

Abstract
Within cardiomyocytes, endothelial nitric oxide synthase (eNOS) and neuronal nitric oxide synthase (nNOS) are thought to modulate L-type calcium channel (LTCC) function and sarcoplasmic reticulum calcium cycling, respectively. However, divergent results from mostly invasive prior studies suggest more complex roles. To elucidate the roles of nNOS and eNOS in vivo, we applied noninvasive cardiac MRI to study wild-type (WT), eNOS−/−, and nNOS−/− mice. An in vivo index of LTCC flux (LTCCI) was measured at baseline (Bsl), dobutamine (Dob), and dobutamine + carbacholamine (Dob + CCh) using manganese-enhanced MRI. Displacement-encoded MRI assessed contractile function by measuring circumferential strain (Ecc) and systolic (dEcc/dt) and diastolic (dEcc/dtdiastolic) strain rates at Bsl, Dob, and Dob + CCh. Bsl LTCCI was highest in nNOS−/− mice (P < 0.05 vs. WT and eNOS−/−) and increased only in WT and eNOS−/− mice with Dob (P < 0.05 vs. Bsl). LTCCI decreased significantly from Dob levels with Dob + CCh in all mice. Contractile function, as assessed by Ecc, was similar in all mice at Bsl. With Dob, Ecc increased significantly in WT and eNOS−/− but not nNOS−/− mice (P < 0.05 vs. WT and eNOS−/−). With Dob + CCh, Ecc returned to baseline levels in all mice. Systolic blood pressure, measured via tail plethysmography, was highest in eNOS−/− mice (P < 0.05 vs. WT and nNOS−/−). Mice deficient in nNOS demonstrate increased Bsl LTCC function and an attenuated contractile reserve to Dob, whereas eNOS−/− mice demonstrate normal LTCC and contractile function under all conditions. These results suggest that nNOS, not eNOS, plays the dominant role in modulating Ca2+ cycling in the heart.
Keywords: magnetic resonance imaging, endothelial nitric oxide synthase, neuronal nitric oxide synthase, contractile function
nitric oxide (no) is a key signaling molecule in both the vasculature and the heart (12, 30). Within healthy cardiomyocytes, two distinct isoforms of NO synthase (NOS), endothelial NOS (eNOS) and neuronal NOS (nNOS), are constitutively expressed (2, 4, 13, 27, 36), whereas an inducible NOS (iNOS) isoform is expressed in the heart under stress (16, 58). Both eNOS and nNOS are spatially localized, with eNOS colocalized with caveolae in close proximity to the L-type Ca2+ channel (LTCC) (4) and nNOS colocalized with the ryanodine receptor (RyR) (21, 51) at the sarcoplasmic reticulum (SR). NO generated in healthy cardiomyocytes by eNOS and nNOS has been shown to modulate cardiac Ca2+ cycling by modulating LTCC and RyR function, thereby modulating contractile function (3, 12, 30, 57). However, the specific role of each NOS isoform in Ca2+ cycling and cardiac function in the healthy heart remains unclear (30). In addition to important roles in the healthy heart, changes in cardiac NO signaling have been associated with arrhythmia (8, 18, 50), diastolic dysfunction (38), and left ventricular (LV) remodeling after myocardial infarction (13, 16, 26, 33, 34).
Prior studies, primarily using in vitro or invasive in vivo techniques in mice, have sought to elucidate the roles of eNOS and nNOS in modulating LTCC function and contractile function. Specifically, several studies have suggested that eNOS plays a dominant role in inhibiting LTCC function either at baseline (4), during β-adrenergic stimulation (50), or during muscarinic cholinergic inhibition of β-adrenergic stimulation (20). nNOS has also been suggested to modulate LTCC function (8, 36), as well as SR Ca2+ cycling (4, 27) and phospholamban (PLB) mediated modulation of diastolic function (53) under various physiological conditions. Divergent findings among prior studies have been attributed to differences in background strains, age of mice, anesthesia, temperature, and dose of β-adrenergic agonist (3, 9, 30). Although prior in vitro studies of LTCC current (ICa,L) in isolated myocytes have used a range of low stimulation frequencies [ranging from 0.2 Hz (50) to 3 Hz (20)], most invasive in vivo studies of contractility have been performed at much higher frequencies [8 Hz (47) to 12 Hz (27)]. No previous study has examined both LTCC function and contractile function in a completely in vivo and minimally invasive manner and at physiological frequencies.
In recent years, cardiac magnetic resonance (CMR) imaging has emerged as an accurate and versatile modality for noninvasively assessing the mouse heart (15, 46). In addition to cine CMR for imaging LV size and function (6, 39), dynamic manganese-enhanced CMR can probe in vivo LTCC function (24), and displacement-encoded (DENSE) CMR can quantify myocardial strain and strain rate (28). We previously used CMR to study nNOS−/− mice and showed that they have normal baseline contractile function but attenuated contractile reserve in response to medium and high doses of β-adrenergic stimulation (47). We also showed that the attenuated contractile reserve was not due to reduced perfusion reserve. In the present study, we extended these investigations using novel in vivo CMR techniques to elucidate the roles of both nNOS and eNOS in excitation contraction coupling. Specifically, we used dynamic Mn-enhanced CMR and DENSE CMR to noninvasively characterize nNOS−/− and eNOS−/− mice for the first time in terms of in vivo LTCC and contractile function, respectively, under varying pharmacological stimulation.
METHODS
Animals.
Ten wild-type (WT), 10 eNOS−/−, and 10 nNOS−/− male mice on a C57BL/6 background (Jackson Laboratory, Bar Harbor, ME) were studied under protocols that conformed to the Declaration of Helsinki as well as the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996); the protocols were approved by the Animal Care and Use Committee at our institution. CMR assessment of LTCC function and contractile function were performed at 10 ± 3 wk of age.
CMR preparation.
Mn-enhanced CMR was performed on a 4.7 T MRI system (Varian, Palo Alto, CA), and DENSE imaging of LV contractile function was performed on a 7T ClinScan MRI system (Bruker, Ettlingen, Germany). Cylindrical birdcage RF coils were used with both magnetic resonance scanners. Mn-enhanced CMR was performed on the 4.7T system and not the 7T system to maintain continuity with our initial studies. Body temperature was maintained at 36.4 ± 0.3°C by circulating thermostated water, and anesthesia was maintained using 1.25% isoflurane in O2 inhaled through a nose cone. Each complete Mn-enhanced CMR study took 2 h, and each CMR study of contractile function took 1.5 h. During both experiments, mice lay prone within the scanner. Heart rate (HR), respiration, and core body temperature were monitored during imaging using a fiber optic, MR compatible system (Small Animal Instruments, Stony Brook, NY). For administration of pharmacological agents or infusion of MnCl2, an indwelling catheter was inserted into the intraperitoneal cavity as part of CMR preparation.
Mn-enhanced cardiac magnetic resonance.
A series of mid-ventricular short-axis T1 weighted images were acquired before, during, and after continuous intraperitoneal infusion of MnCl2 to probe manganese (Mn2+) influx kinetics. Mn2+ ions enter cardiomyocytes through the LTCC in proportion to Ca2+ flux (31), remain sequestered within cardiomyocytes for a period on the order of hours, and shorten the T1 of nearby water in proportion to their concentration (23–25). Representative T1-weighted images (Fig. 1) acquired before, during, and after infusion of MnCl2 in a WT mouse at baseline (A), with dobutamine (B), and in response to nifedipine (C), illustrate the effect of Mn2+ accumulation on signal intensity under different pharmacological conditions. The accumulation of Mn2+, and subsequent changes in myocardial signal intensity, reflects the integrated effects of rapid LTCC currents over the period of MnCl2 infusion, therefore enabling the noninvasive in vivo assessment of LTCC function with CMR (24, 25, 46). Mn-enhanced CMR was performed using an ECG-gated saturation recovery pulse sequence with constant saturation time (Tsat). Before acquisition of Mn-enhanced data, a proton density weighted image was acquired using a Tsat of 5 s. All subsequent T1-weighted images, which were acquired with Tsat of 200 ms, had measurement times of 2 to 3 min, depending on HR. Pulse sequence parameters were chosen to achieve a balance between spatial resolution, signal-to-noise ratio, degree of T1 weighting, and sampling rate for measuring Mn-enhancement kinetics. Specific sequence parameters included a field of view of 25.6 × 25.6 mm2, 1 mm slice thickness, 90° flip angle, and four averages. Images were continuously acquired for 15 min before, during, and for 30 min following a 30-min intraperitoneal infusion of 30 mM MnCl2 (0.42 mg/kg·min), with specific image acquisition times varying slightly based on HR.
Fig. 1.

A–C: midventricular short-axis T1 weighted images from a wild-type (WT) mouse at baseline (A), with dobutamine (B), and with nifedipine (C). Images were acquired over 90 min, whereas MnCl2 was infused only from 20 min until 50 min. Representative images in each series were acquired at ∼15, 30, 50, and 80 min. At baseline, accumulation of Mn2+ in cardiomyocytes results in increasing myocardial enhancement throughout the period of MnCl2 infusion. Myocardial enhancement is increased as a result of heightened L-type calcium channel (LTCC) function in response to dobutamine and decreased as a result of partial LTCC inhibition with nifedipine. D: sample data from a WT mouse demonstrate linear increases in signal-to-noise ratio (SNR) during infusion of MnCl2. The rate of increase in SNR, measured as the slope of a linear fit (dotted black line) over the points corresponding to MnCl2 infusion (filled gray symbols between the start and end of infusion), provides an index of integrated Mn2+ flux through the LTCC [in vivo index of LTCC flux (LTCCI); bold numbers]. When compared with baseline, LTCCI increases in response to β-adrenergic stimulation with dobutamine and decreases in response to nifedipine. AU, arbitrary units.
Image analysis was performed in MATLAB (Mathworks, Natick, MA), where signal-to-noise ratio (SNR) was quantified by dividing mean myocardial signal intensity in each image by the standard deviation of background noise. SNR was then divided by the mean SNR of corresponding proton density weighted images, yielding the normalized SNR. LTCC index (LTCCI) was quantified as the rate of enhancement of the normalized myocardial SNR per 105 heart beats during MnCl2 infusion, as shown in Fig. 1D.
We first examined changes in LTCCI in response to pharmacological manipulation of LTCC function in WT mice. LTCCI was measured at baseline (Bsl; n = 10), during β-adrenergic stimulation with an intraperitoneal infusion of a low dose of dobutamine (Dob; 5 μg/kg·min; n = 10), during concomitant muscarinic cholinergic inhibition of β-adrenergic stimulation with an intraperitoneal infusion of Dob and carbamylcholine chloride (CCh; 3 mg/kg·min) (Dob + CCh; n = 10), in response to the LTCC inhibitor nifedipine (10 mg/kg; n = 3), and in response to increased frequency of contraction (14) induced by the A2A adenosine receptor agonist ATL313 (47) (12.5 μg/kg; n = 4). Finally, to verify a sufficient dynamic range of our dynamic Mn-enhanced CMR technique, LTCCI was measured in response to a higher dose of Dob (Dob 20; 20 μg/kg·min; n = 2). Infusion of Dob and Dob + CCh, or bolus injections of nifedipine and ATL313 occurred through a second indwelling intraperitoneal line. Next, LTCCI was measured in eNOS−/− (n = 9) and nNOS−/− (n = 9) mice at Bsl, Dob, and Dob + CCh. Because eNOS is theorized to be involved in muscarinic cholinergic inhibition of β-adrenergic stimulation in the heart, the effect of CCh was evaluated only in combination with Dob. In all cases, 1 wk elapsed in between experiments to allow time for complete washout of manganese, since Bsl, Dob, and Dob + CCh studies were performed in the same mice. Although the effects of both nifedipine and ATL313 are observed for the entire course of the experiment following bolus injection, Dob and Dob + CCh were applied using constant-rate infusion to maintain a constant level of stimulation.
Cine-DENSE CMR.
One week after conclusion of measurement of LTCCI, contractile function was assessed in the same mice using cine DENSE (28, 54, 55), a technique that is related to, but improves upon, myocardial tagging (47). Cine DENSE data were acquired for two midventricular short-axis slices at Bsl, Dob, and Dob + CCh during the same imaging session, using an intraperitoneal infusion of pharmacological agents as stated previously. Care was taken to ensure that all data were acquired at the midventricle in each mouse. Specific acquisition parameters included field of view = 25.6 mm, matrix = 128 × 128, slice thickness = 1 mm, repetition time = 6.9 ms, echo time = 1 ms, number of averages = 4, number of spiral interleaves = 27, and displacement encoding frequency = 1.1 cycles/mm. The total scan time for a single two-dimensional slice was 6–8 min.
For data analysis, segmentation of the heart used a semi-automated technique (41), and displacement and strain were calculated using the phase unwrapping and motion tracking algorithms described previously (40). Peak midwall circumferential systolic strain (Ecc) and systolic strain rate (dEcc/dt) were used to assess contractile function. Diastolic strain rate (dEcc/dt)diastolic was used to assess diastolic function, as described previously (47).
Systolic blood pressure.
Systolic blood pressure was measured in nonanesthetized, conscious mice. A minimum of five baseline waveforms were obtained for each mouse. Measurements were obtained from each waveform, and then averaged together for each individual mouse, using a non-invasive tail plethysmography system (Model BP 2000; Visitech Systems, Apex, NC).
Statistical analysis.
All statistical analyses were performed using SigmaStat (Systat Software, Point Richmond, CA). Differences in body weight and hemodynamic measurements were evaluated using one-way ANOVA. Differences in LTCCI, Ecc, dEcc/dt, and (dEcc/dt)diastolic at Bsl, Dob, and Dob + CCh between WT, eNOS−/−, and nNOS−/− mice were assessed using one-way repeated-measures ANOVA, assuming compound symmetry. Analysis was performed after confirmation of normal distribution with a statistical power greater than 0.95 unless otherwise noted. Comparisons of LTCCI at Dob20, ATL313, and nifedipine in WT mice were performed using t-test analysis. All values in the text, tables, and graphs are presented as means ± SE.
RESULTS
Body mass and hemodynamics.
Body mass was lower in eNOS−/− and nNOS−/− mice (Table 1), systolic blood pressure was significantly higher in eNOS−/− mice compared with both WT and nNOS−/− mice, and basal HR (statistical power = 0.16) did not differ between groups (Table 1).
Table 1.
Body mass and hemodynamics in wild-type, eNOS−/−, and nNOS−/− mice
| Wild-type | eNOS−/− | nNOS−/− | |
|---|---|---|---|
| n | 10 | 9 | 8 |
| Body mass, g | 29.5 ± 0.8 | 23.9 ± 0.3∗ | 22.8 ± 0.4∗ |
| Systolic blood pressure, mmHg | 110 ± 3 | 131 ± 2∗ | 110 ± 2† |
| Heart rate, beats/min | 520 ± 19 | 481 ± 13 | 494 ± 11 |
Values are means ± SE. eNOS and nNOS, endothelial and neuronal nitric oxide synthase, respectively.
P < 0.05 vs. wild-type;
P < 0.05 vs. eNOS−/−.
LTCC function.
We probed LTCCI with Mn-enhanced CMR under a variety of pharmacological conditions in WT mice. Measurements of LTCCI and HR in WT mice are shown in Fig. 2. In response to β-adrenergic stimulation with a low dose of dobutamine (5μg/kg·min), LTCCI increased by 47% over baseline, reflecting a significant increase in LTCC function. In response to Dob + CCh, LTCCI returned to basal levels, reflecting the effect of muscarinic cholinergic inhibition of β-adrenergic stimulation. LTCCI fell by 26% (P < 0.05 vs. Bsl, statistical power = 0.53) compared with baseline in response to increasing HR with ATL313, mirroring the negative ICa,L_density-frequency relationship seen in mouse cardiomyocytes (1). Finally, in response to partial LTCC inhibition with nifedipine, LTCCI decreased by 42% (P < 0.05 vs. Bsl). HR increased significantly over baseline in response to Dob and ATL313, while it did not change in response to Dob + CCh or nifedipine (Fig. 2B). In response to a higher dose of dobutamine (20μg/kg·min), LTCCI (1.19 ± 0.03 A.U./105 H.B., P < 0.05 vs. Bsl, Dob) and HR (618 ± 58 B.P.M , P < 0.05 vs. Bsl) increased 96% and 27% over baseline, respectively, demonstrating a dynamic range that extends beyond the low-dose dobutamine response measured in WT mice.
Fig. 2.

Measurement of LTCCI (A) and heart rate (HR; B) in WT mice obtained at Bsl, dobutamine (Dob), dobutamine + carbacholamine (Dob + CCh), ATL313, and nifedipine. Dob-induced increases in LTCCI and HR were reversed with Dob + CCh. With adenosine A2a receptor stimulation using ATL313, LTCCI decreased, whereas HR increased, reflecting a negative LTCCI-frequency response. Nifedipine, an LTCC inhibitor, decreased LTCCI significantly while not having any effect on HR. *P < 0.05 vs. Bsl; †P < 0.05 vs. Dob. BPM, beats/min; HB, heartbeat.
Mn-enhanced CMR experiments were performed in eNOS−/− and nNOS−/− mice to elucidate the role of each NOS isoform in modulating LTCC function. The average pre-Mn2+myocardial SNR was similar in WT, eNOS−/−, and nNOS−/− mice (0.28 ± 0.01 WT, n = 27 vs. 0.28 ± 0.01 eNOS−/−, n = 24 vs. 0.29 ± 0.01 nNOS−/−, n = 24, P = NS), as was the SNR of the proton density weighted images (n = 4 per group, 171 ± 3 WT vs. 170 ± 7 eNOS−/− vs. 166 ± 11 nNOS−/−, P = NS). At baseline, LTCCI trended higher in eNOS−/− mice compared with WT mice (P = 0.06) (Fig. 3). However, LTCCI increased significantly over baseline with Dob, and decreased significantly from Dob levels in response to Dob + CCh similarly in eNOS−/− and WT mice. In contrast, basal LTCCI was significantly higher in nNOS−/− mice compared with both WT (P < 0.01) and eNOS−/− (P < 0.05) mice (Fig. 3). LTCCI trended towards a decrease compared with basal levels (P < 0.1 vs. Bsl) in response to Dob and decreased significantly from both Bsl and Dob in response to Dob + CCh in nNOS−/− mice. Despite differences in the responses to Dob and Dob + CCh between nNOS−/− and eNOS−/− mice that reflect differences at baseline, absolute LTCCI values in animals given Dob and Dob + CCh were similar amongst WT, eNOS−/−, and nNOS−/− mice.
Fig. 3.
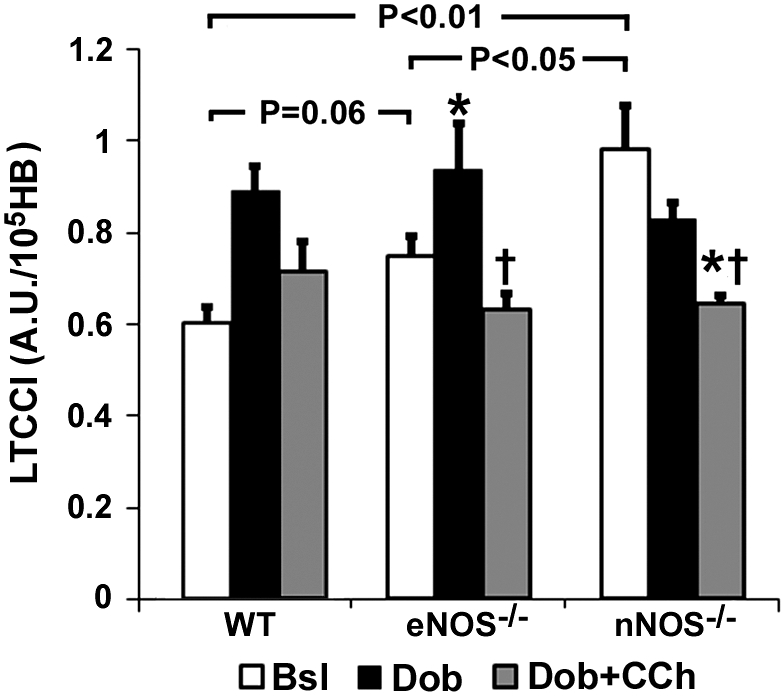
Measurements of LTCCI in endothelial nitric oxide synthase (eNOS)−/− and neuronal nitric oxide synthase (nNOS)−/− mice at Bsl, Dob, and Dob + CCh. Basal LTCCI was highest in nNOS−/− mice compared with WT and eNOS−/− mice (WT results copied from Fig. 2A for purposes of comparison). In response to Dob and Dob + CCh, LTCCI was similar among all mice. *P < 0.05 vs. baseline; †P < 0.05 vs. Dob.
Contractile function.
Basal contractile function as quantified by cine DENSE MRI was similar in WT, eNOS−/− and nNOS−/− mice. Fig. 4 illustrates representative examples of Ecc-time curves from WT, eNOS−/− and nNOS−/− mice at Bsl, Dob, and Dob + CCh. At baseline, peak Ecc was similar in WT, eNOS−/−, and nNOS−/− mice (Fig. 5A). WT and nNOS−/− mice demonstrated similar basal dEcc/dt (−419 ± 16%/s WT, n = 10 vs. −378 ± 13%/s nNOS−/−, n = 9, P = 0.17, statistical power = 0.79), dEcc/dtdiastolic (374 ± 33%/s WT, n = 9 vs. 331 ± 14%/s nNOS−/−, n = 9, P = 0.17, statistical power = 0.76), and HR (540 ± 16 BPM WT, n = 9 vs. 515 ± 11 BPM nNOS−/−, n = 9, P = 0.14, statistical power = 0.60). In contrast, dEcc/dt (−368 ± 11%/s, n = 9, P < 0.05 vs. WT), dEcc/dtdiastolic (296 ± 17%/s, n = 9, P < 0.05 vs. WT), and HR (495 ± 12 BPM, n = 9, P < 0.05 vs. WT) were significantly lower in eNOS−/− mice at baseline. No differences were seen between eNOS−/− and nNOS−/− mice at baseline in terms of dEcc/dt or dEcc/dtdiastolic.
Fig. 4.

Sample midwall circumferential strain (Ecc) vs. time curves from WT (top), eNOS−/− (middle), and nNOS−/− (bottom) mice at baseline and in response to infusion of Dob and Dob + CCh. In WT and eNOS−/− mice, peak Ecc increased significantly from basal levels (dotted gray line) in response to Dob and returned to basal levels following administration of Dob + CCh. In nNOS−/− mice, no change in Ecc was seen in response to either Dob or Dob + CCh.
Fig. 5.

Measurements of contractile function in WT, eNOS−/−, and nNOS−/− mice. A: at baseline, peak Ecc (representing maximal contraction) was similar in all mice. WT and eNOS−/− mice, but not nNOS−/− mice, demonstrated a strong inotropic response to Dob as measured by Ecc (B) and systolic strain rate (dEcc/dt; C), which was reversed in response to Dob + CCh. D: only WT mice demonstrated a strong lusitropic response to Dob, which was reduced with Dob + CCh. *P < 0.05 vs. WT.
In response to Dob, Ecc increased significantly in WT and eNOS−/−, but not nNOS−/− mice (Fig. 5B). Mirroring Ecc results, the increase in strain rate (dEcc/dt) with Dob was significantly lower in nNOS−/− mice compared with WT and eNOS−/− mice (Fig. 5C). However, both eNOS−/− and nNOS−/− mice demonstrated an attenuated lusitropic response to Dob (Fig. 5D). Changes in HR with Dob were similar in WT (10 ± 8 BPM, n = 9) and eNOS−/− mice (26 ± 6 BPM, n = 9, P = 0.12 vs. WT, statistical power = 0.60), but were greater in eNOS−/− compared with nNOS−/− (−4 ± 11 BPM, n = 9, P < 0.01 vs. eNOS−/−) mice. In response to Dob + CCh, WT, eNOS−/−, and nNOS−/− mice all demonstrated no change from baseline in either Ecc or dEcc/dt (Fig. 5B,C). The change compared with baseline in dEcc/dtdiastolic decreased significantly from Dob levels in WT mice, did not change in eNOS−/− mice, and remained lower in nNOS−/− mice compared with WT mice (Fig. 5D). Also, the change in HR with Dob + CCh was significantly lower in nNOS−/− mice (−14 ± 7 BPM, n = 9) compared with both WT (8 ± 6 BPM, n = 9, P < 0.05 vs. nNOS−/−) and eNOS−/− mice (26 ± 6 BPM, n = 9, P < 0.01 vs. nNOS−/−), and trended higher in eNOS−/− compared with WT mice (P = 0.07).
DISCUSSION
Using noninvasive CMR to assess the functional phenotype of eNOS−/− and nNOS−/− mice in vivo, the major findings of this study are that: (i) basal LTCCI is highest in nNOS−/− mice even though basal contractile function is similar to WT mice, (ii) in response to dobutamine, LTCCI decreased from baseline while contractile function did not change in nNOS−/− mice, (iii) in nNOS−/− mice, contractile function remained similar to baseline while LTCCI decreased significantly in response to concomitant β-adrenergic and muscarinic cholinergic stimulation, and (iv) both LTCC function and systolic contractile function are similar in eNOS−/− and WT mice at baseline, in response to β-adrenergic stimulation, and in response to concomitant β-adrenergic and muscarinic cholinergic stimulation. These novel findings represent the first completely in vivo study of modulation of LTCC function and contractile function by eNOS and nNOS in mice, and demonstrate a dominant role for nNOS in modulating LTCC function and contractile function under physiological conditions.
Whereas traditional measurements of ICa,L reflect ultra-fast currents across the plasma membrane of an individual cardiomyocyte, LTCCI measurements represent the integrated effect of Mn2+ accumulation over time and over all cardiomyocytes. A prior study by Masumiya et al. demonstrated that ICa,L kinetics are mirrored by LTCC-Mn2+ current (IMn) kinetics, although at a lower magnitude (approx. 8%) (31). Thus, LTCCI is an analog of integrated ICa,L density per heart beat over the entire heart, and reflects the cumulative effect of ultra-rapid LTCC currents during MnCl2 infusion. It is important to note that most prior Mn-enhanced CMR techniques used only ECG gating for both triggering of the saturation pulse and image acquisition (24). This resulted in variable saturation times, thus making the degree of Mn-enhancement a function of both Mn2+ uptake and HR. In contrast, our modified dynamic Mn-enhanced CMR method adjusted the timing of a non-selective saturation pulse such that a nearly constant saturation time was maintained, thereby making the rate of Mn-enhancement reflect only Mn2+ uptake, and thus enabling quantitative examination of LTCC function. Comprehensive examination of our dynamic Mn-enhanced CMR studies in WT mice indicated that LTCCI measurements are sensitive to pharmacological manipulation of LTCC function, and mirror changes in both IMn (31), and ICa,L in prior patch-clamp experiments performed in isolated myocytes (20). Increases in LTCCI with Dob and decreases with Dob + CCh were similar to changes in ICa,L density in isolated myocytes from WT mice in response to β-adrenergic stimulation with isoproterenol (Iso) (20, 45, 50) and Iso+CCh (20, 45). In response to LTCC inhibition with nifedipine, LTCCI decreased similarly to changes in ICa,L in prior studies in isolated guinea pig ventricular myoctyesc (37). Our findings agree with prior studies which demonstrated that Mn2+ enhancement of myocardium is sensitive to increased LTCC flux following administration of dobutamine (24), and to inhibition with LTCC inhibitors (24). Finally, to probe for an LTCCI-frequency relationship, we used adenosine A2a receptor stimulation with ATL313 and observed a decrease in LTCCI compared with basal levels. Several studies performed in isolated mouse myocytes have demonstrated that both peak ICa,L and integrated ICa,L density, a measure of calcium influx, decrease with increasing pacing frequency (1). Unlike β-adrenergic stimulation, which increases HR and LTCC function simultaneously, A2A receptor stimulation increases HR (14) without increasing LTCC function (22). With ATL313 induced increases in HR, the integrated Mn2+ flux over the period of MnCl2 infusion should increase. However, since LTCCI is an analog of integrated ICa,L density per heart beat, and not per unit time, we interpret our LTCCI results with ATL313 as showing a negative in vivo LTCCI-frequency relationship, in agreement with the negative ICa,L density-frequency relationship in isolated myocytes (1).
Traditionally, nNOS has been considered to modulate calcium cycling at the SR level (4, 13, 29, 47), and not the LTCC level (4, 57). However, our finding of changes in LTCCI without accompanying changes in contractile function in nNOS−/− mice, across different pharmacological stimulation protocols, suggests a role for nNOS in modulating calcium cycling at both the LTCC and SR levels. Previous in vitro studies have demonstrated either elevated basal ICa,L (8, 36) and myocyte shortening (2, 29), or normal ICa,L (4) and myocyte shortening (4, 27) in nNOS−/− myocytes. Since measurement of circumferential strain and strain rate approximates in vivo measurements of myocyte shortening and shortening velocity, respectively (47), one would anticipate increased baseline circumferential strain to accompany our measurement of elevated basal LTCCI in nNOS−/− mice. However, given that ICa,L accounts for only ∼5% of systolic calcium in murine myocytes (compared to ∼30% in humans (7)), the increase in basal LTCC function in nNOS−/− mice may be insufficient to elicit significant changes in contractile function given normal basal SR function, as evidenced by normal baseline SR calcium transients in nNOS−/− mice amongst prior studies (4, 27). In fact, our finding of normal basal contractile function in nNOS−/− mice agrees with most previous in vivo studies (4, 8, 27, 33, 36), including both a prior study by Sears et al. which found increased ICa,L and myocyte shortening but normal basal in vivo contractility in nNOS−/− mice (36), as well as our previous myocardial tagging study (47). In response to Dob, nNOS−/− mice demonstrated a slightly negative LTCCI response (Fig. 3) and no Ecc, dEcc/dt, or dEcc/dtdiastolic response (Fig. 5). As HR increased on average 9%, and LTCCI decreased on average 10% with Dob in nNOS−/− mice, this likely reflects a negative LTCCI-frequency response, similar to that witnessed in WT mice with ATL313, and further suggests that basal LTCC function is already maximal. While these results alone may suggest that the primary role of nNOS is to modulate only LTCC function under basal conditions, our Dob + CCh results suggest a more complex role for nNOS. In WT mice, LTCCI, Ecc, dEcc/dt, and dEcc/dtdiastolic returned to baseline levels in response to Dob + CCh. In contrast, nNOS−/− mice demonstrated a significant reduction in LTCCI without any change in Ecc, dEcc/dt, or dEcc/dtdiastolic. These results suggest that while basal LTCC function was maximal, contractile function remained at baseline levels even with Dob and Dob + CCh in nNOS−/− mice. A number of previous studies have demonstrated reduced SR calcium transients and diastolic SR calcium reuptake in nNOS−/− mice in response to β-adrenergic stimulation (4, 49), further suggesting that compromised SR function in nNOS−/− mice may contribute to altered ventricular function observed in the current study. Recent studies have suggested that nitrosylation decreases LTCC function (8, 43) while increasing RyR function (42, 52). Similarly, NO can modulate Ca2+ cycling through the NO/cGMP signaling cascade (35, 42, 48, 49, 56). Although nNOS is spatially co-localized with the RyR it is possible that, given the small dimensions of the cardiac dyad, NO from nNOS can modulate the function of both the LTCC and the RyR. With this in mind, our findings of increased basal LTCCI and attenuated contractile reserve in nNOS−/− mice may suggest a prominent role for nNOS in modulating contractile function separately from LTCC function, as has been suggested by others (3).
Based on its physical proximity to the LTCC, eNOS has been suggested to modulate LTCC function under a variety of physiological conditions (4, 20, 50). Although basal LTCCI was slightly higher in eNOS−/− compared with WT mice in our study, LTCCI was lower compared with nNOS−/− mice. This suggests that the elevation in basal LTCCI in eNOS−/− compared with WT mice may result from hypertension in eNOS−/− mice (4, 50). Ideally, the impact of hypertension on LTCC function in eNOS−/− mice could be normalized through pharmacological intervention. However, most methods for reducing blood pressure would themselves alter either LTCC function or the β-adrenergic responsiveness in vivo, thus obfuscating the role of eNOS in such studies. Our finding of similar LTCCI in WT and eNOS−/− mice with Dob agrees with most in vitro studies showing similar ICa,L density in response to β-adrenergic stimulation with Iso (5, 17, 20, 45). Only one prior study has demonstrated greater increases in ICa,L density in eNOS−/− mice in response to Iso at a stimulation frequency of 0.5Hz (50). It is possible that, given the negative ICa,L-frequency relationship (1), Wang et al. (50) detected frequency-dependent differences in ICa,L which are not apparent at much higher in vivo frequencies. Finally, in agreement with most (5, 17, 45), but not all (20) prior ICa,L studies, we detected similar LTCCI in WT and eNOS−/− mice in response to Dob + CCh. Previously, only Han et al. (20) have detected increased ICa,L in eNOS−/− mice in response to Iso+CCh, possibly from using higher doses of Iso and CCh than prior studies. However, the concentration of CCh in our study (15μM) was greater than that used by Han et al. (10μM) (20). Furthermore, in WT mice, we observed a ratio of LTCCI with Dob and Dob + CCh similar to that of ICa,L in response to Iso and Iso+CCh in their study (20). These findings suggest that, in vivo, eNOS does not play a significant role in modulating LTCC function.
A few previous studies have suggested that eNOS may also play a role in modulating contractility (4) and β-adrenergic responsiveness (4, 50). Our finding of similar basal Ecc in WT and eNOS−/− mice agrees with the majority of in vivo findings of similar (dP/dt)/Pid (4, 19, 27) and in vitro findings of similar myocyte shortening (4, 17, 20, 27, 29, 45, 50) in WT and eNOS−/− mice. In agreement with the (dP/dt)/Pid findings of Barouch et al. in response to low and medium doses of Iso (4), as well as most in vitro findings of similar myocyte shortening in WT and eNOS−/− mice (4, 20, 29, 45), we observed a normal inotropic contractile response to Dob in eNOS−/− mice. Two studies have demonstrated enhanced β-adrenergic dP/dtmax in eNOS−/− mice with Iso (10, 19). However, dP/dtmax is known to be sensitive to afterload (44), suggesting that the innate hypertension in eNOS−/− mice may have influenced prior results (10, 19). It is also possible that no differences were detected between eNOS−/− and WT mice as a result of the relatively low dose of Dob used. However, Martin et al. demonstrated similar sarcomere shortening in eNOS−/− and control mice in response to several doses of Iso (29). Interestingly, we observed an attenuated lusitropic response to Dob in eNOS−/− mice, which given the recently discovered role of eNOS in preventing early after-depolarizations during diastole (50), may result from the absence of eNOS mediated regulation of Ca2+ handling during diastole. In addition, we also observed greater HR responses in eNOS−/− mice in response to Dob and Dob + CCh, further suggesting that eNOS may be involved in regulating the chronotropic response in the heart. Finally, our finding of normal contractile function in eNOS−/− mice in response to Dob + CCh agrees with previous in vitro findings (17, 29, 45), but not with findings of elevated whole heart contractility in response to Iso+CCh by Champion et al. (10). Whereas Champion et al. infused CCh (30 mg/kg·min) intravenously (10), we found that IP infusion of CCh at the same rate was immediately lethal, suggesting that the dose of CCh may have affected our findings. However, the Ecc response to Dob + CCh in WT mice was similar in magnitude to that of Champion et al. (10). Together, these results suggest that eNOS does not play a significant role in modulating contractile function in the mouse heart, but does play a role in diastolic function.
One limitation of our study was that differences in LTCC and NCX expression between WT and nNOS−/− mice were not investigated, which could affect the rate of Mn-enhancement. However, several prior studies have demonstrated similar expression of both LTCC (18, 36) and NCX (18, 36, 49) proteins in WT and nNOS−/− mice. Also, relative abundances of eNOS and nNOS in knockout mice were not measured, suggesting that potential genetic compensation of the remaining NOS isoform could obfuscate the role attributed to a given NOS based on results in knockout mice. However, prior studies using similar mice have not found compensatory over-expression or translocation of the available NOS under similar experimental conditions (2, 4, 11, 13, 33, 45). In addition, mice used in this study were global knockouts, and experiments were not repeated using selective nNOS inhibitors (such as L-VNIO) or donors in WT mice. An additional limitation was that LTCCI measurements were not validated using conventional patch clamp methods. This suggests that it is possible that increases in LTCCI in nNOS−/− mice with Dob were not detected because of a possible maximum sensitivity in our Mn-enhanced technique. However, LTCCI in nNOS−/− mice at both Bsl and Dob was lower than the maximum LTCCI measured in WT mice at high-dose Dob (20μg/kg·min), suggesting that sensitivity was not a limitation. Also, although measurements of LTCCI and contractile function were made after allowing for IP administration of Dob and Dob + CCh to reach a steady state, prolonged administration of Dob and Dob + CCh could affect abdominal organs, thus producing secondary effects on the heart that are not observed in experiments on isolated myocytes. Additionally, while in vitro experiments typically use a variety of doses of nifedipine to determine a dose-response curve, preliminary dosing experiments with nifedipine proved fatal at doses higher than 10mg/kg. At the chosen dose of nifedipine, we believe that we achieved LTCC inhibition on the order of 40%, in agreement with prior ex vivo data (37). Also, nifedipine is known to be more active on arterial and arteriolar Ca2+ channels than on cardiac LTCCs, raising the possibility that the significant decrease in LTCCI in response to nifedipine may have also reflected a decrease in Mn-enhancement of the cardiac vasculature. However, the water within the wall of the cardiac vasculature represents a very small portion of the water, and therefore MR signal, within each voxel compared with that within cardiomyocytes. Thus, our measurements of LTCCI with nifedipine predominantly reflect the impact of nifedipine on cardiomyocytes. Finally, differences in body weight between WT and knockout mice raise the possibility that differences in heart structure and conduction system may have existed, implying that the regions sampled in CMR studies may have been different between different strains. However, care was taken to acquire data on in midventricular slices in all experiments, thereby ameliorating this concern.
Conclusions
Our results demonstrate that nNOS plays a dominant role in modulating both LTCC and contractile function in vivo. Further, our results suggest that nNOS separately modulates LTCC function at baseline, and contractile function during β-adrenergic stimulation. While other groups have demonstrated that eNOS plays a role in preventing ventricular arrhythmia (50), and in modulating the cardiomyocyte response to stretch (32), our results suggest that, in vivo, the role of eNOS in modulating calcium cycling is less dominant than that of nNOS in the normal heart.
GRANTS
This work was supported by American Heart Association predoctoral grant AHA0815242E (to M. H. Vandsburger), and American Heart Association Established Investigator Award 0540060N and National Institutes of Health R01EB001763 (to F. H. Epstein).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.H.V., B.A.F., X.Z., and F.H.E. conception and design of research; M.H.V. performed experiments; M.H.V. analyzed data; M.H.V., B.A.F., C.M.K., and F.H.E. interpreted results of experiments; M.H.V. prepared figures; M.H.V. drafted manuscript; M.H.V., B.A.F., C.M.K., X.Z., and F.H.E. edited and revised manuscript; M.H.V., B.A.F., C.M.K., X.Z., and F.H.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank R. Jack Roy, Victor Laubach, and Joseph DiMaria for their assistance. Currently, M. H. Vandsburger is affiliated with the Weizmann Institute of Science, Rehovot, Israel.
REFERENCES
- 1. Antoons G, Mubagwa K, Nevelsteen I, Sipido K. Mechanisms underlying the frequency dependence of contraction and [Ca2+]i transients in mouse ventricular myocytes. J Physiol 543: 889–898, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and β-adrenergic contractility in murine ventricular myocytes. Circulation 105: 3011–3016, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Balligand JL. “La Donna e Mobile.” Is cardiac neuronal nitric oxide synthase such a disconcerting enzyme? Circulation 112: 3668–3671, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, Rodriguez ER, Huang PL, Lima JAC, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416: 337–339, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Belevych A, Harvey R. Muscarinic inhibitory and stimulatory regulation of the L-type Ca2+ current is not altered in cardiac ventricular myocytes from mice lacking endothelial nitric oxide synthase. J Physiol 528: 279–289, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berr SS, Roy RJ, French BA, Yang Z, Gilson W, Kramer CM, Epstein FH. Black blood gradient echo cine magnetic resonance imaging of the mouse heart. Mag Res Med 53: 1074–1079, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Bers DM. Calcium cycling and signaling in cardiac myocytes. Ann Rev Physiol 70: 23–49, 2008 [DOI] [PubMed] [Google Scholar]
- 8. Burger DE, Lu X, Lei M, Xiang FL, Hammoud L, Jiang M, Wang H, Jones DL, Sims SM, Feng Q. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 120: 1345–1354, 2009 [DOI] [PubMed] [Google Scholar]
- 9. Casadei B. The emerging role of neuronal nitric oxide synthase in the regulation of myocardial function. Exp Physiol 91: 943–955, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, Kass DA. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ Res 94: 657–663, 2004 [DOI] [PubMed] [Google Scholar]
- 11. Damy T, Ratajczak P, Shah AM, Camors E, Marty I, Hasenfuss G, Marotte F, Samuel JL, Heymes C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 363: 1365–1367, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Danson EJ, Choate JK, Paterson DJ. Cardiac nitric oxide: emerging role for nNOS in regulating physiological function. Pharma & Ther 106: 57–74, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation 112: 3729–3737, 2005 [DOI] [PubMed] [Google Scholar]
- 14. Dhalla AK, Wong MY, Wang WQ, Biaggioni I, Belardinelli L. Tachycardia caused by A2A adenosine receptor agonists is mediated by direct sympathoexcitation in awake rats. J Pharma Exp Thera 316: 695–702, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Epstein FH. MR in mouse models of cardiac disease. NMR in Biomedicine 20: 238–255, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Gilson WD, Epstein FH, Yang Z, Xu Y, Prasad KMR, Toufektsian MC, Laubach VE, French BA. Borderzone contractile dysfunction is transiently attenuated and left ventricular structural remodeling is markedly reduced following reperfused myocardial infarction in inducible nitric oxide synthase knockout mice. J Am Col Cardio 50: 1799–1807, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Godecke A, Heinicke T, Andreij K, Kiseleva I, Strasser RH, Decking UK, Stumpe T, Isenberg G, Schrader J. Inotropic response to beta-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. J Physiol 532: 195–204, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Nat Acad Sci 104: 20612–20617, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol Heart Circ Physiol 278: H971–H981, 2000 [DOI] [PubMed] [Google Scholar]
- 20. Han X, Kubota I, Feron O, Opel DJ, Arstall MA, Zhao YY, Huang P, Fishman MC, Michel T, Kelly RA. Muscarinic cholinergic regulation of cardiac myocyte ICa-L is absent in mice with targeted disruption of endothelial nitric oxide synthase. Proc Nat Acad Sci 95: 6510–6515, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hare JM. Nitric oxide and excitation-contraction coupling. J Mol Cell Cardiol 35: 719–729, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Hove-Madsen L, Prat-Vidal C, Llach A, Ciruela F, Casado V, Lluis C, Bayes-Genis A, Cina J, Franco R. Adenosine A2a receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc Res 72: 292–302, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Hu T, Chuang K, Yanasak N, Koretsky A. Relationship between blood and myocardium manganese levels during manganese-enhanced MRI (MEMRI) with T1 mapping in rats. NMR Biomed 24: 46–53, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu TCC, Pautler RG, MacGowan GA, Koretsky AP. Manganese-enhanced MRI of mouse heart during changes in inotropy. Mag Res Med 46: 884–890, 2001 [DOI] [PubMed] [Google Scholar]
- 25. Hunter DR, Haworth RA, Berkoff HA. Cellular manganese uptake by the isolated perfused rat heart: a probe for the sarcolemma calcium channel. J Mol Cell Cardiol 13: 823–832, 1981 [DOI] [PubMed] [Google Scholar]
- 26. Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer-Crosbie M, Picard MH, Szelid Z, Gillijns H, Van de Werf F, Collen D, Bloch KD. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res 94: 1256–1262, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res 92: 1322–1329, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Kim D, Gilson WD, Kramer CM, Epstein FH. Myocardial tissue tracking with two-dimensional cine displacement-encoded MR imaging: development and initial evaluation. Radiology 230: 862–871, 2004 [DOI] [PubMed] [Google Scholar]
- 29. Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B. Are myocardial eNOS and nNOS involved in the β-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc Res 70: 97–106, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res 93: 388–398, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Masumiya H, Tsujikawa H, Hino N, Ochi R. Modulation of manganese currents by 1,4-dihydropyridines, isoproterenol, and foskolin in rabbit ventricular cells. Pflügers Archiv Euro J Physiol 446: 695–701, 2003 [DOI] [PubMed] [Google Scholar]
- 32. Petroff MGV, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, Sollott SJ. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol 3: 867–873, 2001 [DOI] [PubMed] [Google Scholar]
- 33. Saraiva RM, Minhas KM, Raju SVY, Barouch LA, Pitz E, Schuleri KH, Vandegaer K, Li D, Hare JM. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: role of nitroso-redox equilibrium. Circulation 112: 3415–3422, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation 104: 1286–1291, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Sears C, Choate J, Paterson D. NO-cGMP pathway accentuates the decrease in heart rate caused by cardiac vagal nerve stimulation. J App Physiol 86: 510–516, 1999 [DOI] [PubMed] [Google Scholar]
- 36. Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res 92: e52–e59, 2003 [DOI] [PubMed] [Google Scholar]
- 37. Shen J, Jiang B, Pappano A. Comparison of L-type calcium channel blockade by nifedipine and/or cadmium in guinea pig ventricular myocytes. Pharmacol Exp Ther 294: 562–570, 2000 [PubMed] [Google Scholar]
- 38. Silberman GA, Fan THM, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC., Jr Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121: 519–528, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slawson S, Roman B, Williams D, Koretsky A. Cardiac MRI of the normal and hypertrophied mouse heart. Mag Res Med 39: 980–987, 1998 [DOI] [PubMed] [Google Scholar]
- 40. Spottiswoode B, Zhong X, Hess A, Kramer C, Meintjes E, Mayosi B, Epstein F. Tracking myocardial motion from cine DENSE images using spatiotemporal phase unwrapping and temporal fitting. IEEE Trans Med Imag 26: 15–30 [DOI] [PubMed] [Google Scholar]
- 41. Spottiswoode BS, Zhong X, Lorenz CH, Mayosi BM, Meintjes EM, Epstein FH. Motion-guided segmentation for cine DENSE MRI. Med Image Anal 13: 105–115, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stoyanovsky D, Murphy T, Anno PR, Kim YM, Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium 21: 19–29, 1997 [DOI] [PubMed] [Google Scholar]
- 43. Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res 101: 1155–1163, 2007 [DOI] [PubMed] [Google Scholar]
- 44. Van den Bergh A, Flameng W, Herijgers P. Parameters of ventricular contractility in mice: influence of load and sensitivity to changes in inotropic state. Pflügers Archiv Euro J Physiol 455: 987–994, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Vandecasteele G, Eschenhagen T, Scholz H, Stein B, Verde I, Fischmeister R. Muscarinic and beta-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nat Med 5: 331–334, 1999 [DOI] [PubMed] [Google Scholar]
- 46. Vandsburger M, Epstein F. Emerging MRI methods in translational cardiovascular research. J Cardio Trans Res 4: 477–492, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vandsburger MH, French BA, Helm PA, Roy RJ, Kramer CM, Young AA, Epstein FH. Multi-parameter in vivo cardiac magnetic resonance imaging demonstrates normal perfusion reserve despite severely attenuated β-adrenergic functional response in neuronal nitric oxide synthase knockout mice. Euro Heart J 28: 2792–2798, 2007 [DOI] [PubMed] [Google Scholar]
- 48. Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res 84: 1020–1031, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang H, Kohr MJ, Traynham CJ, Wheeler DG, Janssen PML, Ziolo MT. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am J Physiol Cell Physiol 294: C1566–C1575, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang H, Kohr MJ, Wheeler DG, Ziolo MT. Endothelial nitric oxide synthase decreases β-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am J Physiol Heart Circ Physiol 294: H1473–H1480, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Nat Acad Sci 96: 657–662, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279: 234–237, 1998 [DOI] [PubMed] [Google Scholar]
- 53. Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res 102: 242–249, 2008 [DOI] [PubMed] [Google Scholar]
- 54. Zhong J, Yu X. Strain and torsion quantification in mouse hearts under dobutamine stimulation using 2D multiphase MR DENSE. Mag Res Med 64: 1315–1322, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhong X, Helm P, Epstein F. Balanced multipoint displacement encoding for DENSE MRI. Mag Res Med 61: 981–988, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ziolo MT. The fork in the nitric oxide road: cyclic GMP or nitrosylation? Nitric Oxide 18: 153–156, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ziolo MT, Bers DM. The real estate of NOS signaling: location, location, location. Circ Res 92: 1279–1281, 2003 [DOI] [PubMed] [Google Scholar]
- 58. Ziolo MT, Maier LS, Piacentino V, III, Bossuyt J, Houser SR, Bers DM. Myocyte nitric oxide synthase 2 contributes to blunted β-adrenergic response in failing human hearts by decreasing Ca2+ transients. Circulation 109: 1886–1891, 2004 [DOI] [PubMed] [Google Scholar]