

Abstract
Background
Filopodia, retraction fibers and microvilli, are fragile microextensions of the plasma membrane that are easily damaged by mechanical force during specimen preparation for microscopy. To preserve these structures for electron microscopy glutaraldehyde is generally used, but it often causes antigen masking. By contrast, formaldehyde is generally used for immunofluorescence light microscopy, but few studies have been concerned with the loss of microextensions.
Results
We demonstrate in biochemical experiments that cultured cells needed to be kept in 4% formaldehyde for at least 60 min at room temperature or for 20 min at 37°C to irreversibly crosslink most of the polypeptides. Also, fragmentation of fragile microextensions was observed after Triton X-100 extraction depending on concentration and extent of crosslinking. We also report on a novel fixation procedure that includes the cationic detergent dodecyltrimethylammonium chloride (DOTMAC). Treatment of NIH3T3 cells with DOTMAC resulted in complete removal of membrane lipids and in good preservation of the cytoskeleton in microextensions as well as preservation of ultramicroextensions of <0.05μm in diameter that have not been observed previously unless glutaraldehyde was used. Stress fibers and microextensions of DOTMAC-extracted cells were readily stained with anti-β-actin antibodies, and antibodies to vinculin and moesin stained focal contacts and microextensions, respectively.
Conclusions
Some microextensions were fragmented by the standard Triton X-100 permeabilization method. By contrast, DOTMAC completely extracted membrane lipids while maintaining the cytoskeleton of microextensions. Thus, DOTMAC treatment may provide a valuable new tool for the reliable visualization of previously undetectable or poorly detectable antigens while preserving the actin cytoskeleton of microextensions.
Background
Cellular events are based on specific inter- and intra-molecular interactions of many molecules. To experimentally dissect such interactions, it is important to maintain as closely as possible the cytosolic environment of the living cell. Detergents are frequently employed to disrupt cells for the demonstration and isolation of functionally relevant protein complexes and/or to selectively extract certain proteins. For example, the non-ionic detergents Triton X-100, Tween 20 or Brij, have been widely used for histochemical purposes or in biochemical experiments to separate detergent-soluble from a detergent-resistant residue [1,2,3]. The latter is commonly referred to as the "cytoskeleton" or "membrane skeleton" [4]. During cell activation discrete signaling molecules are known to associate with or to disassociate from the plasma membrane and both, the cytoskeleton and membrane skeleton undergoes significant changes as well [4,5,6,7]. For cytochemical purposes, however, detergents cannot be used before cells have been fixed, as non-ionic detergent treatment causes removal of the cells from their supporting surface (e.g. cover glass). The choice of fixative for a particular cytochemical study is often a difficult task owing to the limited available information about the effects of fixatives on cultured cells.
Considering their effect on proteins, fixatives in general can be divided into two types: "coagulant" or "non-coagulant". Both groups of fixatives have advantages and disadvantages. The coagulant fixatives, including methanol [8, 9], ethanol [10], acetone [11], and trichloroacetic acid [10, 12], denature and precipitate proteins. These fixatives stop biological reactions instantaneously and presumably leave proteins in their original locations. Due to extraction of membrane structures by such fixatives it is not necessary to add detergents during staining with membrane impermeable probes such as antibodies. However, the denaturation of proteins seen with these fixatives has disadvantages for some probes [8, 12] and for visualizing intact cell surfaces. Moreover, it is not known if and to what extent proteins are lost during specimen preparation for microscopic observation (e.g. during incubation or washing procedures). The non-coagulant fixatives are mainly chemical crosslinkers that include glutaraldehyde, formaldehyde, acrolein, and dithiobis (succinimidylpropionate). They are used to preserve surface morphology of cells and to immobilize cellular components before extraction with detergents [8, 13, 14]. However, excess crosslinking frequently masks binding domains for probes while inadequate fixation may not preserve structures to leave them susceptible to mechanical forces during specimen processing. The latter is quite critical especially to preserve fine and fragile cellular microextensions such as filopodia or retraction fibers [15, 16].
We have been interested in the formation and function of microextensions because of their dynamic and universal nature, and since moesin, an actin-binding protein is enriched in these structures [17, 18]. The F-actin-binding activity of moesin is regulated by both phosphorylation and polyphosphoinositides (PIPs)1. Interestingly, PIPs could be substituted for by a cationic detergent, dodecyltrimethylammonium chloride (DOTMAC) in vitro [19]. This result was consistent with biochemical extraction studies, since phosphorylated moesin specifically co-sedimented with DOTMAC-, but not Triton X-100-resistant components, most likely because Triton X-100 interfered with the interaction between moesin and PIPs [19]. This result also suggested that Triton X-100 extraction of cells may not preserve moesin-actin filament complexes.
As expected, we show here that some of microextensions were fragmented by the standard Triton X-100 permeabilization method. By contrast, DOTMAC completely extracted membrane lipids while maintaining the cytoskeleton of microextensions.
We also observed microextensions having a considerably smaller diameter from those commonly seen (<0.05 μm vs. 0. 2 μm). Referred to here as ultramicroextensions, these extended from cell bodies and grew out from the tips of other microextensions. DOTMAC treatment exposed and stabilized their cytoskeleton as well.
Results
Examination of microextensions of NIH3T3 fibroblast by scanning electron microscopy
We used NIH3T3 cells because their morphology and localization of cytoskeletal proteins including actin, tubulin, vinculin, and moesin have been extensively investigated using various fixatives. When removed from stock cultures with trypsin and inoculated into coverslip-containing dishes, NIH3T3 cells readily attached to the serum-coated substrate, began to spread and flattened within one hour (data not shown). Consistent with the report by Rajaraman et al.[20] on human diploid WI-38 cells and Albrecht-Buehler and Goldman [21] on 3T3 fibroblasts, microextensions appeared within 20 min after seeding. Initially during spreading, the cells lost microvilli from their upper cell surface and formed microextensions with a diameter of about 0.2 μm and a maximal length of about 10 μm. Subsequently, the cells continued to spread and to randomly elongate microextensions up to 40 μm. However, closer examination of the fixed cells with a 60° tilt angle showed numerous additional protrusions extending from the cell body with a diameter of only about 0.05 μm (Figure 1). These structures were not observed in cells prior to 12 hours after inoculation. At 48 hour, about 5% (7/130) of the cells had extended such structures that usually were < 0.5 μm in length, and rarely exceeded 1 μm. Because of these unique features, we will use the term ultramicroextensions. Three days after reaching confluency (at about one week in culture), about half of the surface of the cells was covered with ultramicroextensions (Figure 1, D,E,F). When cells were inoculated at high density, ultramicroextensions appeared earlier, but ultramicroextensions were rarely seen even after incubation for prolonged periods in sparse cultures or when the cells did not make contact (data not shown). Ultramicroextensions originated either from microextensions of neighboring cells or from tips of other microextensions in a broom-like fashion (Figure 1G). Ultramicroextensions also originated from the cell surface similar to microvilli (Figure 1H), or as branches from other microextensions and ultramicroextensions (Figure 1I). Identical results were obtained when the cells were grown on poly-L-lysine-coated coverslips (data not shown).
Figure 1.

Scanning electron micrographs of NIH3T3 fibroblasts with a tilt angle 60°. (A-C) Exponentially growing NIH3T3 cells were seeded onto serum-coated glass coverslips at relatively low density (1 × 103 cells/cm2) thereby avoiding cells to contact each other. After 48 hours, cells were fixed with 2% glutaraldehyde in PBS(+), and prepared for SEM as described in Materials and Methods. (A) SEM image of NIH3T3 cell at low magnification. Note several micropodia, but not ultramicroextensions, at this magnification. (B) Higher magnification view of area indicated by square in A. Note that ultramicroextensions can be detected at this magnification. (C) Higher magnification view of area indicated by square in B. Sample was observed from the direction indicated by arrow in B. Note the diameter of ultramicroextensions is less than 0.05 μm and some of the ultramicroextensions appear to originate from a ridge of the cell body (arrowhead). (D-F) Cells were seeded onto serum-coated glass coverslips at 1 × 104 cells/cm2. After 1 week, cells were fixed with 2% glutaraldehyde in PBS(+) and prepared for SEM. (D) SEM image of NIH3T3 cell at low magnification. Note that cells are attached to each other and about half of the cell surface is not smooth. (E and F) Higher magnification view of area indicated by square in D. Note the numerous and parallel rather thin and long microextensions covering some of cell surface. (G-I) High magnification view of an NIH3T3 cell showing the origins of ultramicroextensions. They appear to grow from the tip of microextension in a broom-like fashion (G), from the cell surface like microvilli (H), or branch off from microextensions or ultramicroextensions (I).
Exposure of cytoskeleton in microextensions and ultramicroextensions by DOTMAC extraction
Fixation with formaldehyde followed by extraction with Triton X-100 is a fairly standard method for immunofluorescence microscopy of cells, but microextensions were fragmented and sometimes even lost during this treatment as shown in Figure 2, A,B,C. an alternative method, we extracted cells also with DOTMAC and discovered that fixation and extraction with 1.0 % PFA/0.5 % DOTMAC in PBS(+) on ice for 5 min, followed by fixation with 1.0% PFA in PBS on ice for 20 min (DOTMAC/PFA method), preserved not only micropodia (white arrow, Figure 2, E and 2F), but also microvilli (black arrow, Figure 2E and 2F), and ultramicroextensions (white arrowhead, Figure 2F). This treatment extracted lipids (data not shown) and exposed cytoplasmic filaments giving origin to ultramicroextensions (Figure 2F). At the cell edge the ultramicroextensions are derived from a deeper cytoskeletal meshwork (white arrowhead, Figure 2F) rather than from thick cytoskeletal filaments.
Figure 2.

(A) Scanning electron micrographs of NIH3T3 fibroblast extracted with Triton X-100 or DOTMAC at a tilt angle 60°. (A-C) NIH3T3 cells on glass coverslips were rinsed with PBS(+) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were then extracted with 0.2% Triton X-100 for 5 min at room temperature, and prepared for SEM. (B) Higher magnification view of area indicated by square in A. Note that some of microextensions appeared to be lost. (C) Higher magnification view of area indicated by square in B. Note the fragmentation of microextensions. (D-F) Cells were fixed with the DOTMAC/PFA method and prepared for SEM. (E) Higher magnification view of area indicated by square in D. Note that lipid plasma membrane appeared to be lost and micropodia (white arrow) and microvilli (black arrow) are well preserved. (C) Higher magnification view of area indicated by square in E. Note that origins of ultramicroextensions (arrowhead) were exposed. Some of ultramicroextensions appear to grow out from the inner space of cytoplasm.
We have performed ultrastructural analysis by SEM of cells fixed and/or extracted by the methods listed in Table 1. When the cells were treated with coagulant fixatives, the cytoskeleton was apparently exposed, but the three dimensional structures at the edge of the cells were not preserved as well as those prepared by the DOTMAC/PFA method (data not shown). When the cells were extracted with Triton X-100, NP-40 or saponin after relatively weak fixation with formaldehyde, the cytoskeleton apparently was lost, fragmented, or scraped off depending on the concentration of detergents and the period of incubation (Table 2). Too high a concentration of glutaraldehyde or incubation for extended period of time in formaldehyde, on the other hand, did not allow to expose the cytoskeleton (data not shown).
Comparison of different fixation/extraction methods for preservation and fluorescence labeling of microextensions
We also compared the different fixation and extraction procedures listed in Table 1 with respect to fluorescence intensity and labeling of actin stress fibers and microtubules, and with respect to morphological preservation of cells and microextensions (Table 2). The latter was also assessed from SEM micrographs before and after preparation for fluorescence microscopy and these results are summarized in Table 2 as well.
Staining of actin and tubulin filaments differed depending on the probe and on the fixative. Among the coagulant fixatives, acetone produced the best results with respect to preservation of microextensions, staining of actin with either phalloidin or with monoclonal β-actin antibodies, and staining of microtubules with a monoclonal anti-β-tubulin reagent. After methanol or ethanol fixation, some cells were partially detached and changed their shape, but this did not occur with other coagulant fixatives. Acetone also provided good preservation of microextensions even after preparing the cells for immunolabeling, whereas some of the microextensions were lost, when cells were fixed with other coagulant fixatives (Table 2).
Fixation with chemical crosslinkers, followed by extraction with detergents or organic solvents, are standard procedures for preparing cells for immunofluorescence. First, we investigated effects on the preservation of microextensions by variations in detergent concentration and time of incubation with formaldehyde. As summarized in Table 2, with shorter time periods of fixation and with higher detergent concentrations more cells lost microextensions. For example, when cells were fixed with 3.7% FA for 20 min and extracted with 0.2% TX100 for 5 min (Table 1), stress fibers were stained well with phalloidin, but microextensions were lost (Figure 3A and 3B). The SEM observation before and after preparation for fluorescence microscopy (washing and incubation with probes) indicated that microextensions were lost during these steps of the procedure. On the other hand, microextensions of cells prefixed with glutaraldehyde or DSP prior to extraction were very well preserved indeed (Figure 3E and 3I). However, the cytoskeleton of glutaraldehyde-fixed cells could not be exposed by the detergent and some structures apparently were lost in DSP-Tsb-fixed cells when analysed by SEM (data not shown). Quick extraction with 0.2% Triton X-100 in MTSB (30 seconds, 2 times), followed by fixation with 1% glutaraldehyde also preserved microextensions well indicating that fragmentation or removal of microextensions by extraction with 0.2% Triton X-100 does not occur within 1 min. The DOTMAC/PFA method provided excellent morphological preservation and immunolabeling with high fluorescence intensity of actin stress fibers and microextensions (Figure 3Q and 3S). Although phalloidin staining and immunolabeling of tubulin were poor in cells prepared by the DOTMAC/PFA method (Figure 3R and 3T), this method provided the widest spectrum of reactivity with the probes investigated here (Figure 3 and Table 1). The monoclonal antibody reagent specific for β-actin worked only in TCA or DOTMAC/PFA-treated cells (Figure 3O and 3S).
Figure 3.

Scanning electron and confocal laser scanning micrographs of NIH3T3 cells fixed with various procedures. (A, E, I, M, and Q) SEM image of NIH3T3 cells shown with no tilt angle. Cells were double labeled with TRITC-phalloidin (B, F, J, N, and R) and anti-β-tubulin monoclonal antibodies (T4026), which was detected using a secondary antibody conjugated to FITC (D, H, L, P, and T). Note that the fluorescence intensity of each samples varied as summarized in Table 2. Images are shown at optimum fluorescence intensity. Cells were also stained with anti-β-actin monoclonal antibodies (A5441), followed by FITC-conjugated anti-mouse IgG (C, G, K, O, and S). Cells were rinsed with PBS(+) and fixed with 3.7% FA/0.2% TX100 (see Table 1, A-D), 4% PFA+1% GA/0.2% TX100 (see Table 1, E-H), DSP-Tsb (see Table 1, I-L), TCA (see Table 1, M-P), or DOTMAC/PFA (see Table 1, Q-T). Bars, 10 μm.
Next, we compared the staining patterns of two well-characterized cytoskeletal proteins, vinculin and moesin, in cells prepared by DOTMAC/PFA and other conventional fixation methods (Figure 4). As expected. vinculin localized at focal contacts in cells prepared by either PFA/Triton X-100 or by DOTMAC/PFA (Figure 4, B, J, and 4M). Moesin staining, on the other hand, was primarily at the edge of cells and in microextensions, when cells were fixed with TCA or DOTMAC/PFA (Figure 4G and 4K). DOTMAC/PFA was superior to TCA and the standard Triton X-100 permeabilization procedure to preserve microextensions and for moesin staining (Figure 4, G, K, and 4N,O,P).
Figure 4.

Comparison of fluorescence labeling of actin, vinculin, moesin and threonine558-phospho-moesin in NIH3T3 cells. Cells were rinsed with PBS(+) and fixed with TX100 (3.7% FA/0.2% TX100, see Table 1, A-D), TCA (see Table 1, E-H), or DOTMAC/PFA (see Table 1, I-L). Cells were double labeled with β-actin monoclonal antibodies (A5441) and a secondary antibody reagent conjugated to TRITC (A, E, and I), as well as with moesin polyclonal antibodies and secondary antibodies conjugated to FITC (C, G, and K). Vinculin and threonine558-phospho-moesin were also stained with vinculin monoclonal (B, F, and J) and threonine558-phospho-moesin polyclonal antibodies (D, H, and L), followed by FITC-conjugated anti-rabbit IgG. Note that the fluorescence intensity of each samples varied as summarized in Table 2. Images are shown at optimum fluorescence intensity. (M) Double exposure of J and TRITC-phalloidin-counterstained cells fixed with DOTMAC/PFA method. (N-M) Double exposure of I and K at different level of focus. Bars, 10 μm.
TCA fixation was reported recently to be useful for staining of phosphorylated moesin [12]. Therefore, we tested our anti-phospho-moesin antibodies with this method and, as shown in Figure 4H, the fluorescence intensity was too low to distinguish characteristic structures probably because the amount of phosphorylated moesin is low in unstimulated NIH3T3 cells (cf. Figure 7). Figure 5 demonstrates another example of NIH3T3 cells stained with actin monoclonal and moesin polyclonal antibodies after DOTMAC/PFA fixation. Many branched microextensions were double-stained with these antibodies, but occasionally, part of a microextension or an entire microextension apparently lacked actin [17, 22].
Figure 7.

Determination of detergent-resistant components of NIH3T3 cells before (C) and after treatment with kinase, staurosporine (ST), or phosphatase inhibitors, calyculin A (CA) or pervanadate (PV). The subconfluent cells were rinsed with PBS(+), and detached by treating with 0.02% EDTA in PBS. The suspended cells were incubated with or without 1 μM calyculin A, 1 μM staurosporine, or 100 μM pervanadate in PBS(+) for 10 min at 37°C. The cells were extracted for 5 min on ice with an equal volume of 2× detergent extraction buffer containing 2% of Triton X-100 or DOTMAC. The lysates were centrifuged at 15,800 × g for 5 min at 4°C. The resulting pellets were solubilized in SDS sample buffer and subjected to SDS-PAGE on a 9% polyacrylamide gel electrophoresed. Polypeptides were stained with Coomassie Brilliant blue (CBB). β-Actin, moesin, and threonine558-phospho-moesin were detected by immunoblot analysis.
Figure 5.
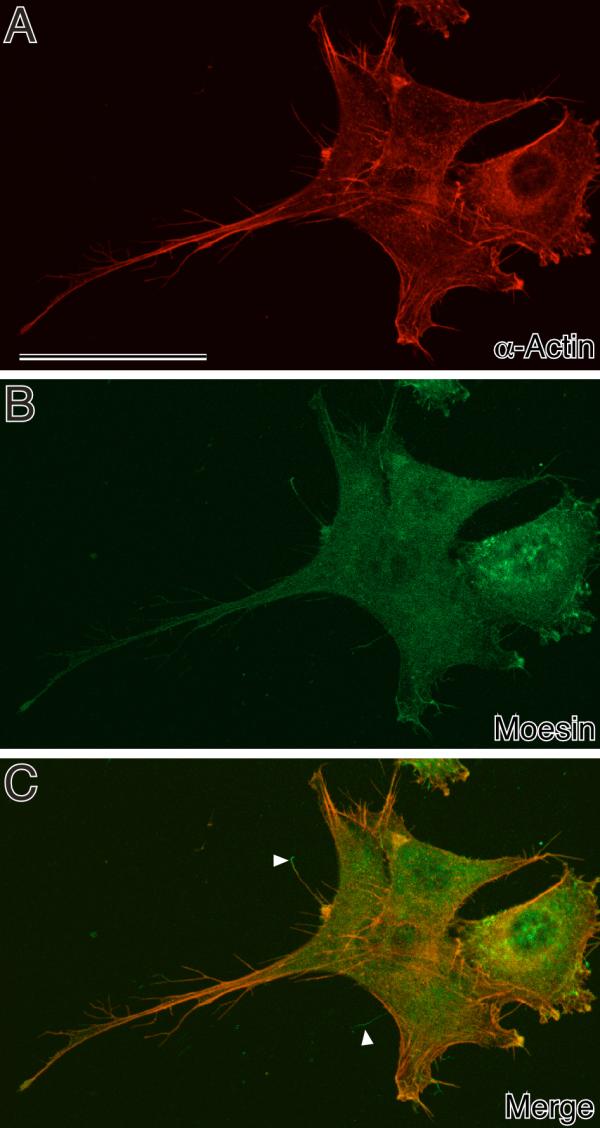
Double staining of actin and moesin in NIH3T3 cells fixed with DOTMAC/PFA shown at high magnification. Note that branched micropodia were well preserved by the DOTMAC/PFA fixative and stained with both β-actin monoclonal and moesin polyclonal antibodies. Some of the moesin-positive micropodia were β-actin-negative (arrowhead). Bars, 50 μm.
Evaluation of crosslinked polypeptides after fixation and extraction
Our results suggested that some of procedures listed in Table 1 were inadequate for preserving fine structural details. Therefore, we re-evaluated the extent of crosslinking of polypeptides by gel electrophoresis and western blotting. The polypeptide banding patterns of extracts from unfixed and fixed cells with SDS sample buffer are shown in Figure 6. Incubation in 0.5% Triton X-100 in PBS detached cells from the dish (Figure 6A, lane 2), but addition of 1 mM magnesium and 1 mM calcium ions in PBS prevented detachment to some extent (Figure 6A, lane 3). Microtubule stabilizing buffer also prevented detachment (Figure 6A, lane 5). On the other hand, most polypeptides were retained on the dish after DOTMAC treatment (Figure 6A, lane 6). Although most of β-actin was retained after 0.5% detergent extraction, moesin was not unless 1% paraformaldehyde was added (Figure 6A, lanes 4 and 7). Under these conditions, moesin and β-actin migrated predominantly as monomeric polypeptides at 45 and 78 kDa positions on the SDS-PAGE gels and only very small amounts of crosslinked moesin and β-actin were detected by western blotting at higher molecular weight positions (data not shown).
Figure 6.

SDS-polyacrylamide gel electrophoresis of extracted polypeptides from NIH3T3 cells after fixation and/or permeabilization. Subconfluent cultures of cells were rinsed with PBS(+), fixed and/or extracted as indicated. After rinsing with PBS, the material remaining on the culture dish was extracted with SDS sample buffer. The dissolved and undissolved material was collected and heated at 95°C for 10 min and resolved by SDS-PAGE on a 9% polyacrylamide gel. Polypeptides were stained with Coomassie Brilliant blue (CBB). β-Actin, moesin, and threonine558-phospho-moesin were detected by immunoblot analysis. (A) Cells were extracted with the extraction buffer as indicated in the figure for 5 min at room temperature. In the last lane to the right, cells were fixed with 1% PFA in PBS for 20 min at 4°C after extraction. (B) Cells were fixed and/or extracted with the fixative indicated in the figure as shown in Table 1 except that 3.7% formalin was used instead of 3.7% formaldehyde in lane 7. (C) Cells were fixed with 4% PFA in PBS(+) for the indicated time period (min) at 25°C or 37°C, rinsed with PBS, and then extracted with SDS sample buffer. Note that moesin is more readily irreversibly crosslinked than actin. Other polypeptides appeared to be same as moesin.
Crosslinking of polypeptides was also evaluated after treatment with several other fixatives that preserved microextensions well and that were found to be useful previously for moesin staining (Figure 6B). Only a small amount of polypeptides was extracted when cells were fixed with glutaraldehyde or DSP-Tsb (Figure 6B, lane 2, 3 and 6) and the cells were morphologically well preserved by these fixatives as observed by SEM, indicating that most proteins were irreversibly crosslinked. On the other hand, when cells were fixed with PLP or LP and treated with 1% saponin in 3% BSA for permeabilization, most of the polypeptides were either not crosslinked by these relatively mild fixatives or crosslinks were reversed by the addition of SDS sample buffer (Figure 6B, lanes 4 and 5). Similar results were obtained when cells were fixed with 3.7% formalin or 4% PFA, followed by permeabilization with 0.1% Triton X-100. Threonine558-phosphomoesin was not dephosphorylated in these fixatives, as determined by western blotting with specific phosphomoesin antibodies (Figure 6B). We examined the effect of incubation time and temperature during fixation with 4% PFA in PBS(+), since conditions for irreversible crosslinking of polypeptides in cultured cells with formaldehyde are unknown. As shown in Figure 6C, fixation under the widely used conditions at room temperature (25°C) for 20 min, is not sufficient to irreversibly crosslink actin, moesin and many other polypeptides. At 37°C, the required time for incubation can be shortened, but most of the actin molecules were not irreversibly crosslinked even after 30 min. Incubation with sodium borohydride after fixation in PFA had no effect either (data not shown) suggesting that reversible bridges may have formed that could not be reduced.
Biochemical analysis of DOTMAC-insoluble materials of NIH3T3 cells
NIH3T3 cells suspended in PBS were treated with 1 μM staurosporine, 1 μM calyculin A or 100 μM pervanadate and fractionated with 0.5% Triton X-100 or DOTMAC. Similar to results in platelets [19], the polypeptide patterns on SDS-PAGE gels differed for DOTMAC- and Triton X-100-extracted cells and more polypeptides were recovered in the DOTMAC- as compared to the Triton X-100-insoluble fraction (Figure 7). Many signaling molecules are known to be sequestered into the Triton X-100-insoluble fraction during activation and/or aggregation of human platelets, and cytoskeletal association has been inferred from this result. While this may be true for some proteins, the postulated cytoskeletal association of threonine558-phosphorylated moesin was maintained only when platelets were extracted with DOTMAC, but not with Triton X-100 [19]. By contrast, phosphorylated moesin was found in the insoluble material of NIH3T3 cells after extraction with both types of detergent (Figure 7). When extracted with 0.5% detergent, a larger amount of insoluble phosphomoesin was detected with Triton X-100 as compared to DOTMAC. Different concentrations of detergents (0.1%, 1% and 2%) were tested and correlated with the amount of moesin released from NIH3T3 cells, but the insoluble fraction of moesin was always higher with Triton X-100 than with DOTMAC (data not shown).
We also analysed for tyrosine-phosphorylated proteins in the insoluble fraction of cells treated with pervanadate (PV), a potent inhibitor of tyrosine phosphatases, extracted with 0.5% detergent. A larger fraction of tyrosine-phosphorylated proteins were recovered in the DOTMAC-insoluble pellet (unpublished observation)
DOTMAC induces bundling of actin filaments and microtubules
The excellent preservation of the cytoskeleton in microextensions of cells fixed with PFA/DOTMAC could be due to stabilization of the cytoskeleton in general or of actin filaments selectively. We, therefore, explored whether DOTMAC has a direct effect in vitro on actin filaments prepared from purified actin. As shown in Figure 8, actin filaments bundled in the presence of DOTMAC (Figure 8B) suggesting that this might contribute to the greater stability of the actin cytoskeleton that we have observed in cells extracted with this detergent. This effect is relatively specific since DOTMAC disrupted microtubules and they appeared as amorphous filaments (Figure 8D).
Figure 8.

Negative staining electron micrographs of rabbit skeletal muscle F-actin (A and B) and bovine microtubules (C and D) in the absence (A and C) and presence (B and D) of DOTMAC. Note that DOTMAC induces bundling of actin filaments and microtubules. Bars: (overviews) 500 nm; (inserts) 200 nm.
Discussion
Detection and definition of micropodia and ultramicroextensions
Extensions of the cell surface were first described as fibrous projections of tissue culture cells by electron microscopy in 1945 [23]. Since then, the terminology has varied in the literature and extensions have been designated as pseudopodia [24], microextensions [25], retraction fibers [25], microspikes [26], filopodia [24, 27, 28], often without a clear definition of these terms. Microvilli are light microscopically distinct from filopodia and retraction fibers [29]. Filopodia or microspikes extend actively from the cell surface, whereas retraction fibers are extensions that have been left behind by a retracting lamellipodium or that are derived from an attached filopodium. We use the term "micropodia" here for morphologically describing filopodia, microspikes, and retraction fibers, and to differentiate these structures from lamellipodia. While the transiently occurring filopodia and retraction fibers can largely be distinguished only by time-lapse video microscopy [22], both, micropodia and lamellipodia, are functionally related to movement of cells. Micropodia were reported to have diameters of about 0.2 μm [29] or 0.08∼ 0.16 μm [20] and lengths ranging from 5 to 30 μm [29]. On the other hand, microvilli are more permanent discrete cylindrical projections about 0.1 μm in diameter and up to 2 μm in length [29] that may serve different purposes, such as in brushborder membranes of the intestinal epithelium or in lymphocytes. Numerous slender extensions running in parallel were observed previously, when rat epithelial (parenchymal) cells adhere to each other [30]. Similar structures also occur in primary keratinocytes mediating intercellular adhesion [31]. However, these extensions appeared to be thicker (estimated as 0.1μm in diameter, although their apparent diameter depended on conditions of coating with metals).
Our experiments were focused on the analysis of microextensions and their cytoskeletal core by SEM and confocal laser scanning microscopy by searching for a new fixation protocol. Here, we clearly show that NIH3T3 cells not only extend microextensions of 0.1∼ 0.2 μm in diameter and of up to ∼ 40 μm in length, but also slender structures of less than 0.05 μm diameter. The latter were previously described in the tumor cell lines K-A31 (Balb/3T3 clone A31 transformed by murine sarcoma virus, [32]), and in A875, a malignant human lung melanoma line [33]. In K-A31 cells, membranous strands with diameters of less than 0.1 μm extended away from and between cells over distances of 10-15 μm. In A875 cells, "filopodia" had diameters of 0.05 μm or less and were often branched. However, details of these structures were not given and not further discussed. In the literature, terms such as slender filopodia [34, 35], or slender microvilli [36, 37] have been used. All filopodia and microvilli are morphologically slender and these descriptive terms in general have not been used to refer to microextensions having a particular dimension. In order to distinguish them from others and to avoid confusion, we propose the term ultramicroextensions for discrete cellular extensions that are less than 0.05 μm in diameter. Ultramicroextensions extend like micropodia from the cell surface, from the tips of micropodia, from microvilli, or they branch out from micropodia or other ultramicroextensions. We strongly believe that these are unique structures and not artificially created by our sample preparation technique. First, unless cells contacted each other, they were rarely seen even after incubation for prolonged periods, suggesting that they are not simply created by the collapse of parts of the cell. Secondly, their shape is rather similar to the generally larger microspikes or microvilli. Future work might show that they can be detected by differential interference-contrast microscopy and electronic image processing or other techniques, which would greatly facilitate functional studies. Ultramicroextensions may be difficult to distinguish by immunofluorescence microscopy from micropodia that also appear as small fluorescent dots on the cell body, or they may remain undetected due to low fluorescence signal owing to their small size. The filopodia of spreading 3T3 cells were extensively studied by Albrecht-Buehler et al [21, 27, 29], but ultramicroextensions were not described presumably because these studies were carried out during early spreading of 3T3 cells. Cells that do not touch other cells rarely display ultramicroextensions even after long periods of incubation. Ultramicroextensions were quite frequent between cells and on the surface of cells grown at confluency. This suggests that their formation is triggered by contact with other cells and that they are not primarily related to cell movement.
Preservation and exposure of cytoskeleton in microextensions and ultramicroextensions
We have successfully exposed the cytoskeleton of microextensions and ultramicroextensions using a newly developed fixative that contains DOTMAC. In order to study the structure of the cytoskeletal network it is necessary to extract cells with a detergent to expose cytoskeletal filaments for electron microscopic imaging. Several detergents were tested for their effects on cytoskeletal structures by Schliwa et al. [3], but the cationic detergent DOTMAC was not included. In addition, past studies on the cytoskeleton of detergent-extracted cells have mainly focused on the cell body and subcortex [3, 8], but not on microextensions. Although extraction can be done before, during, or after fixation, we used simultaneous extraction and fixation, because (i) cytoskeletal molecules may be rearranged or altered during fixation, as irreversible crosslinking of polypeptides is not complete even after 15 min incubation with 4% PFA at 37°C; (ii) although actin was more difficult to extract, moesin and other polypeptides were removed already with 0.5% DOTMAC in the absence of PFA. Addition of 1% PFA was a necessary compromise to visualize the cytoskeleton and its associated molecules no matter which detergent was being used.
Although DOTMAC induced bundling of actin filaments and microtubules, which could have affected ultrastructural features of the cytoskeleton, microextensions of DOTMAC/PFA-extracted cells did not differ from those of non-treated cells. On the contrary, bundling of actin filaments by DOTMAC may have stabilized these actin-containing structures. In SEM, micropodia, ultramicroextensions and microvilli were well preserved, but their lipid membranes apparently were lost. This made it possible to trace the origin of micropodia and ultramicroextensions. While the cytoskeleton of micropodia appeared to connect to the cytoskeleton located at the edge of the cells, by contrast, filaments of ultramicroextensions seemed to extend from or connect to cytoplasmic, rather than subcortical structures.
Triton X-100 is the most widely used detergent to expose the cytoskeleton for biochemical and immunohistological studies, but without prefixation it caused a marked loss of cytoskeletal material [38, 39]. This method also has been criticized for loss of the microtrabecular lattice [3, 40]. This loss is sometimes advantageous to expose certain types of filaments [40], but this was not relevant to our study. Triton X-100 is not useful and even inappropriate for exposing and maintaining the cytoskeleton of microextensions. The fragmentation of microextensions and the amount of material lost with this detergent were time, temperature and concentration dependent. The cytoskeleton tended to be unmasked at low temperatures and at low concentrations of Triton X-100, and the loss of material occurred especially in lamellipodia [40]. These earlier data are consistent with our results concerning lamellipodia, but other microextensions were ignored. Previous authors preferred to use 1% Triton X-100, a relatively high concentration of detergent, because it is critical for rapid solubilization of the cell membrane and cessation of cell activity. However, 1% Triton X-100 destroyed microextensions even in PEM buffer. Another problems with Triton X-100 is that most of the cells detached from the substrate and the remaining cells are usually quite distorted in shape [38]. We have seen similar shape changes with Triton X-100-extracted cells after relatively short prefixation with formaldehyde (Table 2).
The composition of the extraction buffer is an important factor [15]. Although MTSB, which is essentially similar to PHEM, PEM, or MTPB, was shown to give the best preservation of the cytoskeleton, no morphological differences were seen between these buffers and PBS(+), when the cells were extracted with DOTMAC (data not shown). Addition of phalloidin, taxol, or PEG did not make a difference either (data not shown). In our experience, the divalent cations in PBS(+) prevented the cellular retraction seen within a few minutes with PBS and are necessary to maintain interactions between DOTMAC-resistant components and the substrate (data not shown). DOTMAC, on the other hand, caused rapid cessation of all cell activity.
The type of fixative is another important determinant for the analysis of cell shape and structural integrity of the cytoskeleton. Glutaraldehyde is considered the best fixative to preserve these structures, but there are associated disadvantages. Prefixation and postfixation with glutaraldehyde precludes exposure of the cytoskeleton and, also quite frequently, the binding of antibody probes. On the other hand, formaldehyde is a rather weak crosslinker. A crosslink formed by formaldehyde between two functional groups is theoretically and experimentally a non-reducible methylene bridge. Such bridges are readily hydrolysed during washing. The only irreversible reaction would be one that takes place slowly with the aromatic hydrogen [41]. Our studies provide first systematic data that cells should be kept in 4% PFA for at least 60 min at room temperature or for 20 min at 37°C to irreversibly crosslink most of the polypeptides (Figure 6). Thus, enzymatic reactions or rearrangement of components in the cytosol could occur for some time during fixation. In fact, dephosphorylation of moesin occurred in mouse epithelial MTD-1 cells during fixation in 1% or 4% paraformaldehyde at room temperature for 15 min [12]. As shown here, however, moesin apparently was not dephosphorylated in PFA-based fixatives in NIH3T3 cells. These conflicting results could be due to differences of cell type and/or antibody reactivity. We have prepared the formaldehyde solution by dissolving PFA in water, because it was pointed out that methanol included in commercial formaldehyde as a preservative, is enough to cause retraction of ruffles and lamellipodia [15]. However, no morphological difference between PFA and formaldehyde was apparent when cells were extracted with DOTMAC (data not shown).
Evaluation of DOTMAC/PFA method by fluorescent microscopy
Amieva et al. [22] pointed out that some microextensions may be lost during fixation and washing steps, and Nobes and Hall [16] added glutaraldehyde to the fixative in order to stain these delicate structures with phalloidin, but glutaraldehyde had to be omitted for immunostaining. Because the procedure developed here resulted in a comparable preservation of microextensions, we systematically analysed these structures with various probes by immunofluorescence microscopy. The DOTMAC/PFA method provided excellent morphological preservation of actin stress fibers and microextensions and high intensity immunolabeling of actin, while phalloidin staining was poor probably owing to partial denaturation of actin filaments. The DOTMAC/PFA method thus should be considered the method of choice for the preservation and immunostaining of microextensions or when cells fixed by other methods cannot be stained with certain antisera.
Functional significance of fractionation of polypeptides into DOTMAC-resistant components in NIH3T3 cells
We have previously shown that purified threonine558-phosphomoesin selectively co-sedimented with actin filaments in the presence of DOTMAC or PIPs, but excess Triton X-100 disrupted the interaction of moesin with PIPs [19]. Furthermore, the cytoskeletal association of threonine558-phosphomoesin was maintained when platelets were extracted with DOTMAC, but not with Triton X-100. These results were consistent with a model of an allosteric change induced by PIPs (or DOTMAC) that exposed and maintained a cytoskeleton-binding site of moesin. In NIH3T3 cells, however, unlike in platelets, most of threonine558-phosphomoesin was retained in the Triton X-100-insoluble, but only ∼ 20% in the DOTMAC-insoluble fraction. How can this difference be reconciled? One possibility is that moesin in NIH3T3 cells is modified differently. For example, moesin is tyrosine phosphorylated in v-src-transfected MDCK cells [42]. A second possibility is that threonine558-phosphomoesin interacts with non-extractable membrane constituents besides actin filaments in cells other than platelets. For example, the N-terminal domain of moesin lacking the C-terminal F-actin binding domain remained strongly associated with the NP-40-insoluble material of NIH3T3 cells [22]. While this issue needs further work, it is clear that detergent extraction alone cannot be used as the sole criterion for cytoskeletal association of proteins such as moesin as stated frequently in the recent literature.
Materials and Methods
Chemicals and antibodies
DME medium was obtained from Nissui (Tokyo, Japan) or Sigma (St. Louis, MO). FCS was purchased from GIBCO BRL Life Technologies (Paisley, Scotland) or Sigma. Other tissue culture reagents were purchased from GIBCO. All lipids were from Sigma. Glutaraldehyde (70% stock in water, EM grade), paraformaldehyde (PFA), and organic solvents were purchased from Nacalai Tesque (Kyoto, Japan). Anti-β-actin mouse mAb (A5441), anti-β-tubulin mouse mAb (T4026), anti-vinculin mouse mAb (V9131), anti-mouse IgG FITC conjugate (F2012), anti-rabbit IgG FITC conjugate (F1262), anti-rabbit IgG TRITC conjugate (T5268), and TRITC labeled phalloidin were purchased from Sigma. Anti-mouse IgG rhodamine conjugate (AP124R) was obtained from Chemicon International (Temecula, CA). Affinity-purified polyclonal antibodies (95/1) were used for the identification of moesin by immunoblotting and for immunofluorescence. The affinity-purified polyclonal antibodies against threonine558-phospho-moesin were prepared as described previously [43]. Actin was prepared as described previously [19]. Tubulin was purchased from Cytoskeleton (Denver, CO).
Cell culture
NIH 3T3 fibroblasts were grown in DME medium containing 1000 mg/liter glucose, supplemented with 10% FCS, 4 mM L-glutamine, penicillin (100 IU/ml), and streptomycin (100 μg/ml), at 37°C in a humidified atmosphere of 5% CO2 in air. Cells were washed with PBS containing 1 mM CaCl2 and 1 mM MgCl2 (PBS(+)), detached by treating with 0.05% trypsin in PBS or 0.02% EDTA in PBS. The suspended cells were washed with PBS, resuspended in growth medium, and plated on FCS-coated glass coverslips for microscopy, or onto 35-mm or 100-mm Falcon plastic culture dishes (Beckton Dickinson, Lincoln, NJ) or FCS-coated glass culture dishes for biochemical experiments. In some cases, detached cells were resuspended in growth medium without washing.
Scanning electron microscopy
For the observation of cellular surface, cells were rinsed with PBS(+) and fixed with PBS(+) containing 2% glutaraldehyde (dilute 70% glutaraldehyde just before use) at 4°C for 1 hour. In most references, cells were rinsed with PBS (although PBS, in some references, means PBS that contains CaCl2 and MgCl2), but we preferred PBS(+) because retraction of cells were observed in a few minutes when the cells were exposed to PBS even briefly. Samples were then washed with distilled water, dehydrated in a graded series of ethanol, transferred into t-butylalchohol, and dried in Freeze Dryer ES-2030 (Hitachi, Japan). Dried samples were coated with Pt-Pd in Ion Sputter E-1030 (Hitachi, Japan) for 120 seconds, and were examined under an S-4200 scanning electron microscope (Hitachi, Japan) at 15 kV. Micrographs were taken at 0° or 60° tilt.
Fixation and extraction for microscopy
Cells on glass coverslips were rinsed with PBS(+), fixed and/or extracted by one of the procedures listed in Table 1. Methanol-free formaldehyde solution can be obtained from commercial sources, but it has to be used within a week and is more expensive than PFA powder [61]. Although formaldehyde and PFA were used in papers as listed in Table 1 and formaldehyde solutions may have been obtained from commercial sources, for our study a formaldehyde aqueous stock solution was prepared by dissolving 20 g of PFA powder in 90 ml of double distilled water. The mixture was heated to 60-65°C and the pH was adjusted to 7.3 with NaOH. The solution was diluted with water to 100 ml (final concentration 20%) and stored at 4°C for not more than one week. In earlier work, formaldehyde solutions may have been prepared from formalin that contains 37% formaldehyde and 8∼ 15% methanol. Unless noted as formalin, formaldehyde solutions were solely prepared here as described above. Therefore, formaldehyde and PFA are essentially indistinguishable. Buffers were prepared as 2× or 5× stock solutions. Magnesium-and calcium-free PBS was used unless noted otherwise in the references. When detailed descriptions were lacking, specimens were treated as listed in Table 1.
Confocal laser scanning microscopy
Blocking of nonspecific binding sites and incubation with antibodies basically followed procedures detailed in references, but cells were stained as follows when descriptions were lacking. After the cells were soaked in 1% BSA/PBS for 30 min, they were treated with the primary antibodies in 1% BSA/PBS for 1 h. The cells were then washed with PBS three times, followed by incubation with secondary antibodies in 1% BSA/PBS for 1 h. For the detection of actin filaments, 100 ng/ml TRITC-phalloidin was mixed with the second antibody solution. For double staining, secondary antibodies that did not cross-react with each other were chosen. Cells were washed three times in PBS for 5 min and rinsed with water at room temperature, mounted in PermaFluor (Shandon, Pittsburgh, PA) and observed under a laser scanning confocal microscope (MRC-1024, Bio-Rad Laboratories, Richmond, CA). Confocal sections were taken with an iris aperture of 2.0. Images were processed using Adobe Photoshop software.
Evaluation of crosslinking by polypeptide analysis after fixation and extraction
Subconfluent cultures of NIH3T3 cells on 3.5 cm plastic culture dishes, were rinsed with PBS(+), fixed with 4% paraformaldehyde in PBS(+) at room temperature or 37°C, or fixed and/or extracted by one of the procedures listed in Table 1. For some experiments, cells after fixation were treated twice with 0.5 mg/ml NaBH4 in PBS for 10 min at room temperature. After rinsing with PBS, the any material remaining on the dish was extracted with 200 μl of 1× SDS sample buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol, 10% 2-mercaptoethanol, pH 6.8) at room temperature for 30 min in preparation for SDS-PAGE. The dissolved material was recovered and heated at 95°C for 10 min and resolved by SDS-PAGE on a 9% polyacrylamide gel. Polypeptides were stained with Coomassie Brilliant blue or immunologically stained with antibodies after transfer to nitrocellulose or polyvinylidenedifluoride membranes.
Extraction for western blotting
NIH 3T3 cells were detached from the dishes in the presence of 1 μM calyculin A, but not staurosporine and pervanadate. In order to exclude the effect of attachment on the extractability of proteins, suspended cells were used as a control sample. The cells were cultured on 10 cm plastic culture dishes as described above. The subconfluent cells were washed three times with PBS(+), once with PBS, detached by treating with 0.02% EDTA in PBS. The suspended cells were collected, centrifuged at 1,000 × g for 5 min, and resuspended with PBS. The suspensions were divided and incubated with or without 1 μM calyculin A, 1 μM staurosporine, or 100 μM pervanadate in 200 μl of PBS(+) for 10 min at 37°C. The cells were extracted for 5 min on ice with 200 μl of 2× detergent extraction buffer (2% of Triton X-100 or DOTMAC, 50 mM Tris-HCl, 10 mM EGTA, 200 mM NaCl, 2 mM MgCl2, protease inhibitor cocktail [20 μg/ml aprotinin, 20 μM E-64, 200 μM leupeptin, 200 μM p-amidinophenylmethanesulfonyl fluoride], 2 μM calyculin A, 10 mM sodium pyrophosphate, 2 μM staurosporine, 20 μM phalloidin, and 20 μg/ml taxol). The lysates were centrifuged at 15,800 × g for 5 min at 4°C. The resulting pellets were solubilized in 150 μl of 1× SDS sample buffer. Total lysate was prepared by adding 150 μl of 1× SDS sample buffer to the pelleted cells after drug treatment described above. All samples were heated at 95°C, sonicated and resolved by SDS-PAGE on a 9% polyacrylamide gel electrophoresed under reducing conditions as described [18]. β-Actin, moesin, and phosphorylated moesin were detected by immunoblotting and the enhanced chemiluminescence detection system.
Lipid analysis of detergent-resistant components
When NIH 3T3 fibroblasts were treated with Triton X-100 lysis buffer, most of the cells detached from the culture dish. This made it impossible to quantitatively analyze lipids in the detergent-insoluble fraction of attached growing cells. Therefore, the cells were suspended using EDTA before lipid analysis and extracted with detergent extraction buffer as described above. The lysates were centrifuged at 15,800 × g for 5 min at 4°C. The supernatant was discarded and the pellet was washed with PBS. The lipids were extracted with 0.5 ml of chloroform/methanol (2:1, v/v) in an ultrasonic bath for 30 min at 37°C from the pellet. After centrifugation at 15,800 × g for 5 min, the supernatant was concentrated under nitrogen and spotted on a 10 × 10 cm Silica gel thin layer glass plate (LHP-K, Whatman). Phospholipids were separated by chromatography in 100:75:7:4 of chloroform:methanol:acetic acid:water. Plates were stained with 40% sulfuric acid in water. L-α-phosphatidylethanolamine, L-α-phosphatidylcholine, L-α-phosphatidyl-L-serine, L-α-phosphatidylinositol, L-α-phosphatidylinositol 4,5-biphosphate, sphingomyelin were identified by using a standard solution.
When the cells were treated with DOTMAC lysis buffer, they did not detach and, therefore, lipids could be extracted directly from the cells on glass culture dishes with 1 ml of chloroform/methanol (2:1, v/v) after treatment with the detergent extraction buffer. The solvents and residues were collected in a tube and sonicated in an ultrasonic bath as described above. After centrifugation at 15,800 × g for 5 min, lipids were analysed from the supernatant as described above.
Transmission electron microscopy
The phalloidin-stabilized α-actin filaments (1 μM) were incubated in buffer F (5 mM Tris-HCl, pH 7.5, 0.5 mM Na2ATP, 2 mM MgCl2, 140 mM NaCl, 0.2 mM DTT, 0.2 mM CaCl2, 0.005% sodium azide) with or without 0.1 % DOTMAC for 10 min at 25°C. The taxol-stabilized β-tubulin (1 μM) was also incubated in PEM buffer (100 mM PIPES, 5 mM EGTA, 2 mM MgCl2, pH6.8) containing 1 mM GTP with or without 0.1 % DOTMAC for 10 min at 25°C. 10 μl of samples were adsorbed for 60 s to carbon-coated 400 mesh/inch copper grids that were rendered hydrophilic by glow discharge for 15 s in air at low pressure. The grids were washed with 100 mM KCl and stained for 60 s on six drops of 1% uranyl acetate. Microscopy was performed on a HITACHI H-8100 electron microscope at an accelerating voltage of 100 kV.
List of abbreviations
1PIPs, polyphosphoinositides; DOTMAC, dodecyltrimethylammonium chloride; FA, formaldehyde; PFA, paraformaldehyde; SEM, scanning EM; TEM, transmission EM. Other abbreviations are indicated in Table 1.
Supplementary Material
Table 1: Methods of fixation and extraction for immunofluorescent microscopy.
Table 2: Comparison of the effects of different methods of preparation on the fluorescence intensity (Flu. int.), pattern of fluorescent labeling of stress fiber (SF) and morphological preservation of microextensions (ME) by TRITC-phalloidin (phalloidin) and monoclonal anti-β-actin antibody (mAb-β-actin) and pattern of tubulin staining by β-tubulin antibody (mAb-β-tubulin). Morphological preservation of the cells attached on coverslips (Morph.) and of microextensions (ME) were also examined by SEM before and after treatment for fluorescence microscopy. The range is from excellent (+++++) to very poor (0).
Acknowledgments
Acknowledgments
I am grateful to Dr. Heinz Furthmayr (Stanford University) and Dr. Monika Pilichowska (New England Medical Center) for critically reading the manuscript and for valuable suggestions. I also thank Tsuruji Sato (Tohoku University) for excellent technical help on electron microscopy.
References
- Osborn M, Webster RE, Weber K. Individual microtubules viewed by immunofluorescence and electron microscopy in the same PtK2 cell. J Cell Biol. 1978;77:R27–34. doi: 10.1083/jcb.77.3.R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins TE, Smith WL. Subcellular localization of prostaglandin-forming cyclooxygenase in Swiss mouse 3T3 fibroblasts by electron microscopic immunocytochemistry. J Biol Chem. 1980;255:4872–4875. [PubMed] [Google Scholar]
- Schliwa M, van Blerkom J, Porter KR. Stabilization and the cytoplasmic ground substance in detergent-opened cells and a structural and biochemical analysis of its composition. Proc Natl Acad Sci USA. 1981;78:4329–4333. doi: 10.1073/pnas.78.7.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JE. The platelet cytoskeleton. Thromb Haemost. 1993;70:884–893. [PubMed] [Google Scholar]
- Meyer zu Heringdorf D, Liedel K, Kaldenberg-Stasch S, Michel MC, Jakobs KH, Wieland T. Translocation of microfilament-associated inhibitory guanine-nucleotide-binding proteins to the plasma membrane in myeloid differentiated human leukemia (HL-60) cells. Eur J Biochem. 1996;235:670–676. doi: 10.1111/j.1432-1033.1996.00670.x. [DOI] [PubMed] [Google Scholar]
- Liu M, Qin Y, Liu J, Tanswell AK, Post M. Mechanical strain induces pp60src activation and translocation to cytoskeleton in fetal rat lung cells. J Biol Chem. 1996;271:7066–7071. doi: 10.1074/jbc.271.12.7066. [DOI] [PubMed] [Google Scholar]
- Foster LJ, Yaworsky K, Trimble WS, Klip A. SNAP23 promotes insulin-dependent glucose uptake in 3T3-L1 adipocytes: possible interaction with cytoskeleton. Am J Physiol. 1999;276:C1108–1114. doi: 10.1152/ajpcell.1999.276.5.C1108. [DOI] [PubMed] [Google Scholar]
- Bell PBJ, Safiejko-Mroczka B. Improved methods for preserving macromolecular structures and visualizing them by fluorescence and scanning electron microscopy. Scanning Microsc. 1995;9:843–857. [PubMed] [Google Scholar]
- Wagner MC, Barylko B, Albanesi JP. Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol. 1992;119:163–170. doi: 10.1083/jcb.119.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong SP, Cooperband SR, Deckers PJ, Sutherland CM, Cesare JF, Krementz ET. Effect of different fixatives on the localization of human melanoma antigens by immunofluorescence. Oncology. 1979;36:202–207. doi: 10.1159/000225342. [DOI] [PubMed] [Google Scholar]
- Stendahl OI, Hartwig JH, Brotschi EA, Stossel TP. Distribution of actin-binding protein and myosin in macrophages during spreading and phagocytosis. J Cell Biol. 1980;84:215–224. doi: 10.1083/jcb.84.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yonemura S, Matsui T, Tsukita S. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J Cell Sci. 1999;112:1149–1158. doi: 10.1242/jcs.112.8.1149. [DOI] [PubMed] [Google Scholar]
- Lamberts R, Goldsmith PC. Fixation, fine structure, and immunostaining for neuropeptides: perfusion versus immersion of the neuroendocrine hypothalamus. J Histochem Cytochem. 1986;34:389–398. doi: 10.1177/34.3.2419392. [DOI] [PubMed] [Google Scholar]
- King JC, Lechan RM, Kugel G, Anthony EL. Acrolein: a fixative for immunocytochemical localization of peptides in the central nervous system. J Histochem Cytochem. 1983;31:62–68. doi: 10.1177/31.1.6187805. [DOI] [PubMed] [Google Scholar]
- Bell PB., Jr The application of scanning electron microscopy to the study of the cytoskeleton of cells in culture. Scanning Electron Microsc. 1981;2:139–157. [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva MR, Furthmayr H. Subcellular localization of moesin in dynamic filopodia, retraction fibers, and other structures involved in substrate exploration, attachment, and cell-cell contacts. Exp Cell Res. 1995;219:180–196. doi: 10.1006/excr.1995.1218. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Amieva MR, Furthmayr H. Phosphorylation of threonine 558 in the carboxyl-terminal actin-binding domain of moesin by thrombin activation of human platelets. J Biol Chem. 1995;270:31377–31385. doi: 10.1074/jbc.270.20.12319. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Huang L, Pestonjamasp K, Luna EJ, Furthmayr H. Regulation of F-Actin Binding to Platelet Moesin In Vitro by Both Phosphorylation of Threonine 558 and Polyphosphatidylinositides. Mol Biol Cell. 1999;10:2669–2685. doi: 10.1091/mbc.10.8.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaraman R, Rounds DE, Yen SP, Rembaum A. A scanning electron microscope study of cell adhesion and spreading in vitro. Exp Cell Res. 1974;88:327–339. doi: 10.1016/0014-4827(74)90248-1. [DOI] [PubMed] [Google Scholar]
- Albrecht-Buehler G, Goldman RD. Microspike-mediated particle transport towards the cell body during early spreading of 3T3 cells. Exp Cell Res. 1976;97:329–339. doi: 10.1016/0014-4827(76)90624-8. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Litman P, Huang LQ, Ichimaru E, Furthmayr H. Disruption of dynamic cell surface architecture of NIH3T3 fibroblasts by the N-terminal domains of moesin and ezrin: in vivo imaging with GFP fusion proteins. J Cell Sci. 1999;112:111–125. doi: 10.1242/jcs.112.1.111. [DOI] [PubMed] [Google Scholar]
- Porter KR, Claude A, Fullam EF. A study of tissue culture cells by electron microscopy. J Exp Med. 1945;8:233–246. doi: 10.1084/jem.81.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnander H, Gustafson T. Further studies on the cullular basis of gastrulation in the sea urchin larva. Exp Cell Res. 1960;19:278–290. doi: 10.1016/0014-4827(60)90008-2. [DOI] [PubMed] [Google Scholar]
- Taylor AC, Robbins E. Observations on microextensions from the surface of isolated vertebrate cells. Dev Biol. 1963;7:660–673. doi: 10.1016/0012-1606(63)90150-7. [DOI] [PubMed] [Google Scholar]
- Weiss P. From cell to molecules. The molecular control of cellular activity Edited by Allen J. pp. 1-72. New York: McGraw-Hill Book Company; 1961. pp. 1–72.
- Albrecht-Buehler G. Filopodia of spreading 3T3 cells. Do they have a substrate-exploring function? J Cell Biol. 1976;69:275–286. doi: 10.1083/jcb.69.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthmayr H, Lankes W, Amieva M. Moesin, a new cytoskeletal protein and constituent of filopodia: its role in cellular functions. Kidney Int. 1992;41:665–670. doi: 10.1038/ki.1992.102. [DOI] [PubMed] [Google Scholar]
- Albrecht-Buehler G, Lancaster RM. A quantitative description of the extension and retraction of surface protrusions in spreading 3T3 mouse fibroblasts. J Cell Biol. 1976;71:370–382. doi: 10.1083/jcb.71.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazekas I, Bacsy E, Rappay G. Surface topography of cultured anterior pituitary cells of the rat. Acta Anat. 1982;114:88–96. doi: 10.1159/000145582. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/S0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- Porter KR, Todaro GJ, Fonte V. A scanning electron microscope study of surface features of viral and spontaneous transformants of mouse Balb-3T3 cells. J Cell Biol. 1973;59:633–642. doi: 10.1083/jcb.59.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda MA, Aaronson SA, Ellmore N, Zeve VH, Nagashima K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning and transmission electron microscopy. J Natl Cancer Inst. 1976;56:245–263. doi: 10.1093/jnci/56.2.245. [DOI] [PubMed] [Google Scholar]
- Paavola LG, Boyd CO. Surface morphology of macrophages in the regressing corpus luteum, as revealed by scanning electron microscopy. Anat Rec. 1979;195:659–681. doi: 10.1002/ar.1091950407. [DOI] [PubMed] [Google Scholar]
- Isenberg WM, Bainton DF, Newman PJ. Monoclonal antibodies bound to subunits of the integrin GPIIb-IIIa are internalized and interfere with filopodia formation and platelet aggregation. Blood. 1990;76:1564–1571. [PubMed] [Google Scholar]
- Speilberg L, Evensen O, Nafstad P. Liver of juvenile Atlantic salmon, Salmo salar L.: a light, transmission, and scanning electron microscopic study, with special reference to the sinusoid. Anat Rec. 1994;240:291–307. doi: 10.1002/ar.1092400302. [DOI] [PubMed] [Google Scholar]
- Quaroni A, Tian JQ, Goke M, Podolsky DK. Glucocorticoids have pleiotropic effects on small intestinal crypt cells. Am J Physiol. 1999;277:G1027–1040. doi: 10.1152/ajpgi.1999.277.5.G1027. [DOI] [PubMed] [Google Scholar]
- Bell PB, Miller MM, Carraway KL, Revel JP. SEM-revealed changes in the distribution of the triton-insoluble cytoskeleton of Chinese hamster ovary cells induces by dibutyryl cyclic AMP. pp. 899-906. Becker RP, Johari O, ed. Scanning electron microscopy. 1978.
- Knull HR. Extraction of glycolytic enzymes: myo-inositol as a marker of membrane porosity. J Neurochem. 1985;45:1433–1440. doi: 10.1111/j.1471-4159.1985.tb07210.x. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 1998;298:570–592. doi: 10.1016/S0076-6879(98)98045-4. [DOI] [PubMed] [Google Scholar]
- Pearse AGE. The chemistry of fixation, 3rd edn London: Churchill; 1968.
- Takeda H, Nagafuchi A, Yonemura S, Tsukita S, Behrens J, Birchmeier W, Tsukita S. V-src kinase shifts the cadherin-based cell adhesion from the strong to the weak state and beta catenin is not required for the shift. J Cell Biol. 1995;131:1839–1847. doi: 10.1083/jcb.131.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Amieva MR, Hirota C, Mizuno Y, Furthmayr H. Phosphorylation of 558T of moesin detected by site-specific antibodies in RAW264.7 macrophages. Biochem Biophys Res Commun. 1996;226:650–656. doi: 10.1006/bbrc.1996.1410. [DOI] [PubMed] [Google Scholar]
- Aarts LH, Schrama LH, Hage WJ, Bos JL, Gispen WH, Schotman P. B-50/GAP-43-induced formation of filopodia depends on Rho-GTPase. Mol Biol Cell. 1998;9:1279–1292. doi: 10.1091/mbc.9.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma T, Witke W, Stossel TP, Hartwig JH, Kwiatkowski DJ. Gelsolin is a downstream effector of rac for fibroblast motility. EMBO (Eur Mol Biol Organ) J. 1998;17:1362–1370. doi: 10.1093/emboj/17.5.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L, Ingham KC, Hynes RO. Fibronectin regulates assembly of actin filaments and focal contacts in cultured cells via the heparin-binding site in repeat III13. Mol Biol Cell. 1999;10:1521–1536. doi: 10.1091/mbc.10.5.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman MJ, Krstulovic AM, Colin H, Guiochon G, Pajer K. Serum indole-3-acetic acid in control subjects and newly abstinent alcoholics after an oral loading with L-tryptophan: a preliminary study using liquid chromatography with amperometric detection. Anal Biochem. 1984;142:480–486. doi: 10.1016/0003-2697(84)90493-7. [DOI] [PubMed] [Google Scholar]
- Gauthier-Rouviere C, Vignal E, Meriane M, Roux P, Montcourier P, Fort P. RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol Biol Cell. 1998;9:1379–1394. doi: 10.1091/mbc.9.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh JE, Ciesielski-Treska J, Aunis D. A phase-contrast and immunofluorescence study of adrenal medullary chromaffin cells in culture: neurite formation, actin and chromaffin granule distribution. Cell Tissue Res. 1981;218:331–343. doi: 10.1007/BF00210348. [DOI] [PubMed] [Google Scholar]
- Kalman D, Gomperts SN, Hardy S, Kitamura M, Bishop JM. Ras family GTPases control growth of astrocyte processes. Mol Biol Cell. 1999;10:1665–1683. doi: 10.1091/mbc.10.5.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laterra J, Silbert JE, Culp LA. Cell surface heparan sulfate mediates some adhesive responses to glycosaminoglycan-binding matrices, including fibronectin. J Cell Biol. 1983;96:112–123. doi: 10.1083/jcb.96.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarides E, Weber K. Actin antibody: the specific visualization of actin filaments in non-muscle cells. Proc Natl Acad Sci USA. 1974;71:2268–2272. doi: 10.1073/pnas.71.6.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Muller RT, Honnert U, Reinhard J, Bahler M. The rat myosin myr 5 is a GTPase-activating protein for Rho in vivo: essential role of arginine 1695. Mol Biol Cell. 1997;8:2039–2053. doi: 10.1091/mbc.8.10.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullink H, Walboomers JM, Tadema TM, Jansen DJ, Meijer CJ. Combined immuno- and non-radioactive hybridocytochemistry on cells and tissue sections: influence of fixation, enzyme pre-treatment, and choice of chromogen on detection of antigen and DNA sequences. J Histochem Cytochem. 1989;37:603–609. doi: 10.1177/37.5.2467928. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Peranen J, Auvinen P, Virta H, Wepf R, Simons K. Rab8 promotes polarized membrane transport through reorganization of actin and microtubules in fibroblasts. J Cell Biol. 1996;135:153–167. doi: 10.1083/jcb.135.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetsugu S, Miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO (Eur Mol Biol Organ) J. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielkind U, Swierenga SH. A simple fixation procedure for immunofluorescent detection of different cytoskeletal components within the same cell. Histochemistry. 1989;91:81–88. doi: 10.1007/BF00501916. [DOI] [PubMed] [Google Scholar]
- Yamashiro-Matsumura S, Matsumura F. Intracellular localization of the 55-kD actin-bundling protein in cultured cells: spatial relationships with actin, alpha-actinin, tropomyosin, and fimbrin. J Cell Biol. 1986;103:631–640. doi: 10.1083/jcb.103.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley SP, Wang YL. Indirect immunofluorescence microscopy in cultured cells. Methods Cell Biol. 1998;57:313–332. doi: 10.1016/s0091-679x(08)61588-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1: Methods of fixation and extraction for immunofluorescent microscopy.
Table 2: Comparison of the effects of different methods of preparation on the fluorescence intensity (Flu. int.), pattern of fluorescent labeling of stress fiber (SF) and morphological preservation of microextensions (ME) by TRITC-phalloidin (phalloidin) and monoclonal anti-β-actin antibody (mAb-β-actin) and pattern of tubulin staining by β-tubulin antibody (mAb-β-tubulin). Morphological preservation of the cells attached on coverslips (Morph.) and of microextensions (ME) were also examined by SEM before and after treatment for fluorescence microscopy. The range is from excellent (+++++) to very poor (0).