

Background: Conversion of fibronectin from a compact plasma protein to a fibrillar component of extracellular matrix is not understood.
Results: Binding of polypeptides by β-strand addition to N-terminal modules 1–5FNI is linked to changes in distant integrin- and glycosaminoglycan-binding regions.
Conclusion: Ligation of 1–5FNI is sufficient for fibronectin expansion.
Significance: Allosteric interactions among regions of fibronectin control assembly into extracellular fibrils.
Keywords: Extracellular Matrix, Fibronectin, Glycoprotein, Peptide Interactions, Protein Conformation, Bacterial Adhesin
Abstract
How fibronectin (FN) converts from a compact plasma protein to a fibrillar component of extracellular matrix is not understood. “Functional upstream domain” (FUD), a polypeptide based on F1 adhesin of Streptococcus pyogenes, binds by anti-parallel β-strand addition to discontinuous sets of N-terminal FN type I modules, 2–5FNI of the fibrin-binding domain and 8–9FNI of the gelatin-binding domain. Such binding blocks assembly of FN. To learn whether ligation of 2–5FNI, 8–9FNI, or the two sets in combination is important for inhibition, we tested “high affinity downstream domain” (HADD), which binds by β-strand addition to the continuous set of FNI modules, 1–5FNI, comprising the fibrin-binding domain. HADD and FUD were similarly active in blocking fibronectin assembly. Binding of HADD or FUD to soluble plasma FN exposed the epitope to monoclonal antibody mAbIII-10 in the tenth FN type III module (10FNIII) and caused expansion of FN as assessed by dynamic light scattering. Soluble N-terminal constructs truncated after 9FNI or 3FNIII competed better than soluble FN for binding of FUD or HADD to adsorbed FN, indicating that interactions involving type III modules more C-terminal than 3FNIII limit β-strand addition to 1–5FNI within intact soluble FN. Preincubation of FN with mAbIII-10 or heparin modestly increased binding to HADD or FUD. Thus, ligation of FNIII modules involved in binding of integrins and glycosaminoglycans, 10FNIII and 12–14FNIII, increases accessibility of 1–5FNI. Allosteric loss of constraining interactions among 1–5FNI, 10FNIII, and 12–14FNIII likely enables assembly of FN into extracellular fibrils.
Introduction
Fibronectin (FN)2 is a dimeric glycoprotein of blood and extracellular matrix. Each subunit of FN, depicted in Fig. 1A, includes 12 type 1 (FNI), 2 type 2 (FNII), and 15–17 type 3 (FNIII) modules, depending on the differential inclusion of FNIII extra domains A (EDA) and B (1). Subunits are linked by disulfide bonds at the extreme C termini. The N-terminal 70-kDa region (N-9FNI), which is of particular importance in this paper, includes the N-5FNI fibrin- and 6FNI-9FNI gelatin-binding domains and is composed of nine FNI and two FNII modules (Fig. 1A). FNI modules are found only in chordates, and tandem FNI modules are found only in FN (2).
FIGURE 1.

Diagram of FN and FN constructs, schematic of the F1 adhesin, and sequences of HADD versus FUD. A, each subunit of FN consists of 12 FNI modules (ovals), 2 FNII modules (diamonds), and 15 FNIII modules (squares) for the V89 splice variant shown. In plasma FN, one subunit contains a variable region, and the other subunit lacks it. Modules are numbered to facilitate naming recombinant proteins according to modular content. The boundaries of fibrin- and gelatin-binding domains are indicated, as are locations of epitopes for mAbs (asterisks). B, schematic of the F1 adhesin showing the signal sequence (S), four proline-rich repeats (small rectangles), upstream region (UR), numbered FNBRs, downstream region (DR), the wall-spanning region (W), and membrane-spanning region (M). C, five FNBRs and the upstream and downstream nonrepetitive regions of F1 adhesin (SfbI) and relationship of this sequence to FUD and HADD. The tails introduced into FUD or HADD by the cloning strategy are in lowercase. In HADD, 33 residues of the second or fourth FNBR are joined to 16 residues of the downstream region to add the binding sequence for 1FNI. The underlined sequences in HADD are predicted to interact with the indicated FNI modules (22, 24, 35).
Blood plasma FN is produced mainly by hepatocytes and circulates at near micromolar concentrations (3). Rotary shadowing, sedimentation velocity, and dynamic light scattering (DLS) experiments indicate that plasma FN is in a compact conformation that elongates at increased ionic strength or pH (4, 5). FN conformation can be monitored by monoclonal antibody (mAb) mAbIII-10, which recognizes an epitope in 10FNIII that is cryptic in soluble plasma FN at low ionic strength and exposed when FN is adsorbed to a surface, placed in high ionic strength, or incubated with heparin, gelatin, or gangliosides (6). The compact conformation is likely maintained by interactions among distant modules. Several such interactions have been surmised based on studies of FN fragments, including an intra-subunit interaction between 4FNI and 3FNIII (7), an inter-subunit interaction between 2–3FNIII and 12–14FNIII (8), and a less defined interaction between N-5FNI and 12–14FNIII (9, 10). The compact structure has been hypothesized to obscure ligand-binding regions of plasma FN and thereby prevent aberrant interactions in the bloodstream (11, 12).
Diverse cells, including fibroblasts, smooth muscle cells, and adherent platelets, support the assembly of compact, soluble FN into insoluble fibrils that support cell adhesion, growth, and migration (13). FN assembly is a complex process in which FN binds to the cell surface, engages receptors, most notably the α5β1 integrin binding to the RGD motif in 10FNIII, and elongates into fibrils (13, 14). Pieces of FN comprising the fibrin- and gelatin-binding domains or just the fibrin-binding domain, however, bind to cell surface sites of FN assembly and block assembly (15–17). A central question regarding FN assembly is whether FN initially interacts via α5β1 with 10FNIII followed by exposure of N-terminal FNI modules (13) or engages cell surface molecules via N-terminal FNI modules followed by exposure of 10FNIII (17).
Despite its recent appearance in the animal kingdom, FN functions as a modulator of cell adhesion and extracellular matrix formation from early development onward (18), and it is involved in diverse pathophysiologic processes, including vascular disease (3, 19). Remarkably, multiple bacteria target FN and its FNI modules as a vehicle to adhere to tissues and evade host defense (20). The pathophysiologic importance of bacteria-FN interactions is highlighted by the recent finding that Staphylococcus aureus recovered from patients with infected intravascular devices are enriched in polymorphisms of FN-binding protein A that increase the affinity of the protein for FN (21).
FN-binding protein A of S. aureus and the F1 adhesin (allelic variant SfbI) from Streptococcus pyogenes are representative of a large number of bacterial FN-binding proteins found in Gram-positive cocci and spirochetes that interact with the fibrin- and gelatin-binding domains (20, 22). Embedded within F1, going from N to C termini, are the following: a nonrepetitive sequence that binds to 8–9FNI (23, 24), five FN-binding repeats (FNBR) each of which can bind to 2–5FNI (25, 26), and a nonrepetitive sequence that binds to 1FNI (Fig. 1B) (25, 27, 28), and FN-binding protein A contains multiple FNBRs that bind to 2–5FNI (22). “Functional upstream domain” (FUD), a 49-amino acid polypeptide from the F1 adhesin, binds by anti-parallel β-strand addition to discontinuous sets of FNI modules within N-9FNI; the N-terminal nonrepetitive upstream region binds to 8–9FNI of the gelatin-binding domain (23, 24), and the adjoining first FNBR binds to 2–5FNI of the fibrin-binding domain (Fig. 1C) (24, 27). FUD blocks FN assembly (29) and exposes the mAbIII-10 epitope in 10FNIII (30) and thus may mimic cell surface molecules of vertebrate cells that interact with N-terminal modules and expose 10FNIII. It is not known whether the effects of FUD are due to its interaction with 2–5FNI, 8–9FNI, or both 2–5FNI and 8–9FNI. To learn the importance of the fibrin and gelatin binding, we compared FUD to “high affinity downstream domain” (HADD), a 49-residue polypeptide similar to SfbI-5 with its adjacent nonrepetitive sequence, i.e. HADD was designed to bind tightly to N-5FNI by anti-parallel β-strand addition (Fig. 1C).
MATERIALS AND METHODS
Proteins
Plasma FN was purified from a fibrinogen-rich plasma fraction (31). Expression and purification of polyhistidine-tagged monomeric N-5FNI, N-9FNI, and N-3FNIII and dimeric 6FNI-C (starts at residue 291 (numbering from the initiating methionine) and contains V89 version of the variable region without extra domains A or B) were accomplished using recombinant baculovirus and affinity chromatography as described previously (24, 32). The recombinant proteins were secreted into medium at concentrations of 5–20 μg/ml, and after purification were pure and migrated as expected when analyzed by SDS-PAGE without and with prior reduction. The molarity of FN and constructs stated throughout the paper were calculated based on the monomer or subunit, assumed to have an average molecular mass of 250 kDa for the subunits of heterodimeric plasma FN. Rat tail type I collagen (Upstate) and gelatin (Sigma) were purchased.
FUD was expressed and purified as described previously (24). For HADD, PCR-based strategies were used to add DNA encoding the 16 C-terminal residues of SfbI-5 to DNA encoding 33 residues of FNBR SfbI-4 (Fig. 1C). HADD was expressed, purified, and characterized as described previously for FUD (24) except for determination of concentration. Because HADD lacks tyrosine or tryptophan, its concentration could not be estimated by absorbance at 280 nm and instead was determined using the bicinchoninic acid (BCA) assay (Pierce) with FUD as the standard (24). Amino acid analysis of HADD performed at University of California-Davis Proteomics Core facility yielded a value that was within 10% of that estimated by the BCA.
Mouse anti-human monoclonal antibody mAbIII-10 against 10FNIII was described previously (6), as were mouse anti-human mAbs 4D1, 7D5, 5C3, and 9D2 (24, 30).
Labeling
FN was labeled with fluorescein isothiocyanate FN (FITC-FN) as described previously (33) or with Alexa Fluor 488 (Invitrogen) (A488-FN) as per the manufacturer's instructions. Biotinylation of FUD or HADD with N-hydroxysulfosuccinimide-biotin (Pierce) was done as described previously (24); biotinylated probes are designated by the prefix “b-.”
Assembly Assays
Fluorescence microscopic assays of FN assembly were done as described previously (24) except mouse FN−/− cells were allowed to adhere for 2 h and assembled exogenous FITC-FN for 1 h. For quantitative dose-response assays, human foreskin fibroblasts (strain AH1F) were seeded in microplate wells and incubated with 20 nm A488-FN in the presence or absence of 4 nm to 2.5 μm FUD or HADD. After 18 h at 37 °C, monolayers were washed, and fluorescence of assembled A488-FN was quantified (excitation, 485 nm; emission, 535 nm) using the Tecan GeniosPro.
Enzyme-linked Immunosorbent Assays
Wells of high binding plates (Costar 3950) were coated overnight with 40 nm FN, N-5FNI, 6FNI-C, or N-9FNI in 10 mm Tris, 150 mm NaCl, pH 7.4 (TBS), as indicated in the figure legends. Binding of b-HADD or b-FUD to adsorbed proteins was quantified with the alkaline phosphatase-streptavidin complex as described previously for b-FUD (24). Where indicated, binding of b-HADD was compared with separate wells precoated with 1 μg/ml b-FUD as a positive control. The experimental setup was altered as described previously to compare the effect of soluble mAb, 1 mm zinc sulfate, FN, or FN constructs on binding of b-HADD or b-FUD to adsorbed FN (24). In some experiments, unfractionated heparin, 6–30 kDa in size (grade 1A from porcine intestine (Sigma)), was incubated with FN or N-9FNI prior to the addition of b-HADD or b-FUD. In other experiments, purified mAbIII-10 was preincubated with soluble FN or N-9FNI for 1 h prior to the addition of b-HADD or b-FUD. Binding was done in 10 mm Tris, 50–300 mm NaCl, pH 7.4, as indicated in the figure legends. Wells were washed twice with the solution used in the binding step and twice more in 10 mm Tris, 150 mm NaCl, 0.05% Tween, prior to addition of enzyme-linked streptavidin (24). mAb binding to adsorbed FN in the presence of HADD or FUD was quantified as described previously (24).
In experiments looking at the binding of FN to collagen or gelatin, 4 nm FN and 100 nm polypeptide diluted in TBS containing 0.05% Tween and 0.2% bovine serum albumin (BSA) were incubated for 30 min prior to addition and a 2-h incubation with high binding plates precoated at 37 °C overnight with 10 μg/ml collagen or gelatin. Bound FN was detected with 9D2 followed by peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch). Bound conjugate was quantified by addition of SureBlue TMB microwell peroxidase substrate (KPL, Gaithersburg, MD), and the reaction was stopped with the addition of TMB stop solution (KPL). The A450 nm was determined as above.
Competitive Binding Assays with mAbIII-10
High binding plates were coated overnight with 8 nm FN, washed with 10 mm Tris, 50 mm NaCl, 0.05% Tween, pH 7.4, and blocked with 1% BSA in washing buffer for 1 h. In some experiments, HADD or FUD was added to FN in 10 mm Tris, 50 mm NaCl, pH 7.4, plus 0.2% BSA for 30 min. To 150 μl of the FN-containing solution, 25 μl of mAbIII-10 ascites (previously diluted to yield a final dilution of 1:50,000) was added in 10 mm Tris, 50 mm NaCl, pH 7.4, plus 0.2% BSA and 0.35% Tween 20 (final concentration 0.05%), and the mixture was incubated for 1 h prior to transfer to FN-coated wells and incubation for 2 h. Plates were washed two times with 10 mm Tris, 50 mm NaCl, 0.05% Tween, pH 7.4, and two times with TBS containing 0.05% Tween. Alkaline phosphatase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch) at 1:5000 was added to wash buffer and incubated for 1 h. Plates were washed four additional times before the addition of 1 mg/ml 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma) in 10 mm Tris, 150 mm NaCl, pH 9.0. A405 nm was determined using a Tecan Genios Pro.
In experiments comparing purified FN to FN in plasma, exposure of the mAbIII-10 epitope was monitored by a competitive ELISA similar to that described above. Purified FN was diluted in TBS to the concentration of FN determined to be in the plasma sample by competitive ELISA using mAb 9D2, which is not sensitive to conformation as described previously (30). These samples were then diluted further in 10 mm Tris, 50 mm NaCl, pH 7.4, to 233 nm FN without or with FUD or HADD in 2.5-fold molar excess. After 30 min, the mixtures were diluted further, and mAbIII-10 ascites (final concentration after addition of 1:50,000) was added in a volume that brought the concentration of the FN to 200, 100, 50, 25, or 12.5 nm. After 30 min, these mixtures were added to FN-coated wells, which were incubated for 1.5 h. Washes and incubations to determine binding of mAbIII-10 to adsorbed FN were as described above except that the detecting reagent was peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch), and bound conjugate was quantified by addition of SureBlue TMB microwell peroxidase substrate as above.
Dynamic Light Scattering and Isothermal Titration Calorimetry (ITC)
Measurements were performed using Beckman Coulter (Miami, FL) N5 Submicron Particle Size Analyzer instrument in 20 mm Tris, 100 mm NaCl, pH 7.4, at 25 °C. The buffer was degassed and filtered with 0.22-μm Millex-GP filter (Millipore, Cork, Ireland). FN at a concentration of 1 mg/ml (4 μm FN subunit) was titrated with solutions of 200 μm FUD or 165 μm HADD. After each addition, the mixture was allowed to equilibrate for 10 min, and then scattered intensity of light from the He-Ne laser was measured at 90° for 200 s; each measurement was repeated six times. Data were processed by Photon Correlation Spectroscopy software. The mean size of FN or FN-polypeptide complex was computed assuming a refractive index of 1.33 and viscosity of 0.89 poise. Size distribution processor analysis was performed for each sample, and the mean size of the particle was determined according to the volume (weight) distribution format of size distribution processor analysis. Large particles accounted for <5% of total particles.
ITC was performed as described previously (24) with a VP-ITC microcalorimeter (MicroCal, LLC) at 25 °C. The cell contained 1.4 ml of a solution of FN or N-9FNI, and the syringe contained 600 μl of HADD. The titration was performed in 37 injections (1 of 1 μl, four of 4 μl, and 32 of 8 μl) delivered at 120-s intervals. Data from the initial injection were discarded. HADD, N-9FNI, and FN were dialyzed against PBS, pH 7.4, and adjusted to the concentrations given in Table 1. Data were fit by Lavenberg-Marquardt nonlinear regression with Origin 7.0 using the one-site model.
TABLE 1.
ITC analysis of the interaction of HADD with N-9FNI and intact FN
The stoichiometry given for FN is per FN dimer.
| FN in cell | Polypeptide in syringe | ΔH | ΔS | Kd | N | |
|---|---|---|---|---|---|---|
| μm | μm | kcal mol−1 | cal mol−1degree−1 | nm | ||
| FN-HADD | 2.5 | 35 | −27.6 | −56.4 | 12.9 | 1.5 |
| N-9FNI-HADD | 3.8 | 35 | −48.1 | −122 | 2.4 | 0.8 |
RESULTS
HADD Binds to N-5FNI
Binding of FUD to 2–5FNI and 8–9FNI of soluble FN blocks FN assembly and induces a conformational change that exposes the epitope to mAbIII-10, which is otherwise partially cryptic at low ionic strength (24, 29, 30). We have speculated that binding of FUD blocks assembly and exposes the mAbIII-10 epitope by breaking the electrostatic interaction between 4FNI and 3FNIII or coupling of binding to 8–9FNI with conformational changes in adjacent 1–3FNIII and linkage of changes in 3FNIII to changes in 10FNIII (24). Learning the contribution of 2–5FNI requires a polypeptide that binds by β-strand addition to 2–5FNI with an affinity that is comparable with the affinity of binding of FUD to N-9FNI. Polypeptides based on individual FNBRs of F1 adhesin (e.g. SfbI-2 or -4) bind to N-9FNI with >10-fold looser affinity than FUD (34). We therefore designed, expressed, and purified HADD, which contains SfbI-4 and the downstream region of SfbI-5 (Fig. 1B). Based on studies of SfbI-5 and its adjacent downstream region or a homologous construct from the Streptococcus dysgalactiae adhesin (28, 35), we anticipated that HADD would bind to N-9FNI with an affinity equivalent to FUD because the favorable energy of binding to 1FNI substitutes for loss of the favorable energy of binding to 8–9FNI (Fig. 1C). Indeed, KD values for binding of HADD to N-9FNI and intact FN were 2.4 and 12.6 nm, respectively, as measured by ITC in 150 mm sodium chloride at 25 °C (Table 1); these affinities are comparable with ITC measurements of binding of FUD to N-9FNI and intact FN (24). Furthermore, the interactions were driven by ΔH values favorable enough to overcome unfavorable ΔS values (Table 1), as in the case of binding to SfbI-5 by β-strand addition to N-5FNI (35).
The specificity of HADD for N-5FNI was assessed by five additional assays. When we examined the ability of b-HADD to bind adsorbed FN, N-5FNI, or 6FNI-C, there was similar binding of b-HADD to FN and N-5FNI and no binding to 6FNI-C (Fig. 2A). As with b-FUD binding (24), mAb 4D1 to 2FNI slightly decreased b-HADD binding to coated FN, whereas 7D5 to 4FNI decreased binding of both polypeptides considerably (Fig. 2B). In contrast, 5C3 to 9FNI decreased b-FUD binding to adsorbed FN but increased binding of b-HADD (Fig. 2B). In the reciprocal experiment, FUD or HADD increased binding of 4D1 and decreased binding of 7D5 to coated FN, whereas FUD decreased, and HADD had no effect on binding of 5C3 to coated FN (Fig. 2C). Thus, the cross-competition studies indicate that HADD binds to 2FNI and 4FNI but not 9FNI. Zn2+ binds to and grossly alters the global fold of 8FNI (36) and decreases the ability of FUD to bind to adsorbed FN (24). Consistent with the expected binding specificity of HADD versus FUD, 1 mm Zn2+ had no effect on binding of b-HADD to adsorbed FN under conditions in which binding of b-FUD was decreased (Fig. 2D). Collagenous sequences bind to an extended site on the gelatin-binding domain that includes 8–9FNI (37). Consistent with this, preincubation of FN with soluble FUD, but not HADD, blocked binding of FN to adsorbed gelatin or collagen (Fig. 2E).
FIGURE 2.

HADD binds to FN via N-5FNI. A, enzyme-linked assay of increasing concentrations of biotinylated-HADD (b-HADD) binding to wells coated with 40 nm FN (□), N-5FNI (▴), or 6FNIII-C (▾). The amount bound was normalized to a positive control, and wells were coated with b-FUD at 1 μg/ml. B, binding relative to no mAb of 0.3 nm b-HADD or b-FUD to coated FN in the presence of 30 μg/ml 4D1 to 2FNI, 7D5 to 4FNI, or 5C3 to 9FNI. C, binding relative to no peptide of 4D1 (1:50,000 ascites), 7D5 (1:50,000 ascites), or 5C3 (1:30,000 ascites) in the presence of 175 nm HADD or FUD. D, binding relative to no Zn2+ of 0.3 nm b-HADD or b-FUD incubated with coated FN in the absence or presence of 1 mm Zn2+. E, binding of FN to collagen or gelatin in the presence or absence of 100 nm HADD or FUD as detected by 9D2. Values are mean ± S.D. of three (A–D) or two experiments. Significance of the differences from the indicated 100% controls were calculated by a t test. B–E, differences of p < 0.05 from 100% controls are indicated by asterisks.
HADD, Like FUD, Blocks FN Assembly
Deletion of N-5FNI from FN prevents the formation of FN fibrils (38), and exogenous N-9FNI (15), FUD (29), or monoclonal antibody to 4FNI or 9FNI (24) blocks the assembly of FN. To test whether ligation of 1–5FNI by β-strand addition is sufficient to block FN assembly, we asked if HADD blocks FN assembly. FN−/− cells adherent to adsorbed laminin were incubated with 20 nm FITC-FN for 1 h with or without 50 or 500 nm HADD. There was a decrease in FN fibrils when the concentration of HADD was 50 nm and a loss of FN fibrils at 500 nm (Fig. 3A). Dose-response curves of inhibition were performed on deposition of soluble 20 nm A488-FN over 18 h by monolayers of human foreskin fibroblasts using fluorescence as a read-out (Fig. 3B). HADD and FUD were similarly active and had near maximum effects at 50 nm.
FIGURE 3.

HADD inhibits FN assembly by fibroblasts. A, mouse FN−/− cells adherent to laminin-coated coverslips were given 20 nm FITC-FN in the absence (NA) or presence of 50 or 500 nm HADD. Following incubation for 1 h, cells were washed, fixed, and imaged via fluorescence microscopy. Photomicrographs were taken at the exposure time determined for NA control and manipulated similarly. Bar, 10 μm. Shown are typical images from multiple fields in two different experiments. Similar results were seen in additional experiments not shown on FN−/− cells adherent to FN or to FN lacking the N-9FNI region. B, dose-dependent inhibition of A488-FN incorporation into fibroblast matrices by HADD or FUD. A488-FN (20 nm) in 2% calf serum was incubated for 18 h with monolayers of human foreskin fibroblasts in the presence or absence of the indicated concentrations of FUD or HADD. Following washes in PBS, fluorescence intensity (F) was measured in a microplate reader. In the experiment shown, the fluorescence of cells not treated with A488-FN was 3900 and subtracted from each of the values. The results are typical of four experiments done with FUD and two experiments done with HADD.
Binding of HADD, Like Binding of FUD, Causes Exposure of the mAbIII-10 Epitope and Expansion of Soluble FN
To determine whether HADD, like FUD, causes conformational change in soluble FN, we monitored exposure of the mAbIII-10 epitope using a competitive ELISA in which binding of mAbIII-10 to substrate-bound FN is inhibited by increasing concentrations of soluble purified FN with or without a 2.5-fold molar excess of the polypeptide. The assay was done in low salt (50 mm NaCl) in which the mAbIII-10 epitope is hidden (6). HADD or FUD caused FN to compete many-fold better than FN alone (Fig. 4A). The competition curves for FN alone were complex, steeper at lower concentrations than at higher concentrations, although not as steep as when HADD or FUD was present. Purified FN has been demonstrated previously to contain multimers that interact preferentially with mAbIII-10; such multimers could account for complexity of the inhibition curve (6). To ascertain if the complexity of the inhibition curves is a result of the purification procedure, inhibition was performed with plasma with known FN concentration. The same complexity was found for unfractionated plasma as for purified FN, and enhancement of competition by addition of HADD or FUD to plasma was similar to enhancement of competition in purified FN (Fig. 4A). Thus, FN exists as a variety of conformers in plasma before purification, and some of the conformers compete for mAbIII-10 binding better than others and all or almost all conformers are susceptible to further conformational change upon complex formation with HADD or FUD.
FIGURE 4.
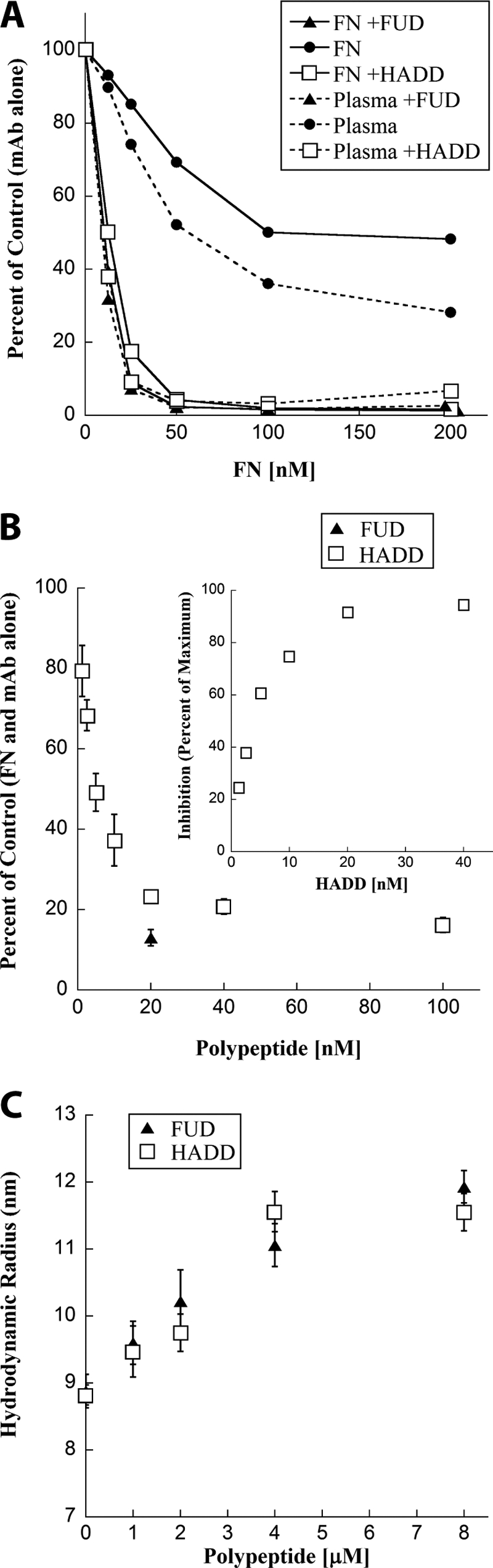
HADD is similar to FUD in exposing the mAbIII-10 epitope of purified FN or FN in plasma and expanding purified FN. A, effect of HADD or FUD on the exposure of the mAbIII-10 epitope in purified FN and FN in plasma as determined by competitive ELISA. Purified FNs (solid lines) or FNs in diluted plasma (dotted lines), 233 nm, were incubated without (●) or with 580 nm FUD (▴) or HADD (□) for 30 min. Prior to the assay, the concentration of FN in neat plasma was found to be 0.65 mg/ml (2600 nm) by competition ELISA with a mAb that is not conformation-sensitive. The six samples were then diluted to the indicated FN concentrations; mAbIII-10 was added, and competition by soluble FN for mAbIII-10 binding to coated FN was determined. Data are expressed as percent of mAbIII-10 binding alone (no FN or polypeptide added) and are representative of two experiments. B, ELISA of competition of binding of mAbIII-10 to coated FN by 20 nm soluble FN alone or preincubated with the indicated concentrations of HADD (□) or with 20 nm FUD (▴). Values are expressed relative to 20 nm soluble FN with mAbIII-10 but no polypeptide and represent mean ± S.D. of three experiments. The inset replots the HADD titration in comparison with the maximum inhibition found with 40 nm HADD. C, HADD (□) or FUD (▴) were titrated into FN solution separately, and the hydrodynamic radius of 4 μm FN or 4 μm FN plus the indicated concentration of polypeptide was calculated. Measurements were performed at 25 °C in 20 mm Tris, 100 mm sodium chloride, pH 7.4. Error bars indicate the standard deviation of six measurements on each sample. The experiment was repeated twice with the same result.
When the concentration of soluble FN was 20 nm and exposure of the mAbIII-10 epitope was measured as function of increasing concentrations of HADD, a similarly complex curve was obtained with a near maximal effect when the ratio of HADD/FN subunit was 1:1 (Fig. 4B). To relate epitope exposure to another measure of conformational change, hydrodynamic radius was determined by DLS as 4 μm FN was titrated with HADD or FUD (Fig. 4C). The radius increased linearly as a function of polypeptide concentration, changing from ∼9 to ∼11.5 nm and reaching a maximum at polypeptide/FN subunit ratios of 1:1. These results indicate that HADD or FUD each forms a tight complex with FN in which FN assumes an expanded conformation in which the mAbIII-10 epitope is exposed.
Soluble N-9FNI and N-3FNIII Compete Better Than Soluble FN for Binding of HADD to Substrate-bound FN
Soluble N-9FNI competes better than soluble FN for binding of b-FUD to coated FN (24). We hypothesized that this difference is due to intramolecular interactions within FN occluding the FUD-binding site. To determine whether the electrostatic interaction between 4FNI and 3FNIII (7) is responsible for the difference in ability of FN and N-9FNI to compete with b-FUD or b-HADD, we tested competition by soluble N-3FNIII, in which the interaction has been demonstrated (7), for binding of b-FUD or b-HADD to adsorbed FN. Because the 4FNI-3FNIII interaction is sensitive to ionic strength (7), binding assays were done in buffer containing 50 mm NaCl. Increasing concentrations of N-3FNIII competed for binding of b-FUD or b-HADD to adsorbed FN to nearly the same extent as N-9FNI, and both competed ∼10-fold better than soluble FN (Fig. 5, A and B).
FIGURE 5.
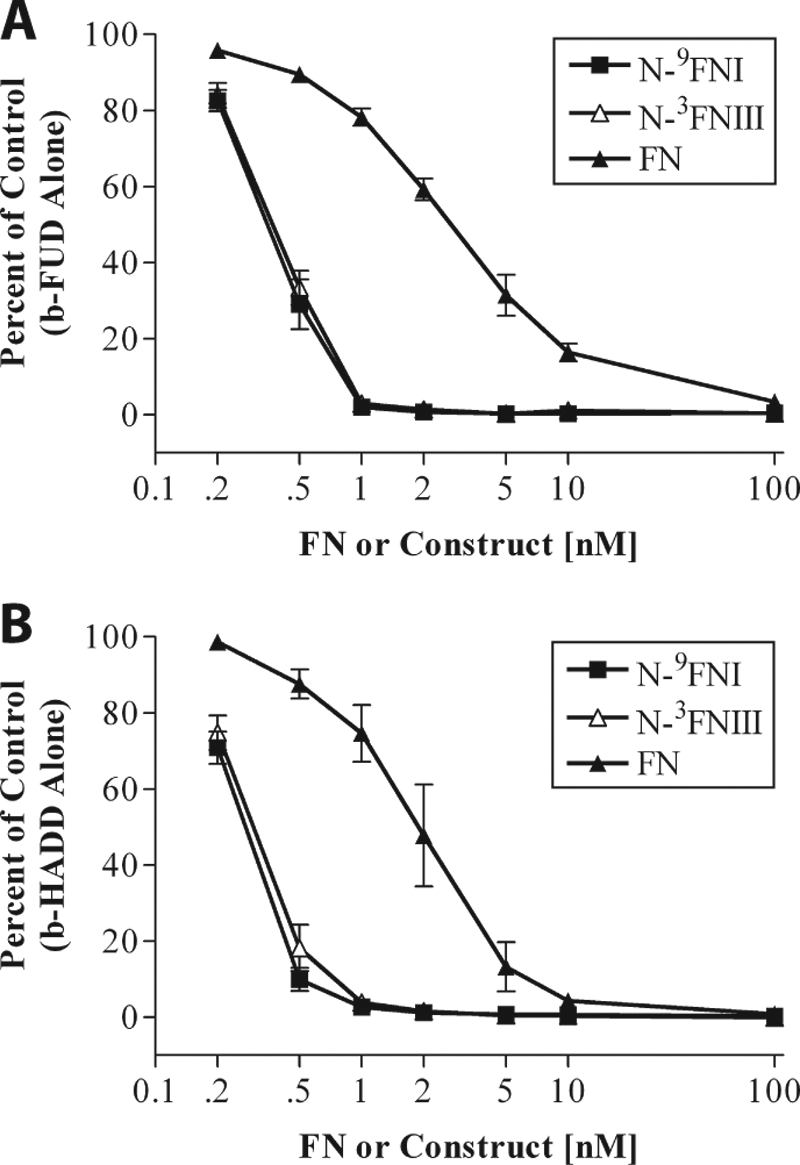
N-9FNI and N-3FNIII compete similarly for FUD or HADD binding to adsorbed FN. Binding of 0.3 nm b-FUD (A) or 0.3 nm b-HADD (B) to coated FN in the presence of increasing concentrations of soluble N-9FNI (■), N-3FNII (△), or FN (▴). Assays were in Tris buffer, pH 7.4, containing 50 mm NaCl. Values are expressed relative to biotinylated polypeptide alone and represent mean ± S.D. of three experiments.
mAbIII-10 or Heparin Increases the Ability of Soluble FN to Bind to b-FUD or b-HADD
The observations that HADD or FUD causes exposure of the mAbIII-10 epitope and N-9FNI and N-3FNIII both compete better than soluble FN for binding of FUD or HADD suggest that the 4FNI-3FNIII interaction is part of a larger network that limits β-strand addition to 1–5FNI. We therefore compared FN and FN-mAbIII-10 1:1 complex as competitors for binding of b-FUD or b-HADD to adsorbed N-9FNI. Complex formation with mAbIII-10 increased the ability of FN to compete for binding of b-FUD (Fig. 6A) or b-HADD (Fig. 6B) to N-9FNI. The effect was small, 2-fold for FUD and ∼1.5-fold for HADD, but reproducible.
FIGURE 6.

Complex formation with mAbIII-10 or heparin increases competition by soluble FN for binding of FUD or HADD to adsorbed N-9FNI or FN. A and B, competition for binding of 0.3 nm b-FUD (A) or b-HADD (B) to coated N-9FNI by increasing concentrations of soluble FN (□) or FN plus mAbIII-10 (present at a ratio of 1 IgG per FN subunit) (▴). Assays were done in Tris buffer, pH 7.4, containing 300 mm NaCl. C and D, competition for binding of 0.3 nm b-FUD (C) or b-HADD (D) to coated FN in the presence of N-9FNI (△), N-9FNI plus 0.25 mg/ml heparin (▴), FN (□), or FN plus 0.25 mg/ml heparin (▾). Assays were done in Tris buffer, pH 7.4, containing 150 mm NaCl. Values are expressed relative to biotinylated polypeptide alone and represent mean ± S.D. of 3 (A), 2 (B), 3 (C), or 2–3 (D) experiments.
Heparin binds to 12–14FNIII (1, 39) and is known to expose the mAbIII-10 epitope (6). To investigate the possibility that heparin binding causes a coupled conformational change encompassing both 10FNIII and the N-terminal FNI modules, we looked at the effect of heparin on the ability of soluble FN to compete with b-FUD or b-HADD for binding to adsorbed FN (Fig. 6, C and D). Using the type of competitive ELISA shown in Fig. 4, heparin, 0.25 mg/ml, ∼10 μm, caused increased exposure of the mAbIII-10 epitope as in published experiments (results not shown) (6). Although it had no effect on competition by soluble N-9FNI for binding of b-FUD (Fig. 6C) or b-HADD (Fig. 6D) to substrate-bound FN, this concentration of heparin caused ∼2-fold enhancement of the ability of soluble FN to compete for binding of b-FUD or b-HADD. The differences between the effects of heparin on the competition by soluble N-9FNI and FN for FUD or HADD binding indicate that the small but reproducible effect of heparin on the mAbIII-10 exposure assay is due to distant binding of heparin to 12–14FNIII rather than direct binding by heparin to the N-9FNI region.
DISCUSSION
FN assembly by adherent vertebrate cells and F1 adhesin/SfbI-mediated entrance of S. pyogenes into host cells utilize common features of FN, i.e. interactions involving the N-terminal N-9FNI region of FN and binding of integrins to FNIII modules, usually α5β1 to 10FNIII (13, 40). To investigate how ligation of the N terminus of FN leads to exposure of 10FNIII for processes including assembly and internalization, we utilized a competitive binding assay to monitor accessibility of the mAbIII-10 epitope in 10FNIII that is cryptic in soluble FN at low ionic strength (6). Previous work showed that the mAbIII-10 epitope becomes available upon incubation of soluble FN with FUD, which binds to 8–9FNI and 2–5FNI by β-strand addition (24) or denatured collagen (gelatin) (6, 30). In addition, expansion of plasma FN is seen upon binding of cyanogen bromide fragment 7 (CB7) of the α1(I) chain of type I collagen (41). CB7 contains a sequence that binds by β-strand addition to 2FNII-9FNI (37). To determine whether ligation of 8–9FNI is necessary for exposure of the mAbIII-10 epitope, we designed a polypeptide, HADD, that mimics SfbI-5 in binding to 1–5FNI (28, 35). HADD exposed the mAbIII-10 epitope and caused expansion of FN as assessed by DLS, indicating that ligation of 8–9FNI is not necessary for FN expansion. Exposure of mAbIII-10 epitope by HADD demonstrates that ligation of the fibrin-binding region alone is sufficient to disrupt intramolecular interactions and cause long range conformational changes that result in the exposure of 10FNIII.
Plasma FN is a heterodimer of subunits that differ in whether the variable region is present (1). The conformations assumed by the 58 modules of plasma FN are presumably controlled by “head-to-tail” interactions between consecutive modules and longer range interactions among nonadjacent modules. Candidate long range interactions have been identified between 4FNI and 3FNIII of the same subunit (7), between 2–3FNIII and 12–14FNIII of different subunits (8), and a less characterized interaction between N-5FNI and 12–14FNIII (9, 10). Soluble FN as compared with soluble N-9FNI has decreased ability to compete for binding of HADD to adsorbed FN. Experiments showing that N-9FNI and N-3FNIII compete equally well for HADD suggest that disruption or loss of the interface between 4FNI and 3FNIII is not sufficient to explain why soluble FN competes better for mAbIII-10 in the presence of FUD or HADD and indicates the involvement of FN modules C-terminal to 3FNIII. This finding is compatible with published ITC experiments demonstrating little difference in KD values or enthalpies for binding of a FUD-like polypeptide to N-9FNI compared with N-3FNIII (34). Fig. 7 describes a “beads on a string” (5) model for compact FN drawn to emphasize the interactions described above. The intra-subunit 4FNI-3FNIII and inter-subunit 2–3FNIII-12–14FNIII interactions constitute a pair of junctions at the core of the molecule. We speculate that disruption of weak 4FNI-3FNIII interactions in both subunits by binding of FUD or HADD may be coupled to more energetically profound disruptions of inter-subunit interactions between 2–3FNIII and 12–14FNIII. These disruptions result in extension of the two subunits, shown at its most extreme in Fig. 7, and exposure of 10FNIII.
FIGURE 7.

Diagram of how FUD and HADD may cause expansion of plasma FN. Plasma FN in a conceptual compact conformation (top) or extended conformation (bottom). One subunit is drawn with completely filled symbols, the other with outlined symbols. The complete dimer is shown in the compact conformation, and one subunit and part of the second is shown for the extended conformation. Only selected modules are numbered. The subunits are held together by disulfides at the C termini and interactions of 12–14FNIII with 2–3FNIII (red three-dimensional box). Each subunit is further constrained by the 4FNI-3FNIII interaction (pink diamond). FUD or HADD, which by themselves are random coils, form β-zippers with the indicated FNI modules, resulting in unfolding and expansion of the quaternary structure. The epitope for mAbIII-10 used to monitor conformational change is in 10FNIII (red dot).
Our experiments and the literature yield additional, albeit limited, insight about the impact of HADD or FUD binding on conformation of a ligated FN subunit versus the overall dimer. Expansion of FN as assessed by DLS was a linear function of the HADD or FUD concentration that reached completion at a polypeptide/FN subunit ratio of 1:1, indicating that ligation of an individual FN subunit results in expansion of that subunit independent of the second subunit. In contrast, the dose response for exposure of the mAbIII-10 epitope by FUD (30) or HADD (Fig. 4B) was curvilinear and ∼75% complete at a polypeptide/FN dimer ratio of 1:1 (polypeptide/FN subunit ratio of 0.5:1). This finding suggests that ligation of a single subunit of plasma FN is sufficient to allow binding of mAbIII-10. Published electron microscopic images of FN-mAbIII-10 complexes revealed FN in a V-shaped configuration with mAbIII-10 in the angle of the “V” with Fab arms binding to each subunit of FN (6). Univalent binding of one Fab arm of mAbIII-10 to one FN subunit therefore may initiate cooperative events to form a complex in which mAbIII-10 and FN are bivalent in relation to each other. Such cooperativity is consistent with the stronger binding of preformed mAbIII-10-FN complexes to HADD or FUD.
HADD, like FUD, blocks FN assembly, suggesting that the fibrin-binding domain of FN mediates the first interaction with the cell surface during FN assembly (15–17, 29). Despite this, there appear to be instances in which ligation of 10FNIII dominates over exposure of the FN N terminus (13). Using competitive ELISAs, we show that binding of mAbIII-10 to FN modestly but significantly increased the ability of soluble FN to interact with FUD or HADD. Heparin, which like FUD or HADD increases exposure of the mAbIII-10 epitope (6), also increased the ability of soluble FN to interact with FUD or HADD. As described above, the modules that bind heparin with high affinity, 12–14FNIII (39), have been deduced to interact with 2–3FNIII in the opposite subunit (8) and with N-5FNI (9, 10). Thus, binding of heparan sulfate to 12–14FNIII may control accessibility of both the RGD cell adhesive sequence in 10FNIII and the N-terminal FNI modules during assembly. The present findings therefore support the existence of an allosteric network among the fibrin-binding domain 1–5FNI, 10FNIII, and 12–14FNIII in dimeric plasma FN. Such an allosteric network would allow for controlled exposure to binding sites in FN during FN assembly and bacterial host cell invasion.
The allosteric network among the fibrin-binding domain, 10FNIII, and 12–14FNIII is likely different in cellular FN containing differentially spliced EDA and extra domain B FNIII modules (1). For instance, in the 7–14FNIII constructs lacking 2–3FNIII, the presence of EDA between 11FNIII and 12FNIII resulted in dimers because of interaction of EDA with 12–14FNIII, suggesting that the presence of EDA in intact FN may favor an open conformation by competing with 2–3FNIII for binding to 12–14FNIII (8). Such an open conformation may account for the unique ability of EDA-positive FN to be assembled into extensive fibrous networks by CHO cells (42). Interestingly, FN assembly associated with lymphatic valves in mice has been shown to require interaction of α9β1 integrin with the α9β1-recognition sequence in EDA rather than interaction of α5β1 integrin with 10FNIII (43).
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL021644 (to D. F. M.), Grants T32 AG000213 and T32 GM008692 (predoctoral support to L. M. M.), and Grant R21 NS07647 (to B. T. J.).
- FN
- fibronectin
- FNI
- FN type I
- FNII
- FN type II
- FNIII
- FN type III
- EDA
- extra domain A
- FUD
- functional upstream domain
- HADD
- high affinity downstream domain
- DLS
- dynamic light scattering
- ITC
- isothermal titration calorimetry
- FNBR
- FN-binding repeat.
REFERENCES
- 1. Pankov R., Yamada K. M. (2002) Fibronectin at a glance. J. Cell Sci. 115, 3861–3863 [DOI] [PubMed] [Google Scholar]
- 2. Tucker R. P., Chiquet-Ehrismann R. (2009) Evidence for the evolution of tenascin and fibronectin early in the chordate lineage. Int. J. Biochem. Cell Biol. 41, 424–434 [DOI] [PubMed] [Google Scholar]
- 3. Maurer L. M., Tomasini-Johansson B. R., Mosher D. F. (2010) Emerging roles of fibronectin in thrombosis. Thromb. Res. 125, 287–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Odermatt E., Engel J. (1989) Physical Properties of Fibronectin in Fibronectin (Mosher D. F., ed) pp. 29–45, Academic Press, Inc., San Diego, CA [Google Scholar]
- 5. Rocco M., Infusini E., Daga M. G., Gogioso L., Cuniberti C. (1987) Models of fibronectin. EMBO J. 6, 2343–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ugarova T. P., Zamarron C., Veklich Y., Bowditch R. D., Ginsberg M. H., Weisel J. W., Plow E. F. (1995) Conformational transitions in the cell binding domain of fibronectin. Biochemistry 34, 4457–4466 [DOI] [PubMed] [Google Scholar]
- 7. Vakonakis I., Staunton D., Ellis I. R., Sarkies P., Flanagan A., Schor A. M., Schor S. L., Campbell I. D. (2009) Motogenic sites in human fibronectin are masked by long range interactions. J. Biol. Chem. 284, 15668–15675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson K. J., Sage H., Briscoe G., Erickson H. P. (1999) The compact conformation of fibronectin is determined by intramolecular ionic interactions. J. Biol. Chem. 274, 15473–15479 [DOI] [PubMed] [Google Scholar]
- 9. Bultmann H., Santas A. J., Peters D. M. (1998) Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J. Biol. Chem. 273, 2601–2609 [DOI] [PubMed] [Google Scholar]
- 10. Homandberg G. A., Erickson J. W. (1986) Model of fibronectin tertiary structure based on studies of interactions between fragments. Biochemistry 25, 6917–6925 [DOI] [PubMed] [Google Scholar]
- 11. Balbona K., Tran H., Godyna S., Ingham K. C., Strickland D. K., Argraves W. S. (1992) Fibulin binds to itself and to the carboxyl-terminal heparin-binding region of fibronectin. J. Biol. Chem. 267, 20120–20125 [PubMed] [Google Scholar]
- 12. Pearlstein E. (1978) Substrate activation of cell adhension factor as a prerequisite for cell attachment. Int. J. Cancer 22, 32–35 [DOI] [PubMed] [Google Scholar]
- 13. Singh P., Carraher C., Schwarzbauer J. E. (2010) Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26, 397–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geiger B., Bershadsky A., Pankov R., Yamada K. M. (2001) Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell. Biol. 2, 793–805 [DOI] [PubMed] [Google Scholar]
- 15. McKeown-Longo P. J., Mosher D. F. (1985) Interaction of the 70,000-molecular weight amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J. Cell Biol. 100, 364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quade B. J., McDonald J. A. (1988) Fibronectin's amino-terminal matrix assembly site is located within the 29-kDa amino-terminal domain containing five type I repeats. J. Biol. Chem. 263, 19602–19609 [PubMed] [Google Scholar]
- 17. Tomasini-Johansson B. R., Annis D. S., Mosher D. F. (2006) The N-terminal 70-kDa fragment of fibronectin binds to cell surface fibronectin assembly sites in the absence of intact fibronectin. Matrix Biol. 25, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. George E. L., Georges-Labouesse E. N., Patel-King R. S., Rayburn H., Hynes R. O. (1993) Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119, 1079–1091 [DOI] [PubMed] [Google Scholar]
- 19. To W. S., Midwood K. S. (2011) Plasma and cellular fibronectin: distinct and indendent functions during tissue repair. Fibrogenesis Tissue Repair 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henderson B., Nair S., Pallas J., Williams M. A. (2011) Fibronectin. A multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol. Rev. 35, 147–200 [DOI] [PubMed] [Google Scholar]
- 21. Lower S. K., Lamlertthon S., Casillas-Ituarte N. N., Lins R. D., Yongsunthon R., Taylor E. S., DiBartola A. C., Edmonson C., McIntyre L. M., Reller L. B., Que Y. A., Ros R., Lower B. H., Fowler V. G., Jr. (2011) Polymorphisms in fibronectin-binding protein A of Staphylococcus aureus are associated with infection of cardiovascular devices. Proc. Natl. Acad. Sci. U.S.A. 108, 18372–18377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz-Linek U., Höök M., Potts J. R. (2006) Fibronectin-binding proteins of Gram-positive cocci. Microbes Infect. 8, 2291–2298 [DOI] [PubMed] [Google Scholar]
- 23. Atkin K. E., Brentnall A. S., Harris G., Bingham R. J., Erat M. C., Millard C. J., Schwarz-Linek U., Staunton D., Vakonakis I., Campbell I. D., Potts J. R. (2010) The streptococcal binding site in the gelatin-binding domain of fibronectin is consistent with a nonlinear arrangement of modules. J. Biol. Chem. 285, 36977–36983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maurer L. M., Tomasini-Johansson B. R., Ma W., Annis D. S., Eickstaedt N. L., Ensenberger M. G., Satyshur K. A., Mosher D. F. (2010) Extended binding site on fibronectin for the functional upstream domain of protein F1 of Streptococcus pyogenes. J. Biol. Chem. 285, 41087–41099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwarz-Linek U., Pilka E. S., Pickford A. R., Kim J. H., Höök M., Campbell I. D., Potts J. R. (2004) High affinity streptococcal binding to human fibronectin requires specific recognition of sequential F1 modules. J. Biol. Chem. 279, 39017–39025 [DOI] [PubMed] [Google Scholar]
- 26. Bingham R. J., Rudiño-Piñera E., Meenan N. A., Schwarz-Linek U., Turkenburg J. P., Höök M., Garman E. F., Potts J. R. (2008) Crystal structures of fibronectin-binding sites from Staphylococcus aureus FnBPA in complex with fibronectin domains. Proc. Natl. Acad. Sci. U.S.A. 105, 12254–12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwarz-Linek U., Höök M., Potts J. R. (2004) The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol. Microbiol. 52, 631–641 [DOI] [PubMed] [Google Scholar]
- 28. Schwarz-Linek U., Werner J. M., Pickford A. R., Gurusiddappa S., Kim J. H., Pilka E. S., Briggs J. A., Gough T. S., Höök M., Campbell I. D., Potts J. R. (2003) Pathogenic bacteria attach to human fibronectin through a tandem β-zipper. Nature 423, 177–181 [DOI] [PubMed] [Google Scholar]
- 29. Tomasini-Johansson B. R., Kaufman N. R., Ensenberger M. G., Ozeri V., Hanski E., Mosher D. F. (2001) A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J. Biol. Chem. 276, 23430–23439 [DOI] [PubMed] [Google Scholar]
- 30. Ensenberger M. G., Annis D. S., Mosher D. F. (2004) Actions of the functional upstream domain of protein F1 of Streptococcus pyogenes on the conformation of fibronectin. Biophys. Chem. 112, 201–207 [DOI] [PubMed] [Google Scholar]
- 31. Mosher D. F., Johnson R. B. (1983) In vitro formation of disulfide-bonded fibronectin multimers. J. Biol. Chem. 258, 6595–6601 [PubMed] [Google Scholar]
- 32. Xu J., Bae E., Zhang Q., Annis D. S., Erickson H. P., Mosher D. F. (2009) Display of cell surface sites for fibronectin assembly is modulated by cell adherence to (1)F3 and C-terminal modules of fibronectin. PLoS One 4, e4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu J., Maurer L. M., Hoffmann B. R., Annis D. S., Mosher D. F. (2010) iso-DGR sequences do not mediate binding of fibronectin N-terminal modules to adherent fibronectin-null fibroblasts. J. Biol. Chem. 285, 8563–8571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marjenberg Z. R., Ellis I. R., Hagan R. M., Prabhakaran S., Höök M., Talay S. R., Potts J. R., Staunton D., Schwarz-Linek U. (2011) Cooperative binding and activation of fibronectin by a bacterial surface protein. J. Biol. Chem. 286, 1884–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Norris N. C., Bingham R. J., Harris G., Speakman A., Jones R. P., Leech A., Turkenburg J. P., Potts J. R. (2011) Structural and functional analysis of the tandem β-zipper interaction of a streptococcal protein with human fibronectin. J. Biol. Chem. 286, 38311–38320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Graille M., Pagano M., Rose T., Ravaux M. R., van Tilbeurgh H. (2010) Zinc induces structural reorganization of gelatin binding domain from human fibronectin and affects collagen binding. Structure 18, 710–718 [DOI] [PubMed] [Google Scholar]
- 37. Erat M. C., Schwarz-Linek U., Pickford A. R., Farndale R. W., Campbell I. D., Vakonakis I. (2010) Implications for collagen binding from the crystallographic structure of fibronectin 6FnI1–2FnII7FnI. J. Biol. Chem. 285, 33764–33770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwarzbauer J. E. (1991) Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 113, 1463–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sachchidanand, Lequin O., Staunton D., Mulloy B., Forster M. J., Yoshida K., Campbell I. D. (2002) Mapping the heparin-binding site on the 13–14F3 fragment of fibronectin. J. Biol. Chem. 277, 50629–50635 [DOI] [PubMed] [Google Scholar]
- 40. Ozeri V., Rosenshine I., Mosher D. F., Fässler R., Hanski E. (1998) Roles of integrins and fibronectin in the entry of Streptococcus pyogenes into cells via protein F1. Mol. Microbiol. 30, 625–637 [DOI] [PubMed] [Google Scholar]
- 41. Williams E. C., Janmey P. A., Ferry J. D., Mosher D. F. (1982) Conformational states of fibronectin. Effects of pH, ionic strength, and collagen binding. J. Biol. Chem. 257, 14973–14978 [PubMed] [Google Scholar]
- 42. Abe Y., Bui-Thanh N. A., Ballantyne C. M., Burns A. R. (2005) Extra domain A and type III connecting segment of fibronectin in assembly and cleavage. Biochem. Biophys. Res. Commun. 338, 1640–1647 [DOI] [PubMed] [Google Scholar]
- 43. Bazigou E., Xie S., Chen C., Weston A., Miura N., Sorokin L., Adams R., Muro A. F., Sheppard D., Makinen T. (2009) Integrin-α9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 17, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]