

Abstract
We addressed the link between excessive exposure to insulin and mitochondrion-derived oxidative stress in this study and found that prolonged exposure to insulin increased mitochondrial cholesterol in cultured hepatocytes and in mice and stimulated production of reactive oxygen species (ROS) and decreased the reduced glutathione to glutathione disulfide ratio in cultured hepatocytes. Exposure of isolated hepatic mitochondria to cholesterol alone promoted ROS emission. The oxidative stress induced by the prolonged exposure to insulin was prevented by inhibition of cholesterol synthesis with simvastatin. We further found that prolonged exposure to insulin decreased mitochondrial membrane potential and the increased ROS production came from mitochondrial respiration complex I. Finally, we observed that prolonged exposure to insulin decreased mitochondrial membrane fluidity in a cholesterol synthesis-dependent manner. Together our results demonstrate that excess exposure to insulin causes mitochondrion-derived oxidative stress through cholesterol synthesis in hepatocytes.
Mitochondrion-derived oxidative stress (mtROS) plays a critical role in the development of insulin resistance (1–4). Insulin resistance is a precursor or key component of various health problems caused by the positive energy imbalance due to overeating and/or lack of physical activity. These major health problems include obesity, metabolic syndrome, cardio- and cerebrovascular diseases, nonalcoholic fatty liver, Alzheimer's disease, asthma, some cancers, and aging (5–13). We have recently shown that excess exposure to insulin increases mtROS production and leads to insulin resistance (14–17). However, the exact mechanism by which excess exposure to insulin increases mtROS production has not been established.
Normally, electrons are transferred through a series of coupled acceptors in a relay that ends in a reaction with oxygen and conjugated into H2O at the respiratory complex IV in the inner mitochondrial membrane, and very few of them leak out from complexes I and/or III to activate oxygen into superoxide anions (O2−), which are promptly converted into H2O2 by the superoxide dismutases (SOD) (18, 19). However, electron leak and mtROS production will increase when the mitochondrial respiration chain is either overloaded by electrons from excess nicotinamide adenine dinucleotide hydrogen or blocked (18, 19). Therefore, both the quantity (mass) and quality of mitochondria can influence the level of mtROS production. Many studies have previously shown altered mitochondrial mass in subjects with oxidative stress and insulin resistance/hyperinsulinemia (20, 21). We have also shown that excess exposure to insulin can affect mitochondrial mass through two different ways. First, excess exposure to insulin can inhibit production of new mitochondria by suppressing transcription of some key mitochondrial biogenic genes (14). Second, excess exposure to insulin can inhibit the autophagy-dependent removal of aged/damaged mitochondria and consequently reduces the quality of mitochondria (22). Others have shown similar results (23). Additionally, the quality or function of the mitochondrial respiration chain can be influenced by many other factors. For example, availability of key substrates such as ADP for converting the H+ gradient into ATP can greatly influence levels of the mitochondrial membrane potential and mtROS production (24). The transportation of ATP out of mitochondria and ADP into mitochondrial matrix depends on the adenine nucleotide translocator, which can be inhibited by activated long-chain acyl CoA (25–29). Insulin is known to activate free fatty acids into long-chain acyl CoA (17, 30). We have recently shown that excess exposure to insulin decreases mitochondrial ADP content (17). Additionally, it is known in cancer cells that increased cholesterol content in mitochondria can decrease the fluidity of mitochondrial inner membrane and permeability to proton, leading to increased mitochondrial membrane potential, which usually leads to elevated mtROS production (31, 32). Insulin is known to be a potent stimulator of cholesterol synthesis, and we have recently shown that the basal level of insulin signaling is elevated in the presence of insulin resistance and hyperinsulinemia (16). In this study, we examine whether excess exposure to insulin can increase mtROS production through stimulating cholesterol synthesis.
Materials and Methods
Reagents and antibodies
Hepa1c1c7 cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cholesterol, simvastatin, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1), 2′,7′-dichlorofluorescein diacetate (DCF-DA), rotenone, and coenzyme Q10 (CoQ10) were from Sigma (St. Louis, MO). Antibodies against phosphorylated Akt or total Akt were from Cell Signaling Technologies Inc. (Beverly, MA). Antibodies against β-actin, mouse IgG, and rabbit IgG were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The ample red cholesterol assay kit was obtained from Invitrogen (Paisley, UK). The assay kit for measuring reduced glutathione (GSH) and glutathione disulfide (GSSG) was obtained from OxisReaserch (Manhattan Beach, CA). The apoptosis assay kit was from Roche (Indianapolis, IN; catalog no. 11774425001). Other materials were all obtained commercially and are of analytical quality.
Cell cultures
Hepa1c1c7 cells were cultured in DMEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, penicillin (100 U/ml), and streptomycin (100 ug/ml) (Invitrogen) at 37 C in a 5% CO2 humidified incubator at 2 × 105 cells/ml density. Cells were split every 2 d.
Animal experiment
C57BL/6 (B6) mice (male, 6–9 wk old) were treated with a long- and slow-acting insulin reagent, glargine (50 U/kg body weight, ip injection, once a day) for 8 wk. Mice were fed the regular chow diet ad libitum and kept at 12-h light, 12-h dark cycles. After an 8-h fast at the end of the 8-wk experiment, liver mitochondria were isolated with a standard approach. Mitochondrial content of cholesterol was quantified as detailed in Measurement of cholesterol below. All animal studies were approved by the Institutional Animal Care and Use Committee of The University of North Carolina at Chapel Hill and fully complied with the guidance from the National Institutes of Health.
Preparation and culture of mouse primary hepatocytes
Mouse primary hepatocytes were isolated from male C57BL6 (B6) mice (6–9 wk) by using the modified in situ collagenase perfusion as we previously described (14, 33–35). The viable parenchymal hepatocytes were suspended in Williams' E media (Invitrogen) containing antibiotics (50 μg/ml penicillin, 50 μg/ml streptomycin, and 100 μg/ml neomycin). Throughout this study, cells were seeded at 3.5 × 105 cells in a 100-mm petri dish (BD Falcon, Oxnard, CA) that were precoated with 0.01% collagen type I. Hepatocytes were incubated at 37 C under 5% CO2, and the media were supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum.
Preparation of mitochondria
Mitochondria were isolated as previously described (36). All subsequent steps were performed at 4 C. Cells (∼2 × 107) in 15-ml conical tubes were precipitated by centrifugation (500 × g, 10 min). Supernatants were carefully removed. The precipitated cells were resuspended and washed with 1 ml 0.9% NaCl solution. Cells were lysed with 1 ml ice-cold lysis buffer by pipetting up and down several times, incubated for 10 min on ice on a shaker, and then precipitated by centrifugation (1000 × g, 10 min). Supernatants were removed and pellets were resuspended in 1.5 ml MSE buffer (containing 225 mm mannitol; 75 mm sucrose; 1 mm EGTA; 5 mm HEPES, pH 7.4; and 1 mg/ml BSA), followed by centrifugation at 1000 × g for 3 min. The supernatants were transferred to a clean 1.5-ml tube, followed by centrifugation at 6000 × g for 10 min. Supernatants were removed and the pellets, mitochondria, were resuspended with 1 ml MSE media containing EGTA. It is important to resuspend mitochondria in a buffer containing EGTA because even a residue amount of calcium in the buffer will severely inhibit mitochondrial reactive oxygen species (ROS) production. Concentrations of mitochondrial proteins were by determined by using a standard approach. To isolate mitochondria from fresh liver, liver samples were collected promptly and homogenized (60 mg/liver). Isolation of mitochondria liver was performed similarly as detailed above.
Measurement of cholesterol
Cells, tissues, or mitochondria were collected, lysed with ice-old lysis buffer (18 mm Tris-HCl; 300 mm mannitol; 50 mm EGTA, pH 7.6, with protease inhibitors), and then sonicated for 10 1-sec pulses with a microtip. Lipids were extracted as described by Folch et al. (37). Briefly, methanol and chloroform [1:2 (vol/vol)] were added into lysates and mixed by vortexing, followed by a 2-h violent shaking. KCl (0.8 ml, 0.15 m) was added and mixed by vortexing and the mixtures were centrifuged for 5 min at 750 rpm (3000 × g) to precipitate lipids. The top aqueous phase was removed, and the bottom phase (organic solvent) was evaporated in a vacuum to achieve lipid pellets. Isopropanol with 10% Triton X-100 was used to dissolve lipids (100–200 μl in total) that contain cholesterol methyl ether. Cholesterol was quantified with the Ample red cholesterol assay kit (Carslbad, CA) in 96-well format. Meanwhile, protein levels in the same samples were measured as the normalization standards.
Immunoblotting
Cells (4 × 105/ml) in six-well plates were treated as noted and then lysed on ice for 30 min in 35 μl lysis buffer [20 mm Tris, pH 7.4; 150 mm NaCl; 1 mm EDTA; 1% Nonidet P-40; 0.1% sodium dodecyl sulfate; 0.5% sodium deoxycholate; 1 mm sodium orthovanadate; 1 mm sodium fluoride; 1 mm phenylmethylsulfonyl fluoride; and proteinase inhibitor cocktail (Roche)]. Cell debris was pelleted by centrifugation, and supernatants were collected and stored at −80 C until assayed. Thirty micrograms of total cellular proteins were denatured at 95 C for 5 min in loading buffer (60 mm Tris, 2.5% sodium dodecyl sulfate, 10% glycerol, 5% mercaptoethanol, 0.01% bromphenol-blue) and subjected to 10% SDS-PAGE gels. Proteins in gels were transferred to polyvinyl difluoride, which were blocked with Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and 5% nonfat milk for 1 h. After being washed with TBS-T, membranes were probed with specific first antibodies against Akt (rabbit) or β-actin (mouse) (1:1000) (Cell Signaling) overnight at 4 C. Membranes were then washed with TBS-T and incubated with polyclonal secondary goat antirabbit or rabbit antimouse antibodies (1:5000; Santa Cruz Biotechnology) for 1 h at room temperature. After three washes with TBS-T, membranes were developed using ECF substrates (GE Healthcare, Indianapolis, IN), according to the manufacturer's protocol (Thermo Scientific, Swedesboro, NJ). Fluorescent bands were visualized and then quantified by densitometry analysis using ImageQuant version 5.2 software from GE Healthcare.
Measurement of ROS from cultured cells
Intracellular ROS was detected by using fluorescent DCF-DA as previously described (36, 38). This nonfluorescent dye freely permeates into cells, in which it is deesterified to form the ionized free acid (dichlorofluorescein) and reacts with ROS to form fluorescent 2′,7′-dichlorofluorescein (DCF). After the noted treatments, cells were washed with PBS three times and then loaded with DCF-DA (20 μm) for 30 min at 37 C to allow for complete deesterification of the dye. DCF fluorescence was quantified by using a fluorescence plate reader (Spectramax Gemini EM; Molecular Devices, Eugene, OR) at the excitation and emission wavelengths of 490 and 530 nm, respectively.
Measurement of ROS release from isolated mitochondria
Mitochondrial generation of ROS was measured by quantifying DCF-DA as detailed above. Mitochondria (0.1 mg protein per milliliter) were incubated at 37 C in the KCL buffer (125 mm KCL; 2 mm K2HPO4; 1 mm MgCl2; and 20 mm HEPES, pH 7.4). Cholesterol (10 μm) was added to isolated mitochondria. As noted, rotenone (0.5 μm) was added to inhibit complex I, and CoQ10 was added to facilitate the electron transportation from complexes I and II to complex III. After the noted treatment, the isolated mitochondria were loaded with DCF-DA (20 μm) for 30 min at 37 C to allow for complete deesterification of the dye. The DCF fluorescence was analyzed using a fluorescence plate reader at excitation and emission wavelengths of 490 and 530 nm, respectively.
Incubation of mitochondria with cholesterol
As previously described (39), cholesterol-BSA aggregates were made by dissolving 50 mg of cholesterol in 5 ml of absolute ethanol and then diluted with 5 ml of double-distilled water. BSA (2 g) was added to the dilution subsequently. The pH of the dilution was adjusted to 7.3. Finally, the dilution was centrifuged at 12000 × g at 4 C for 10 min, and the supernatant was collected as the cholesterol-BSA aggregates. The cholesterol-BSA aggregates were added to mitochondria at noted concentration at 4 C with 50 mg of mitochondria protein for 5 min. Mitochondria were diluted with cold MSE buffer and immediately recovered by centrifugation and washed three times to eliminate the unbound cholesterol.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was determined by flow cytometry using J-aggregate forming lipophilic cationic probe JC-1 according to the protocol from the manufacturer (Sigma). In brief, JC-1 was dissolved in dimethyl sulfoxide (5 mg/ml), and 50 μl of the solution was added to the cells (1 × 105 cells) on six-well plate s(each well contained 2 ml media) for 15 min at 37 C and then washed twice with PBS. Some of these cells were used to visualize the density of JC-1 staining under fluorescence microscopy as previously described (40). Some cells were detached, washed with 0.5 ml of PBS, and transferred to black 96-well plates for quantifying the JC-1 density at excitation/emission wavelengths of 485/530 nm and red fluorescence at excitation/emission wavelengths of 550/595 nm, respectively, as previously described (41).
Measurement of mitochondrial membrane fluidity
Fluidity of mitochondrial membranes was evaluated by fluorescence anisotropy of mitochondrial-bound dyes, 1-[4-(trimethylamino)phenyl]-6-phenyl-1,3,5-hexatriene (TMA)-1,6-diphenyl-1,3,5-hexatriene (DPH) and DPH, as previously described (39). TMA-DPH (3 μm) was added to isolated mitochondria (suspended at 0.5 mg/ml), followed an incubation at 37 C for 30 min. Similarly, dihydropyridine (200 μm) were added to isolated mitochondria in the place of TMA-DPH. Fluorescence polarization was measured in a spectrofluorometer at wavelengths of 366 nm for excitation and 425 nm for emission for TMA-DPH or DPH.
Results
Prolonged exposure to insulin increases mitochondrial content of cholesterol in cultured hepatocytes
To determine the effect of prolonged exposure to insulin on mitochondrial content of cholesterol, Hepa1c1c7 hepatoma cells were incubated with an increasing amount of insulin for 16 h, followed by isolation of mitochondria and measurement of cholesterol in the isolated mitochondria. As shown in Fig. 1A, the level of cholesterol was significantly increased from approximately 120 μg/mg protein (0.31 μm) in control to 190–250 μg/mg protein (0.48–0.68 μm) in cells treated with insulin at 5–10 nm, which are typical plasma levels of insulin in animals with insulin resistance and hyperinsulinemia (16). To determine the levels of the classical Akt-dependent insulin signaling in the cells exposed to insulin, levels of phosphorylated and total Akt and β-actin in the similarly treated cells were evaluated. As shown in Fig. 1B, phosphorylation of Akt was stimulated by insulin. Together these results show that cholesterol was increased in mitochondria by the prolonged exposure to insulin.
Fig. 1.
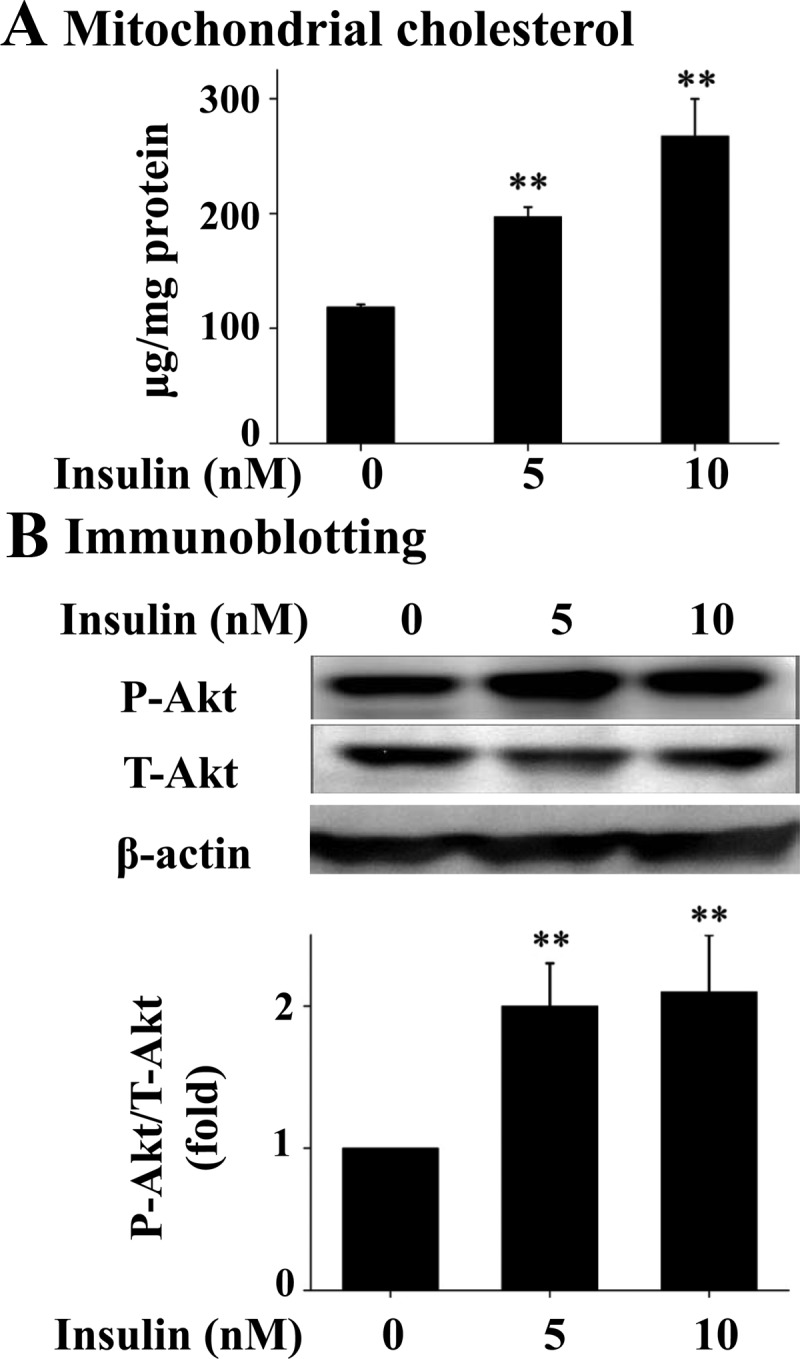
Prolonged exposure to insulin increases mitochondrial content of cholesterol in hepatocytes. Hepa1c1c7 cells were incubated with insulin for 16 h (n = 3). Mitochondria were then isolated and mitochondrial content of free cholesterol was quantified (A). Levels of phosphorylated (P-Akt), total Akt (T-Akt), and β-actin in these same cells were evaluated by immunoblotting with specific antibodies (B). Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no insulin.
Chronic exposure to excess insulin increases mitochondrial content of cholesterol in mice
To determine the effect of excess exposure to insulin on mitochondrial content of cholesterol in animals, B6 mice were treated with the long-acting insulin reagent, glargine, for 8 wk, followed by isolation of hepatic mitochondria and measurement of mitochondrial cholesterol. As shown Fig. 2, the level of cholesterol in hepatic mitochondria was increased significantly by the chronic exposure to excess insulin. It should be noted that animals did not show any sign of hypoglycemia during the whole course of glargine treatment; instead they had increased fasting blood glucose level starting at wk 1 (data not shown). Treatment with glargine did not alter food intake, body weight, and plasma levels of triglyceride and free fatty acids but did increase plasma level of cholesterol significantly (data not shown).
Fig. 2.
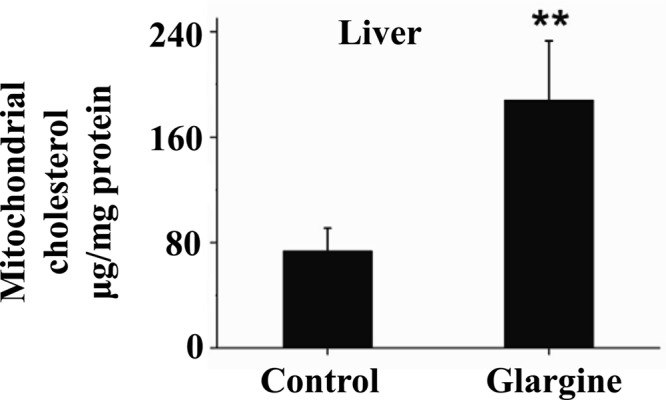
Prolonged exposure to insulin increases mitochondrial content of cholesterol in liver of mice. B6 mice were treated with the long- and slow-acting insulin (glargine) for 8 wk as detailed in Materials and Methods. Liver mitochondria were immediately isolated after animals were killed. Mitochondrial content of cholesterol was quantified. Results represent mean ± sd of six mice per group. **:, P < 0.01.
Prolonged exposure to insulin induces oxidative stress in cultured hepatocytes
To determine the effect of the prolonged exposure to insulin on oxidative stress, Hepa1c1c7 cells and primary hepatocytes were incubated with increasing amount of insulin for 16 h, followed by measurements of ROS level and GSH to GSSG ratio. As shown in Fig. 3, prolonged incubation with insulin stimulated ROS production and decreased the GSH to GSSG ratio. These results show that prolonged exposure to insulin induces oxidative stress in hepatocytes.
Fig. 3.

Prolonged exposure of cultured hepatocytes to insulin increases ROS production. Hepa1c1c7 cells (A and B) or primary mouse hepatocytes (C and D) were incubated with insulin as noted for 16 h (n = 3), followed by incubation with carboxy-H2-DCF-DA for 30 min. Levels of ROS, GSH, and GSSG in the cells were quantified as detailed in Materials and Methods. Results represent Mean ± se of three independent experiments.*, P < 0.05, **, P < 0.01 vs. no insulin.
Exposure of isolated hepatic mitochondria to cholesterol promotes ROS production
To determine the effect of cholesterol on mitochondrial production of ROS, mitochondria were isolated from primary hepatocytes and were incubated with cholesterol bound to BSA, followed by evaluations of ROS production. As shown in Fig. 4, cholesterol stimulated ROS production in isolated hepatic mitochondria when its concentration reached 5 μm or higher concentrations.
Fig. 4.

Exposure of isolated mitochondria to cholesterol increases ROS production. Mitochondria isolated from primary hepatocytes were incubated with cholesterol bound to BSA at noted concentration for 5 min and then with carboxy-H2-DCF-DA for 30 min. ROS levels were subsequently quantified as detailed in Materials and Methods. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no cholesterol.
Oxidative stress induced by the prolonged exposure to insulin is cholesterol synthesis dependent
To determine the effect of cholesterol synthesis on the oxidative stress induced by the prolonged exposure to insulin, Hepa1c1c7 cells and primary mouse hepatocytes were treated with insulin for 16 h in the presence or absence of the cholesterol synthesis inhibitor, simvastatin, followed by measurements of ROS level and activity of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA-R). As shown in Fig. 5, A and B, ROS production was increased by the incubation with insulin in both Hepa1c1c7 cells and primary hepatocytes, but the increase was prevented by simvastatin. Activity of HMG-CoA-R was stimulated by insulin but also inhibited by simvastatin (Fig. 5C). Together these results show that prolonged exposure to insulin promotes ROS production in a cholesterol synthesis-dependent manner.
Fig. 5.

ROS production induced by the prolonged exposure to insulin is cholesterol synthesis dependent in hepatocytes. Hepa1c1c7 cells (A) or primary mouse hepatocytes (B) were treated with either the vehicle solution or insulin (5 nm) in the presence or absence of simvastatin (10 nm) for 16 h as indicated (n = 3). Cells were incubated with carboxy-H2-DCF-DA for 30 min, followed by quantification of ROS level. C, Levels of HMG-CoA-R activity in the cells described in A were quantified by using an HMG-CoA-R assay kit as detailed in Materials and Methods. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no insulin; #, P < 0.05 and ##, P < 0.01 vs. insulin alone.
Prolonged exposure to insulin decreases mitochondrial membrane potential in cultured hepatocytes
To determine the mechanism by which prolonged exposure to insulin induces oxidative stress, mitochondrial membrane potential was visualized by microscopy and quantified by fluorescence density in Hepa1c1c7 cells incubated with insulin for 16 h in the presence or absence of simvastatin. As shown in Fig. 6A, prolonged exposure to insulin turned the color of mitochondria from yellow reddish color into a green color, suggesting that mitochondrial membrane potential was decreased by the prolonged exposure to insulin. However, the effect of insulin was largely reversed when cholesterol synthesis inhibitor simvastatin was added. Similarly, mitochondrial membrane potential measured by quantification of fluorescence density was also decreased by the prolonged incubation with insulin, and the reduction was reversed by simvastatin. To determine whether the reduced membrane potential would lead to cell death, apoptosis level was measured. The prolonged incubation with insulin did not increase apoptotic activity (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Together, these results show that mitochondrial membrane potential was reduced by the prolonged exposure to insulin.
Fig. 6.

Prolonged exposure of hepatocytes to insulin decreases mitochondrial membrane potential in a cholesterol synthesis-dependent manner. Hepa1c1c7cells (A and B) or primary mouse hepatocytes (C) were treated with either the vehicle solution or insulin (5 nm) in the presence of simvastatin (10 nm) for 16 h as indicated (n = 3). Mitochondrial membrane potential was then either visualized by using fluorescent microscopy (A) or quantification of fluorescence density (B and C) as detailed in Materials and Methods. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no insulin control. #, P < 0.05 and ##, P < 0.01 vs. insulin alone.
Cholesterol increases mitochondrial ROS production at complex I
Mitochondrion-derived ROS is usually generated at complex I and/or III (42). To determine the source of ROS caused by cholesterol, complex I inhibitor, rotenone, was first used. As shown in Fig. 7A, induction of ROS by cholesterol was prevented by addition of rotenone. Because coenzyme Q (ubiquinone) plays an essential role in shuttling electrons from complexes I and II to complex III, addition of CoQ10 should increase electron movement to complex III and consequently promote ROS production at complex III if the cholesterol-mediated ROS production comes from complex III. Nevertheless, CoQ10 did not alter the effect of cholesterol on ROS production, as shown in Fig. 7B. Together these results suggest that ROS induced by cholesterol was generated at complex I.
Fig. 7.
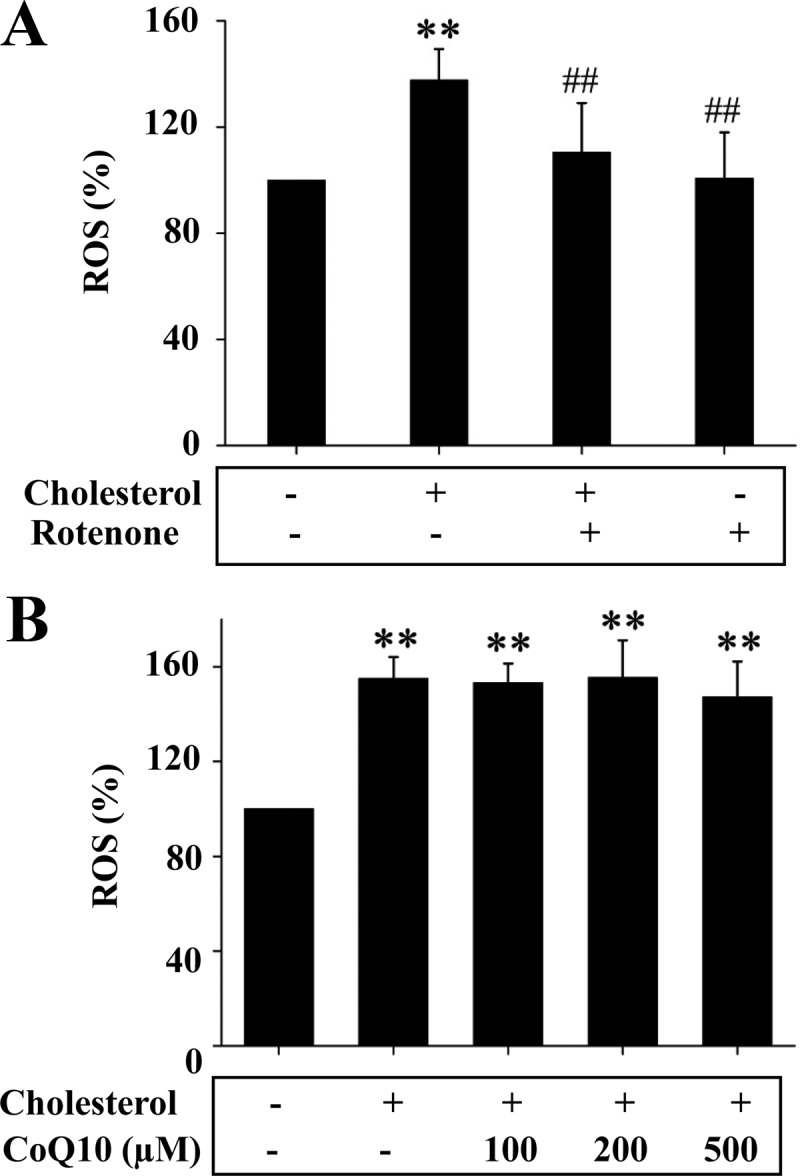
ROS production induced by cholesterol was prevented by the inhibition of mitochondrial respiration complex I. A, Mitochondria isolated from Hepa1c1c7 cells were incubated with cholesterol (10 μm) bound to BSA in the presence or absence of rotenone (10 nm) for 5 min as noted. Carboxy-H2-DCF-DA was then added to the mitochondria for 30 min, followed by measurements of ROS as detailed in Materials and Methods. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no cholesterol control; ##, P < 0.01 vs. cholesterol alone. B, Mitochondria isolated from Hepa1c1c7 cells were incubated with cholesterol (10 μm) bound to BSA in the presence of CoQ10 at noted concentration for 5 min (n = 3) and were then incubated with carboxy-H2-DCF-DA for 30 min, followed by quantifications of ROS as detailed in Materials and Methods. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. no cholesterol control; ##, P < 0.01 vs. cholesterol alone.
Prolonged exposure to insulin decreases mitochondrial membrane fluidity in a cholesterol synthesis-dependent manner
Mitochondrial membrane fluidity is known to regulate the motility and functions of mitochondrial membrane proteins including the respiratory complexes (39, 43). To further determine the mechanism by which prolonged exposure to insulin induces ROS production, the effect of cholesterol on the fluidity of mitochondrial membrane was determined in isolated mitochondria from Hepa1c1c7 cells in two different methods. As shown in Fig. 8, A and B, prolonged exposure to insulin increased mitochondrial absorption of TMA-DPH, suggesting that mitochondrial membrane from cells exposed to insulin were more hydrophobic or less fluid. The effect of insulin on mitochondrial membrane fluidity was prevented by simvastatin. Similarly, mitochondria from cells exposed to insulin absorbed more DPH, also suggesting that mitochondria from cells exposed to insulin were less fluid. Again, the effect of insulin on mitochondrial membrane fluidity was prevented by simvastatin. To further determine the effect of cholesterol on mitochondrial membrane fluidity, isolated mitochondria from cells that were not treated with insulin were exposed to cholesterol, followed by evaluation of mitochondrial membrane fluidity. As shown in Fig. 8, C and D, mitochondrial fluidity measured by either TMA-DPH or DPH was decreased by the direct exposure to cholesterol. Together these results demonstrate that prolonged exposure to insulin alters the fluidity of mitochondrial membrane in a cholesterol synthesis-dependent manner.
Fig. 8.

Prolonged exposure to insulin decreases mitochondrial membrane fluidity in a cholesterol synthesis-dependent manner. A and B, Hepa1c1c7cells were treated with either the vehicle solution or insulin (5 nm) in the presence or absence of simvastatin (10 nm) for 16 h as indicated (n = 3). Mitochondria were then isolated from these cells and labeled with either TMA-DPH or DPH. Fluorescence density was quantified at 366 nm (emission = 440 nm) using polarizing filters in excitation and emission planes and normalized to mitochondrial protein level. C and D, Mitochondria (50 mg proteins) isolated from Hepa1c1c7 cells were incubated cholesterol-BSA complex for 5 min at 4 C (n = 3), washed three times to eliminate the free cholesterol, and labeled with TMA-DPH or DPH. Fluorescence density was quantified at 366 nm (emission = 440 nm) using polarizing filters in excitation and emission planes and normalized to mitochondrial protein level. Results represent mean ± se of three independent experiments. **, P < 0.01 vs. control; ##, P < 0.01 vs. insulin alone.
Discussion
The increased exposure to mtROS and insulin is currently a major health problem caused by the positive energy imbalance due to overeating and/or lack of physical activity. Specifically, the increased exposure to both mtROS and insulin plays a fundamental role in the development of insulin resistance (1–4) and its numerous association health problems (5–13). Nevertheless, it is currently unclear how the increased exposure to insulin is connected with the increased production of mtROS. In this study, we addressed this important issue and made several key findings.
First, our results show that prolonged exposure of hepatocytes or mice to insulin increases mitochondrial content of cholesterol in hepatocytes or liver. Specifically, mitochondrial content of cholesterol is significantly increased by the prolonged exposure of cultured hepatocytes to insulin. Similarly, treatment of mice with long-acting insulin (glargine) also increased mitochondrial content of cholesterol in liver. Excess exposure to insulin is commonly seen in overweight or obese subjects with insulin resistance and hyperinsulinemia. Increased levels of cholesterol synthesis and plasma cholesterol are also frequently seen in these same subjects (44). Numerous studies have shown an association of the increased cholesterol level with the development of various cardiovascular disorders, which are tightly connected to mitochondrion-derived oxidative stress (19, 45). It has been shown in cancer cells that increased mitochondrial content of cholesterol is associated with increased mitochondrial membrane potential (31, 32), which usually leads to increased production of mtROS. However, the level of mitochondrial cholesterol and its impact on mitochondrial function such as mtROS production have not been evaluated in metabolic disease previously. Thus, we have first observed that prolonged exposure to insulin is associated with increased mitochondrial content of cholesterol in cultured hepatocytes and liver.
Second, our results show that prolonged exposure of hepatocytes to insulin stimulates mtROS production by increasing mitochondrial cholesterol. It has previously been shown that insulin is involved in oxidative stress in endothelial cells and hepatocytes. The mechanism by which insulin induces oxidative stress remains unestablished. In endothelial cells, the insulin-induced oxidative stress may be associated with increased nitric oxide because insulin is known to be a potent stimulator of nitric oxide production in endothelials (46, 47). In hepatocytes, previous studies have implicated that excess exposure to insulin may promote oxidative stress through inhibition of mitochondrial biogenesis and mitophagy and increasing mitochondrial long-chain acyl CoA (14, 17, 22). In this study, our results show that the mtROS production induced by the prolonged exposure to insulin is completely prevented by inhibition of cholesterol synthesis in hepatocytes. It is noteworthy that mtROS may play an essential role in the development of insulin resistance. For example, neutralization of hepatic mtROS by overexpression of manganese SOD can prevent or reverse insulin resistance induced by a high-fat diet or genetic obesity in ob/ob mice (1). It should be noted that manganese SOD neutralizes the superoxide from complex I, whereas copper/zinc-SOD neutralizes the superoxide from complex III (42). Neutralization of mtROS can also prevent insulin resistance in various metabolism-active cells (adipocytes, hepatocytes, and myocytes) (3, 4). Interestingly, it has been widely shown that reduction of blood cholesterol level with some cholesterol synthesis inhibitors, statins, can improve insulin sensitivity (48). However, the direct effects of cholesterol synthesis inhibition on mtROS production and insulin sensitivity have not been recognized due to the diverse nonspecific effect of statins. For example, pravastatin can improve insulin sensitivity and stimulate adiponectin secretion (48, 49). Thus, the effect of pravastitin on improving insulin sensitivity has been assumed to be mediated by adiponectin (48). Simvasatin can influence not only adiponectin secretion but also insulin secretion (48, 50). As a result, the direct effect of simvastatin on insulin sensitivity varies a lot from increase, no change, to decrease (48). Our results indicate that increased mitochondrial cholesterol content plays a primary role in mtROS production in hepatocytes. Together with our previous finding that insulin signaling is increased at the basal level and may stimulate cholesterol synthesis continuously, these new results strongly suggest that inhibition of cholesterol synthesis itself may be a viable approach to improve insulin sensitivity through inhibition of mtROS production in subjects with insulin resistance.
Third, our results show that prolonged exposure to insulin/cholesterol increases mtROS production at respiratory complex I through decreasing the fluidity of mitochondrial membrane. It has been shown in cancer cells that increased mitochondrial content reduces the permeability of inner mitochondrial membrane to protons, contributing to increased mitochondrial membrane potential (31, 32). However, our results show that mitochondrial membrane potential is decreased while mitochondrial cholesterol is increased by the prolonged exposure to insulin in hepatocytes, suggesting that the increased mtROS production is not due to increased mitochondrial membrane potential. ROS from mitochondria is usually generated at complexes I and III of mitochondrial respiration chain (42). To determine the source of mtROS, we inhibited respiratory complex I with rotenone in isolated mitochondria exposed to cholesterol and found that inhibition of complex I prevented the mtROS production induced by cholesterol. Besides, supplementation of CoQ10, which transports electrons from complexes I and II to complex III, does not alter the cholesterol-mediated ROS production from mitochondria. Supplementation of CoQ10 should increase the cholesterol-induced ROS production if the ROS comes from complex III. Thus, it appears that prolonged exposure to insulin/cholesterol stimulates mtROS production at complex I, likely through increasing electron leak at complex I. It is known that mitochondrial membrane fluidity can regulate the functions of the proteins localized in the mitochondrial membrane such as the respiratory complexes, and the fluidity of mitochondrial membrane can alter the folding of mitochondrial membrane proteins such as respiratory complexes and can be differentially altered by the membrane contents of lipids including cholesterol and saturated and unsaturated free fatty acids. Thus, the increased electron leak from complex I is likely due to the decreased mitochondrial membrane fluidity. Together our results show that prolonged exposure of hepatocytes to insulin and exposure of isolated mitochondria to cholesterol can both decrease the fluidity of mitochondrial membrane, and the decreased membrane fluidity is a major contributor to the increased mtROS production in hepatocytes.
In summary, we have identified cholesterol synthesis as the link between the excess exposure to insulin and the increased mtROS production in hepatocytes, which plays a critical role in the development of insulin resistance and its many associated major health problems. Therefore, our results will likely provide more specific targets to prevent or reverse many major health problems caused by mtROS.
Supplementary Material
Acknowledgments
This work was supported by the American Diabetes Association Grant 7-09-BS-27 (to W.C.), and National Institutes of Health Grant R01DK076039 (to W.C.).
Disclosure Summary: The authors have no conflict of interest to declare.
Footnotes
- CoQ10
- Coenzyme Q10
- DCF
- 2′,7′-dichlorofluorescein
- DCF-DA
- DCF diacetate
- DPH
- 1,6-diphenyl-1,3,5-hexatriene
- GSH
- glutathione
- GSSG
- glutathione disulfide
- HMG-CoA-R
- 3-hydroxy-3-methylglutaryl coenzyme A reductase
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide
- MSE
- buffer containing mannitol, sucrose, EGTA, HEPES, and BSA
- mtROS
- mitochondrion-derived oxidative stress
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- TBS-T
- Tris-buffered saline containing Tween 20
- TMA
- 1-[4-(trimethylamino)phenyl]-6-phenyl-1,3,5-hexatriene.
References
- 1. Kumashiro N, Tamura Y, Uchida T, Ogihara T, Fujitani Y, Hirose T, Mochizuki H, Kawamori R, Watada H. 2008. Impact of oxidative stress and peroxisome proliferator-activated receptor γ coactivator-1α in hepatic insulin resistance. Diabetes 57:2083–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, Mikus CR, Laye MJ, Laughlin MH, Booth FW, Ibdah JA. 2010. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol 52:727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, Stocker R, Van Remmen H, Kraegen EW, Cooney GJ, Richardson AR, James DE. 2009. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci USA 106:17787–17792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Houstis N, Rosen ED, Lander ES. 2006. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440:944–948 [DOI] [PubMed] [Google Scholar]
- 5. Accili D. 2004. Lilly lecture 2003: the struggle for mastery in insulin action: from triumvirate to republic. Diabetes 53:1633–1642 [DOI] [PubMed] [Google Scholar]
- 6. Burns JM, Donnelly JE, Anderson HS, Mayo MS, Spencer-Gardner L, Thomas G, Cronk BB, Haddad Z, Klima D, Hansen D, Brooks WM. 2007. Peripheral insulin and brain structure in early Alzheimer disease. Neurology 69:1094–1104 [DOI] [PubMed] [Google Scholar]
- 7. Cole GM, Frautschy SA. 2007. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's disease. Exp Gerontol 42:10–21 [DOI] [PubMed] [Google Scholar]
- 8. Crowell JA, Steele VE, Fay JR. 2007. Targeting the AKT protein kinase for cancer chemoprevention. Mol Cancer Ther 6:2139–2148 [DOI] [PubMed] [Google Scholar]
- 9. Moreira PI, Santos MS, Seiça R, Oliveira CR. 2007. Brain mitochondrial dysfunction as a link between Alzheimer's disease and diabetes. J Neurol Sci 257:206–214 [DOI] [PubMed] [Google Scholar]
- 10. Al-Shawwa BA, Al-Huniti NH, DeMattia L, Gershan W. 2007. Asthma and insulin resistance in morbidly obese children and adolescents. J Asthma 44:469–473 [DOI] [PubMed] [Google Scholar]
- 11. Sterry W, Strober BE, Menter A. 2007. Obesity in psoriasis: the metabolic, clinical and therapeutic implications. Report of an interdisciplinary conference and review. Br J Dermatol 157:649–655 [DOI] [PubMed] [Google Scholar]
- 12. Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. 2007. The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 48:751–762 [DOI] [PubMed] [Google Scholar]
- 13. Okazaki R. 2008. Links between osteoporosis and atherosclerosis: beyond insulin resistance. Clin Calcium 18:638–643 [PubMed] [Google Scholar]
- 14. Liu HY, Yehuda-Shnaidman E, Hong T, Han J, Pi J, Liu Z, Cao W. 2009. Prolonged exposure to insulin suppresses mitochondrial production in primary hepatocytes. J Biol Chem 284:14087–14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu HY, Cao SY, Hong T, Han J, Liu Z, Cao W. 2009. Insulin is a stronger inducer of insulin resistance than hyperglycemia in mice with type 1 diabetes mellitus (T1DM). J Biol Chem 284:27090–27100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu HY, Hong T, Wen GB, Han J, Zhuo D, Liu Z, Cao W. 2009. Increased basal level of Akt-dependent insulin signaling may be responsible for the development of insulin resistance. Am J Physiol Endocrinol Metab 297:E898–E906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ning J, Hong T, Yang X, Mei S, Liu Z, Liu HY, Cao W. 2011. Insulin and insulin signaling play a critical role in fat induction of insulin resistance in mouse. Am J Physiol Endocrinol Metab 301:E391–E401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pamplona R, Barja G. 2006. Mitochondrial oxidative stress, aging and caloric restriction: the protein and methionine connection. Biochim Biophys Acta 1757:496–508 [DOI] [PubMed] [Google Scholar]
- 19. Madamanchi NR, Runge MS. 2007. Mitochondrial dysfunction in atherosclerosis. Circ Res 100:460–473 [DOI] [PubMed] [Google Scholar]
- 20. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. 2002. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23:599–622 [DOI] [PubMed] [Google Scholar]
- 21. Widlansky ME, Wang J, Shenouda SM, Hagen TM, Smith AR, Kizhakekuttu TJ, Kluge MA, Weihrauch D, Gutterman DD, Vita JA. 2010. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl Res 156:15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. 2009. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem 284:31484–31492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. 2010. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loschen G, Flohé L, Chance B. 1971. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett 18:261–264 [DOI] [PubMed] [Google Scholar]
- 25. Wojtczak L, Zaluska H. 1967. The inhibition of translocation of adenine nucleotides through mitochondrial membranes by oleate. Biochem Biophys Res Commun 28:76–81 [DOI] [PubMed] [Google Scholar]
- 26. Lerner E, Shug AL, Elson C, Shrago E. 1972. Reversible inhibition of adenine nucleotide translocation by long chain fatty acyl coenzyme A esters in liver mitochondria of diabetic and hibernating animals. J Biol Chem 247:1513–1519 [PubMed] [Google Scholar]
- 27. Shug AL, Shrago E, Bittar N, Folts JD, Koke JR. 1975. Acyl-CoA inhibition of adenine nucleotide translocation in ischemic myocardium. Am J Physiol 228:689–692 [DOI] [PubMed] [Google Scholar]
- 28. Pande SV, Blanchaer MC. 1971. Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J Biol Chem 246:402–411 [PubMed] [Google Scholar]
- 29. Shrago E, Shu A, Elson C. 1976. Regulation of cell metabolism by mitochondrial transport systems. In: Hanson RW, eds. Gluconeogenesis: its regulation in mammalian species. New York: John Wiley, Sons; 221–238 [Google Scholar]
- 30. Li LO, Klett EL, Coleman RA. 2010. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta 1801:246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baggetto LG, Testa-Parussini R. 1990. Role of acetoin on the regulation of intermediate metabolism of Ehrlich ascites tumor mitochondria: its contribution to membrane cholesterol enrichment modifying passive proton permeability. Arch Biochem Biophys 283:241–248 [DOI] [PubMed] [Google Scholar]
- 32. Baggetto LG, Clottes E, Vial C. 1992. Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res 52:4935–4941 [PubMed] [Google Scholar]
- 33. Cao W, Collins QF, Becker TC, Robidoux J, Lupo EG, Jr, Xiong Y, Daniel KW, Floering L, Collins S. 2005. p38 mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem 280:42731–42737 [DOI] [PubMed] [Google Scholar]
- 34. Xiong Y, Collins QF, An J, Lupo E, Jr, Liu HY, Liu D, Robidoux J, Liu Z, Cao W. 2006. p38 mitogen-activated protein kinase plays an inhibitory role in hepatic lipogenesis. J Biol Chem 282:4975–4982 [DOI] [PubMed] [Google Scholar]
- 35. Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W. 2007. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5′-AMP-activated protein kinase. J Biol Chem 282:30143–30149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palmer JW, Tandler B, Hoppel CL. 1977. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252:8731–8739 [PubMed] [Google Scholar]
- 37. Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509 [PubMed] [Google Scholar]
- 38. D'Alessandro S, Basilico N, Corbett Y, Scaccabarozzi D, Omodeo-Salè F, Saresella M, Marventano I, Vaillant M, Olliaro P, Taramelli D. 2011. Hypoxia modulates the effect of dihydroartemisinin on endothelial cells. Biochem Pharmacol 82:476–484 [DOI] [PubMed] [Google Scholar]
- 39. Colell A, García-Ruiz C, Lluis JM, Coll O, Mari M, Fernández-Checa JC. 2003. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J Biol Chem 278:33928–33935 [DOI] [PubMed] [Google Scholar]
- 40. Szilágyi G, Simon L, Koska P, Telek G, Nagy Z. 2006. Visualization of mitochondrial membrane potential and reactive oxygen species via double staining. Neurosci Lett 399:206–209 [DOI] [PubMed] [Google Scholar]
- 41. Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. 1993. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun 197:40–45 [DOI] [PubMed] [Google Scholar]
- 42. Wallace DC, Fan W. 2010. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10:12–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rockett BD, Franklin A, Harris M, Teague H, Rockett A, Shaikh SR. 2011. Membrane raft organization is more sensitive to disruption by (n-3) PUFA than nonraft organization in EL4 and B cells. J Nutr 141:1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paramsothy P, Knopp RH, Kahn SE, Retzlaff BM, Fish B, Ma L, Ostlund RE., Jr 2011. Plasma sterol evidence for decreased absorption and increased synthesis of cholesterol in insulin resistance and obesity. Am J Clin Nutr 94:1182–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ballinger SW. 2005. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med 38:1278–1295 [DOI] [PubMed] [Google Scholar]
- 46. Joshua IG, Zhang Q, Falcone JC, Bratcher AP, Rodriguez WE, Tyagi SC. 2005. Mechanisms of endothelial dysfunction with development of type 1 diabetes mellitus: role of insulin and C-peptide. J Cell Biochem 96:1149–1156 [DOI] [PubMed] [Google Scholar]
- 47. Muniyappa R, Iantorno M, Quon MJ. 2008. An integrated view of insulin resistance and endothelial dysfunction. Endocrinol Metab Clin North Am 37:685–711, ix-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koh KK, Sakuma I, Quon MJ. 2011. Differential metabolic effects of distinct statins. Atherosclerosis 215:1–8 [DOI] [PubMed] [Google Scholar]
- 49. Takagi T, Matsuda M, Abe M, Kobayashi H, Fukuhara A, Komuro R, Kihara S, Caslake MJ, McMahon A, Shepherd J, Funahashi T, Shimomura I. 2008. Effect of pravastatin on the development of diabetes and adiponectin production. Atherosclerosis 196:114–121 [DOI] [PubMed] [Google Scholar]
- 50. Yada T, Nakata M, Shiraishi T, Kakei M. 1999. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet β-cells. Br J Pharmacol 126:1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.