. This disagreement probably arises because those studies did not include pairs of closely related species, and therefore missed the leftmost part of the graph, which is highly significant for the sigmoid shape.
Within this observed global trend, several bacterial species present small deviations from the average. Although such deviations are small, in some cases they are indicative of evolutionary processes shaping the genomes.
An interesting case is that of Buchnera. Figure 1b shows that the degree of gene order conservation in Buchnera is greater than expected according to the phylogenetic position of this bacterium, as previously observed [18, 19]. As an endosymbiont, Buchnera is experiencing extensive gene loss due to reductive processes, and consequently, lower levels of gene order conservation could be expected. However, many gene rearrangement processes are dependent on RecA activity [20, 21], which could not be found in Buchnera [18]. As a result, it is likely that the genome of this bacterium has experienced few rearrangement events. Lateral gene transfer also seems negligible in this case [22], and therefore gene loss remains as the only process capable of altering gene order in the Buchnera genome. With the exception of lost genes, the Buchnera genome might reflect the gene order it had when the bacterium became an endosymbiont and lost recA. Accordingly, it could be used as a convenient reference point in studies on gene order.
Deep-branching species on the bacterial tree, such as Aquifex and Synechocystis, also deviate from the average. These species have the lowest values for gene order conservation among the Bacteria (Figure 1b). This agrees with classical molecular phylogenies as well as with genomic phylogenies based on whole-proteome analysis [3], in which these species are also the most divergent within the Bacteria.
To study whether a common trend in conservation of gene order occurs within prokaryotes, I also included archaeal species in the comparisons. According to Figure 1a, the trend observed in Bacteria is not found in the Archaea. Conservation of gene order between archaea is less than between bacteria, even for very closely related species (Pyrococcus horikoshii and Pyrococcus abyssi), and the point at which only residual conservation persists is reached much faster. I think that this difference is probably artifactual, and due to anomalous measurement of the phylogenetic distances between organisms. Brinkmann and Philippe [23] argued that SSU rRNAs of bacteria evolve faster than those of archaea, thus resulting in an underestimation of the phylogenetic distances between archaea. The distances between archaea are thus probably higher than shown in Figure 1a and, consequently, gene order conservation would fit well into the overall trend found for the Bacteria, although the lack of points on the left-hand side of the graph makes it difficult to extract a conclusion. Moreover, measures of phylogenetic distances between bacteria and archaea should also be higher, which would shift the Bacteria-Archaea points to the right in the plot, thus eliminating the surprising artificial overlap between Bacteria-Archaea and Bacteria-Bacteria points.
This is a good example of the difficulties encountered when using molecular phylogenies. Phenomena such as unequal mutation rates and lateral gene transfer, or artifacts such as long-branch attraction may produce biased results [1]. Here, I show that these problems seem surmountable with the aid of genomic methods. The unequal mutation rate in SSU rRNA, detectable only by careful comparison of different molecular phylogenies, can be readily discovered by looking at gene order conservation. Hence, gene order conservation could be used as an alternative measure of distances between organisms, especially when such distances are small.
Conservation of gene order between bacteria and archaea is much lower than within each domain, and is even nonexistent in some cases. There is one exception: gene order conservation between the hyperthermophilic bacterium Thermotoga maritima and archaea is higher than the rest, and much higher than between Aquifex and archaea, even though the SSU rRNA distances between bacteria and archaea are approximately equal. The existence of extensive lateral gene transfer between Thermotoga maritima and archaea has been claimed [24]. This possibility is of great importance, as it suggests lateral gene transfer can occur between different domains. Thermotoga thus provides a nice example of conservation of gene order produced via lateral gene transfer.
Molecular phylogenies of universally conserved genes for better estimating distances
A different set of phylogenetic distances can be extracted by averaging those obtained from the molecular phylogenies of universally conserved genes (see Materials and methods). The results are shown in Figure 2. Distances between organisms seem to be more accurately estimated using this set of genes, and thus gene order conservation within the Archaea follows more closely the trend observed in the Bacteria. As the agreement between the two distributions is still not complete, however, I conjecture that the estimates of distances are still not entirely correct. It is likely that there are no differences between the amount of gene order conservation among the Bacteria and among the Archaea, and therefore the trend of conservation of gene order could be approximately the same for both domains.
Figure 2.
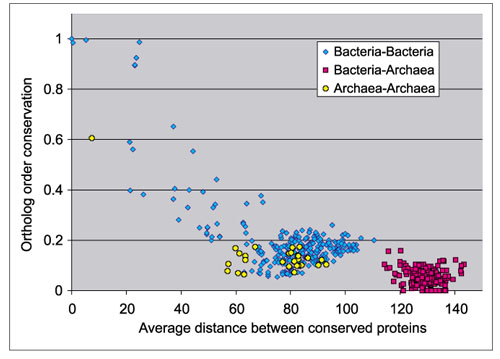
Conservation of gene order between all prokaryotic species in relation to phylogenetic distance, estimated by means of phylogenies of universal conserved proteins. Each point represents a pair of species.
Common gene content and gene order conservation
Realizing the difficulty of estimating the relationships between organisms using molecular phylogenies, some authors have proposed a genomic method based on the common gene content of the genomes [2, 3]. This method of estimating distances is claimed to be more accurate as it is not affected by the drawbacks of molecular phylogenies. I used common gene content as an additional estimation of distance between genomes, and compared the resulting distances with gene order conservation. The results are shown in Figure 3.
Figure 3.
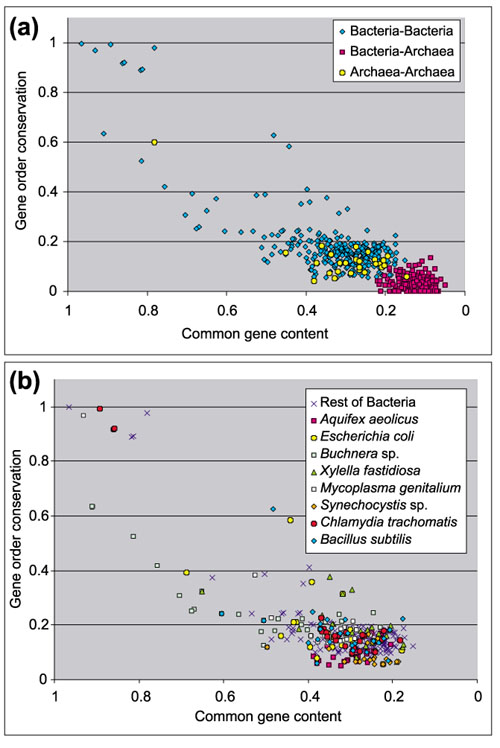
Conservation of gene order in relation to common gene content within and between prokaryotic domains. Each point represents a pair of species. (a) Results for all species. (b) Same plot as in (a), but with Archaea removed and values for some bacterial species highlighted.
When using common gene content as a measure of phylogenetic distance, gene order conservation in the Archaea follows a similar trend to that in the Bacteria (Figure 3a). Even if common gene content has some biases, as I will illustrate below, such biases are expected to be the same for the Bacteria as for the Archaea. This reinforces the hypothesis that both domains have a similar trend in the conservation of gene order.
In a more general sense, common gene content seems to be a noisy measure, as it is affected by factors such as the different lifestyles of the organisms. For example, Xylella fastidiosa is a proteobacterium, and one of its closest relatives in this study is Pseudomonas aeruginosa. Nevertheless, their common gene content is low, less than 40%. Between E. coli and Haemophilus influenzae, with a comparable phylogenetic distance, common gene content is around 70%. The fact is that X. fastidiosa has a very high number of open reading frames (ORFs) with no known relatives in other species (unique genes). The number of unique genes is as high as 40% for X. fastidiosa [25], and it is also very high for some other species [26]. As a result, distances between X. fastidiosa and other bacteria are overestimated by using common gene content. This is often the case for closely related bacteria with different lifestyles, such as E. coli and Vibrio cholerae, which share less than half of their genes because their different environments require different adaptations and different systems. Common gene content thus has disadvantages as a measure for estimating phylogenetic distances. In contrast, gene order conservation defines much more precisely the course of evolution of genomes, as it is not affected by the presence of particular sets of genes in individual genomes.
Regions of conservation and non-conservation of gene order in the genome
The second object of this study was to determine how the conservation of gene order is distributed along the genome. Are the conserved regions uniformly spread, or are there instead well-defined regions of high and low conservation? The latter answer seems to be the right one. Figure 4 shows conservation of gene order using the genomes of E. coli and X. fastidiosa as references. The rest of the genomes are sorted according to their phylogenetic distance (estimated by SSU rRNA substitutions) to the reference genomes. The gradual loss of gene order is easily seen, and it is apparent that regions of high gene order conservation coexist with regions in which no conservation can be found.
Figure 4.
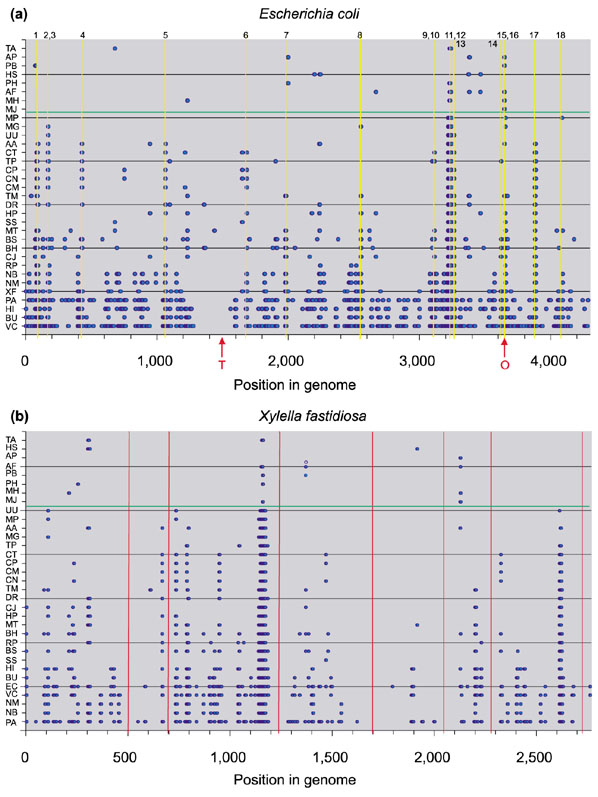
Gene order conservation in the species studied, using (a) Escherichia coli and (b) Xylella fastidiosa as a reference. Position in the reference genome means number of genes from minute zero. Individual species are plotted in the y axis and are ordered according to their phylogenetic distance (estimated by SSU rRNA substitutions) to the reference species. The more closely related species are shown lower down and more distantly related species higher up the axis. Species names are listed in Table 2. Blue dots indicate genes belonging to conserved runs for each species. A horizontal green line separates Bacteria from Archaea. (a) For E. coli, yellow lines show the regions with especially high conservation of gene order. A detailed study of these regions can be found in Table 1. The origin and terminus of replication are marked O and T, respectively, at the bottom of the graph. (b) For X. fastidiosa, red lines indicate regions of high frequency of unique genes [25]. A low degree of gene order conservation was found in these regions.
Regions with no trace of gene order conservation are not rare, even between closely related organisms. They represent either regions in which active rearrangement processes occur, or regions with a majority of unique genes. The first case is illustrated in Figure 4a for E. coli, in which the terminus of replication, which is a recombination hotspot, has no gene order conservation because of the extensive rearrangement in this region. An example of the second case is shown in Figure 4b for the genome of X. fastidiosa, in which regions where unique genes are prevalent are easily detected because of their lack of gene order conservation.
At the other extreme, regions of high gene order conservation exist in all the genomes. Figure 1 shows that there is a remnant of gene order conservation even between distantly related organisms, in both the Bacteria and the Archaea. These regions of special conservation can be thought of as being subject to selective processes for keeping genes together. I analyzed the functional composition of these regions.
To find out whether the conserved regions are related to any functional characteristics, the proteins encoded by the genes in these regions were functionally classified using the EUCLID system [27]. I also explored the correspondence of the runs of genes with experimentally determined operons, as found in the RegulonDB database [28]. The most conserved runs are shown in Table 1. No apparent preferences for particular functional classes were found (apart from the translation class, over-represented because of ribosomal proteins). The runs are composed of genes for proteins involved in many different types of processes, from metabolic-related classes to information-related ones. With some exceptions, every run is preferentially composed of ORFs belonging to the same functional class. The selective forces acting to keep these genes together could indeed be different when the run is composed of different functional classes. For instance, the conservation of gene order in metabolic-related runs is often related to their coding for enzymes that act sequentially in a pathway, forming multifunctional complexes in several cases. For the runs related to cellular processes and information management, the selective scenario might be more complex [7].
Table 1.
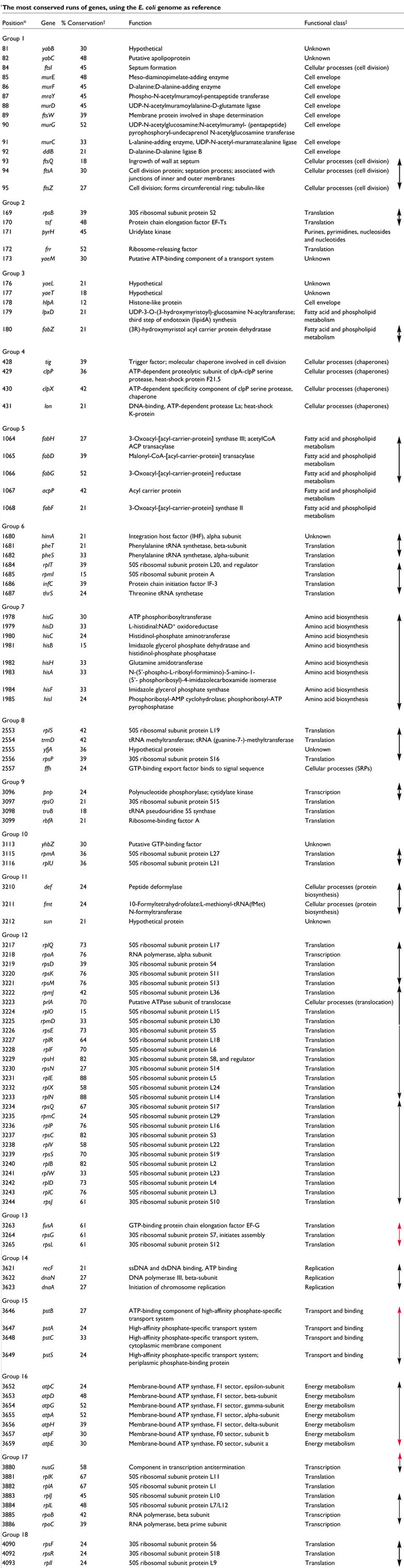
*Location of the gene in the genome, expressed in absolute number of genes from minute zero. †Percentage of conservation of gene order with respect to other genomes, expressed as the ratio between the number of times that the gene is conserved in the run and the total number of times that the gene is present. ‡The functional class is a general assignment of function as provided by the EUCLID system. Arrows in the right part of the figure indicate operons. Red tips in the arrows indicate that the operon continues in that direction, therefore containing genes not included in the run. Only operons for which experimental evidence is available are considered.
The conserved runs of genes usually correspond to operons in E. coli, and combinations of two or even three operons are common. If we consider the proposal that operons are unstable structures [9], the maintenance of gene order within the operons would be striking in itself, but the conservation of combinations of operons points to additional factors, other than common regulation, acting in the conservation of gene order. Lateral gene transfer could play a part in such a process [13], even if it is not easy to envisage how it could explain such extensive conservation. It is too early to say whether the assumption that operons are independent units [29] is challenged. Additional research on these conserved structures is needed in order to elucidate the factors acting in each case.
Conclusions
Gene order is a labile genomic characteristic. The level of conservation is high when organisms are phylogenetically closely related, but conservation is lost rapidly, probably to a higher degree than other genetic or genomic features [8]. Thus, the instances in which gene order is conserved between phylogenetically distant organisms may indicate that strong selection pressures are keeping them together, in the cases in which lateral gene transfer is unlikely to be the origin of the conservation. Selection could be because the operon controls the assembly of a multifunctional enzymatic complex or the performance of an important stage in a metabolic pathway. But in some cases, other explanations should be considered, in which the gene order could influence the phenotype. The existence of conserved units bigger than operons seems to argue in favor of other explanations [12, 16].
Gene order conservation can be valuable for establishing the relationships between organisms as it is not influenced by parameters that affect other genomic measures, such as the content of unique genes, that are ultimately dependent on the lifestyle of the species. Genomic properties have been proposed as alternatives to classical molecular phylogenies as they measure global features of the genomes. So far, no genomic property by itself can represent that alternative, and integration of information on different properties is desirable. In this perspective, the information offered by gene order conservation is crucial.
Materials and methods
Sequences, positions and orientations of genes and corresponding proteins in complete prokaryotic genomes (Table 2) were obtained from NCBI [30]. Where the genome is composed of several chromosomes and/or plasmids, the sequences were linearized and concatenated.
Table 2.
Species used in this study
| Bacteria | Archaea |
| AA: Aquifex aeolicus | AF: Archaeoglobus fulgidus |
| BB: Borrelia burgdorferi | AP: Aeropyrum pernix |
| BH: Bacillus halodurans | HS: Halobacterium sp. |
| BS: Bacillus subtilis | MJ: Methanococcus jannaschii |
| BU: Buchnera sp. | MH: Methanobacterium |
| thermoautotrophicum | |
| CJ: Campylobacter jejuni | PB: Pyrococcus abyssi |
| CM: Chlamydia muridarum | PH: Pyrococcus horikoshii |
| CP: Chlamydia pneumoniae strain | TA: Thermoplasma acidophilum |
| CWL029 | |
| CN: Chlamydia pneumoniae strain AR39 | |
| CT: Chlamydia trachomatis | |
| DR: Deinococcus radiodurans | |
| EC: Escherichia coli | |
| HI: Haemophilus influenzae Rd | |
| HP: Helicobacter pylori strain 26695 | |
| MG: Mycoplasma genitalium | |
| MP: Mycoplasma pneumoniae | |
| MT: Mycobacterium tuberculosis | |
| NM: Neisseria meningitidis serogroup A | |
| NB: Neisseria meningitidis serogroup B | |
| PA: Pseudomonas aeruginosa | |
| RP: Rickettsia prowazekii | |
| SS: Synechocystis sp. | |
| TM: Thermotoga maritima | |
| TP: Treponema pallidum | |
| UU: Ureaplasma urealyticum | |
| VC: Vibrio cholerae | |
| XF: Xylella fastidiosa |
Homologs and orthologs between genomes were detected by BLAST [31] similarity searches. For two ORFs to be considered as homologous, their alignment should include at least 75% of the length of both ORFs, and the expected value (E-value) must be less than 10-5. I will refer to this homology relationship as bidirectional hits (BHs). ORFs related in this way are not necessarily orthologous, however, as paralogous genes may exist and are also identified. Therefore one gene can have more than one BH, which may introduce a bias in the count of related genes and conserved blocks of genes between two genomes. For identifying real orthologs, I look for best bidirectional hits (BBHs), such that one ORF is the closest relative of the other and vice versa. All the results shown in this article were obtained using BBHs, but the use of BHs does not alter the tendencies, as only minor quantitative differences were found.
The position of each gene in the genome is converted into a linear scale, from one to the total number of genes in the genome, and the information on either BHs or BBHs between two genomes is used to extract 'runs' - clusters of genes in which order is conserved. A run cannot comprise genes from different strands; hence a change of coding strand implies the termination of the run. I introduce two parameters setting the minimum length of the run and the maximum length of gaps (inserted genes) within it. For the purposes of this article, these parameters were set to a minimum length of three genes, allowing gaps of three genes as well. As gene duplications may exist, duplicated runs are also possible. Duplicated runs are taken into account if, and only if, they are present in both genomes. Otherwise the duplication is discarded. By definition, duplicated runs do not exist when working with BBHs.
The measure of gene order conservation between two genomes used here is the ratio between the number of genes located in conserved runs and the total number of related genes (BHs or BBHs).
Molecular phylogenetic methods have been widely used to determine the degree of relationship between organisms. The genes of choice are those universally conserved, especially SSU rRNA. The classical molecular phylogeny of SSU rRNA was obtained from the RDP database [32], and was used to estimate distances between the organisms on the basis of the number of substitutions between the sequences. The distances were computed using different correction methods (Jukes-Cantor, Jin-Nei and Kimura two-parameter methods), by means of the program 'distances' of the GCG package [33]. The differences using different correction methods were found to be very small (less than 5%), and did not influence this study.
Averaging molecular phylogenies of different universally conserved genes has been proposed as a way of alleviating the problems of individual phylogenies, by compensating for the different tendencies found in single genes [24, 34]. By a systematic search, 24 genes conserved in all the genomes used in this study were found. Molecular phylogenies of these collections of genes were constructed using neighbor-joining and maximum likelihood methods, extracting 24 sets of distances. A unique set of distances was obtained by averaging the 24 sets and used as an additional measure of divergence between species.
Common gene content between two organisms is proposed as a genomic estimation of distances between them. Common gene content is defined as the ratio between the number of orthologous genes found between the two species and the maximum number of possible orthologs (the number of genes in the smaller genome).
Therefore, three different estimations of distances between organisms were used in this study: distances based on SSU rRNA phylogeny; averaged distances of molecular phylogenies of universally conserved genes; and common gene content between the species.
Acknowledgments
Acknowledgements
I thank Carlos Briones (Centro de Astrobiología) and Alfonso Valencia (Centro Nacional de Biotecnología) for fruitful discussions.
References
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2128. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Lazcano A, Dujon B. The genomic tree as revealed from whole proteome comparisions. Genome Res. 1999;9:550–557. [PMC free article] [PubMed] [Google Scholar]
- Fitz-Gibbon S, House CH. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999;27:4218–4222. doi: 10.1093/nar/27.21.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen MA, Snel B, Lathe W, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J, Ouzounis C, Casari G, Valencia A. Conserved clusters of functionally related genes in two bacterial genomes. J Mol Evol. 1997;44:66–73. doi: 10.1007/pl00006122. [DOI] [PubMed] [Google Scholar]
- Huynen MA, Bork P. Measuring genome evolution. Proc Natl Acad Sci USA. 1998;95:5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Takemoto K, Mori H, Gojobori T. Evolutionary instability of operon structures disclosed by sequence comparisions of complete microbial genomes. Mol Biol Evol. 1999;16:332–346. doi: 10.1093/oxfordjournals.molbev.a026114. [DOI] [PubMed] [Google Scholar]
- Nikolaichik YA, Donachie WD. Conservation of gene order amongst cell wall and cell division genes in Eubacteria, and ribosomal genes in Eubacteria and eukaryotic organelles. Genetica. 2000;108:1–7. doi: 10.1023/a:1004077806910. [DOI] [PubMed] [Google Scholar]
- Ayala JA, Garrido T, de Pedro MA, Vicente M. New Comprehensive Biochemistry, Vol 27: Bacterial Cell Wall London; Elsevier Science: 1994. pp. 73–101.
- Lathe WC, Snel B, Bork P. Gene context conservation of a higher order than operons. Trends Biochem Sci. 2000;25:474–479. doi: 10.1016/s0968-0004(00)01663-7. [DOI] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Roth JR. Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996;143:1843–1860. doi: 10.1093/genetics/143.4.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin A, Guerdoux-Jamet P, Moszer I, Nitschke P. Mapping the bacterial cell architecture into the chromosome. Phil Trans R Soc Lond B. 2000;355:179–190. doi: 10.1098/rstb.2000.0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J, Gonzalez-Moreno M, Mingorance J, Valencia A, Vicente M. Bringing gene order into bacterial shape. Trends Genet. 2001;17:124–126. doi: 10.1016/s0168-9525(00)02212-5. [DOI] [PubMed] [Google Scholar]
- Huynen MA, Snel B. Gene and context: integrative approaches to genome analysis. Adv Protein Chem. 2000;54:345–379. doi: 10.1016/s0065-3233(00)54010-8. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocelullar bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Andersson JO. Is Buchnera a bacterium or an organelle? Curr Biol. 2000;10:R866–R868. doi: 10.1016/s0960-9822(00)00816-2. [DOI] [PubMed] [Google Scholar]
- Roth JR, Benson N, Galitski T, Haack K, Lawrence JG, Miesel L. Rearrangements of the bacterial chromosome: formation and applications. In Escherichia coli and Salmonella typhimurium Edited by Neidhardt FC, Curtiss R, Ingraham JL, Lin ECC, Brooks Low K, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE Washington DC: ASM Press; 1996.
- Hughes D. Evaluating genome dynamics: the constraints on rearrangements within bacterial genomes. Genome Biology. 2000;1:reviews0006.1–0006.8. doi: 10.1186/gb-2000-1-6-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N, Munson MA, Baumann P, Ishikawa H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc R Soc Lond B. 1993;253:161–171. [Google Scholar]
- Brinkmann H, Philippe H. Archaea sister group of Bacteria? Indications from tree reconstruction artifacts in ancient phylogenies. Mol Biol Evol. 1999;16:817–825. doi: 10.1093/oxfordjournals.molbev.a026166. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickley EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Simpson AJ, Reinach FC, Arruda P, Abreu FA, Acencio M, Alvarenga R, Alves LM, Araya JE, Baia GS, Baptista CS, et al. The genome sequence of the plant pathogen Xylella fastidiosa. Nature. 2000;406:151–159. doi: 10.1038/35018003. [DOI] [PubMed] [Google Scholar]
- Iliopoulos I, Tsoka S, Andrade MA, Janssen P, Audit B, Tramontano A, Valencia A, Leroy C, Sander C, Ouzounis C. Genome sequences and great expectations. Genome Biology. 2000;2:interactions0001.1–0001.3. doi: 10.1186/gb-2000-2-1-interactions0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J, Ouzounis C, Casari G, Valencia A. EUCLID: Automatic classification of proteins in functional classes by their database annotations. Bioinformatics. 1996;14:542–543. doi: 10.1093/bioinformatics/14.6.542. [DOI] [PubMed] [Google Scholar]
- Salgado H, Santos-Zavaleta A, Gama-Castro S, Millan-Zarate D, Diaz-Peredo E, Sanchez-Solano F, Perez-Rueda E, Bonavides-Mar-tinez C, Collado-Vides J. RegulonDB (version 32): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res. 2001;29:72–74. doi: 10.1093/nar/29.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolaeva MD, White O, Salzberg SL. Prediction of operons in microbial genomes. Nucleic Acids Res. 2001;29:1216–1221. doi: 10.1093/nar/29.5.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnological Information http://www.ncbi.nlm.nih.gov
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RDP database http://www.cme.msu.edu/RDP/html/index.html
- GCG http://www.gcg.com
- Eisen JA. Assessing evolutionary relationships among microbes from whole-genome analysis. Curr Opin Microbiol. 2000;3:475–480. doi: 10.1016/s1369-5274(00)00125-9. [DOI] [PubMed] [Google Scholar]