

Background: JNK1β1 has a role in diabetes and unique substrate interactions could affect function.
Results: JNK1β1 showed lower affinity and capacity to phosphorylate ATF2 than other variants. ATP binding and JNK1β1 activation affected substrate interaction rates and affinities.
Conclusion: JNK1β1 activation and ATP binding affects interactions with substrates.
Significance: First kinetic and biochemical characterization of a β JNK splice variant.
Keywords: Enzyme Kinetics, Jun N-terminal Kinase (JNK), Kinetics, MAP Kinases (MAPKs), Signal Transduction
Abstract
c-Jun N-terminal kinase (JNK) is a stress signal transducer linked to cell death, and survival. JNK1 has been implicated in obesity, glucose intolerance, and insulin resistance. In this study we report the kinetic mechanism for JNK1β1 with transcription factors ATF2 and c-Jun along with interaction kinetics for these substrates. JNK1β1 followed a random sequential mechanism forming a ternary complex between JNK-substrate-ATP. Km for ATF2 and c-Jun was 1.1 and 2.8 μm, respectively. Inhibition studies using adenosine 5′-(β,γ-methylenetriphosphate) and a peptide derived from JNK interacting protein 1 (JIP1) supported the proposed kinetic mechanism. Biolayer interferometry studies showed that unphosphorylated JNK1β1 bound to ATF2 with similar affinity as it did to c-Jun (KD = 2.60 ± 0.34 versus 1.00 ± 0.35 μm, respectively). The presence of ATP increased the affinity of unphosphorylated JNK1β1 for ATF2 and c-Jun, to 0.80 ± 0.04 versus 0.65 ± 0.07 μm, respectively. Phosphorylation of JNK1β1 decreased the affinity of the kinase for ATF2 to 11.0 ± 1.1 μm and for c-Jun to 17.0 ± 7.5 μm in the absence of ATP. The presence of ATP caused a shift in the KD of the active kinase for ATF2 to 1.70 ± 0.25 μm and for c-Jun of 3.50 ± 0.95 μm. These results are the first kinetic and biochemical characterization of JNK1β1 and uncover some of the differences in the enzymatic activity of JNK1β1 compared with other variants and suggest that ATP binding or JNK phosphorylation could induce changes in the interactions with substrates, activators, and regulatory proteins.
Introduction
c-Jun N-terminal kinase is one of the Ser/Thr kinases in the last tier of the mitogen-activated protein kinase (MAPK)2 cascade. It is activated by environmental and cellular stresses such as UV light (1, 2), γ-irradiation (3), osmotic shock, cytokines, and oxidative stress (4, 5). Phosphorylation of the Thr183 and Tyr185 residues of the Thr-Pro-Tyr motif in the activation loop of JNK activates this kinase triggering responses like cell death among others (6).
There are three genes that encode for JNK isoforms: jnk1 (2), jnk2 (7, 8), and jnk3 (9). These genes can undergo alternative splicing to produce 10 variants that differ in the subdomain IX (α and β) and the C-terminal end, which is shorter in variants 1 and have an additional 43 amino acids in variants 2. JNK1 and JNK2 can be spliced into four variants each, whereas JNK3 is spliced into two variants (10). Studies using knock-out or knockdown cells and organisms, as well as the use of JNK-specific inhibitors have suggested that JNK isoforms display both redundant and non-redundant functions. For example, phosphorylation of the most well known substrate of JNK, c-Jun, seems to be equally accomplished by all isoforms upon activation by certain stimuli (11); JNK1 is directly involved in insulin resistance by phosphorylating Ser307 of insulin receptor substrate 1 (IRS1) (12, 13) and is required for normal brain cytoarchitecture (14) and T cell receptor-mediated TH cell proliferation, apoptosis, and differentiation (15). JNK1, and not JNK2, is preferentially activated during TNFα-induced cell death (16). JNK2 is a negative regulator of cell proliferation, whereas JNK1 seems to be a positive regulator (17). Finally, JNK3 deficiency protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic cell loss (18), kainic acid-induced seizures (19), stroke (20), oxidative stress (21, 22), nerve growth factor deprivation (21), and β-amyloid-induced neuronal death (23).
Conversely, very little is known regarding the biochemical and functional differences of JNK splice variants, hence the need to address this question. First, Gupta et al. (10) qualitatively demonstrated differences in JNK variants capacity to bind and activate transcription factors c-Jun, ATF2, and Elk-1 using an immune complex kinase assay. Among JNK1 variants, the β forms (JNK1β1 and JNK1β2) showed the highest level of c-Jun and ATF2 binding. All JNK2 variants were capable of binding c-Jun and ATF2, but the −1 variants, JNK2α1, and JNK2β1, showed the highest c-Jun and ATF2 binding, respectively. α Forms of JNK2 were better at binding c-Jun, whereas β forms showed higher affinity for ATF2. Both JNK3 variants showed a comparable low capacity to bind the transcription factors assayed by the authors. RNA and protein analysis of various human cell lines has shown that the 54- and 46-kDa bands observed in Western blots, correspond mostly to JNK2 and JNK1, respectively (24). Interestingly, the most abundant splice variant of the JNK1 isoform is JNK1β1, whereas JNK2α2 is the most prevalent of the JNK2 splice variants (24). Lastly, Yang et al. (25) showed that higher expression levels of scaffold protein JIP1 in tissues and cell lines correlated with higher levels and increased stability toward degradation of the short (46 kDa) but not the long (54 kDa) variants of JNK. Moreover, using Western blots, these authors showed that JIP1 inducible HEK-293 cells had 4-fold higher protein expression levels of JNK1β1 than any other splice variant. Couple this with the finding that a missense mutation in JIP1 (S59N), which reduced its ability to inhibit JNK, segregated with type II diabetes mellitus in one French family (26), provide genetic and biochemical evidence that JNK1 and perhaps JNK1β1 plays a significant role in type II diabetes mellitus. Hence the relevance of an exhaustive biochemical characterization of JNK1β1, which would not only contribute to the understanding of the differences between splice variants; but may also facilitate the design of specific small molecule inhibitors for therapeutic purposes.
To date, only three of the 10 JNK splice variants have been biochemically characterized: JNK1α1 (27), JNK3α1 (28), and JNK2α2 (29). This study presents an in vitro characterization of the JNK1β1 kinetic mechanism and the kinetics of the interactions with substrates c-Jun and ATF2. Here we report the first exhaustive biochemical characterization of the most well known substrate of JNK, c-Jun. JNK1β1 phosphorylated ATF2 and c-Jun via the formation of a ternary complex with non-interacting binding sites for the protein substrate and ATP. JNK1β1 showed similar affinity for ATF2 and c-Jun. Both the phosphorylation state of the kinase and the presence of ATP significantly affected the kinetics of association and dissociation of JNK1β1 and its substrates, and therefore the KD for the interactions. Our results reveal some distinctive features of JNK1β1 and support the idea of different roles for JNK splice variants.
EXPERIMENTAL PROCEDURES
Reagents and Supplies
All reagents, chemical compounds, and materials used were purchased from Fisher Scientific, Sigma, Invitrogen, or Pierce, unless a different company is specified.
Protein Production and Purification
Expression and purification of Bioease-FLAG tag ATF2 construct (2–115, B-F-ATF2) has been previously reported (27, 28). cDNA of full-length human JNK1β1(1–384) (EC 2.7.11.24) was cloned into a modified pET19 vector using BamHI and EcoRI restriction enzymes. This plasmid was used to generate N-terminal His10-tagged full-length JNK1β1. The N-terminal portion of c-Jun(1–89) was cloned using the Gateway® technology into the vector pDEST544 developed by D. Esposito (Addgene). The latter plasmid allowed the bacterial expression of His6-tagged NusA c-Jun fusion protein (1–89, H-N-cJun). Another plasmid, pEL124–544, for expression of His6-tagged NusA (H-N) followed by the 8-amino acid attB1 peptide and a stop codon (a kind gift of Dr. Esposito) was used to produce a negative control substrate for JNK activity and binding assays. Escherichia coli strain BL21(DE3) was used for protein production in Luria broth and induction of expression was done during log phase with 0.25 mm isopropyl β-d-thiogalactopyranoside for 3–5 h at 30 °C. Cells were collected by centrifugation and lysed on ice with B-Per reagent containing lysozyme and benzonase (Novagen). Lysates were clarified by centrifugation at 20,000 × g for 60 min at 4 °C. Supernatants were filtered and loaded onto the 15-ml cobalt column HisPur. The column equilibration buffer used was 50 mm sodium phosphate, pH 8, 300 mm NaCl, 5 mm imidazole, and the elution buffer was 50 mm sodium phosphate, pH 8, 300 mm NaCl, 250 mm imidazole. Quality and purity of proteins was assessed by SDS-PAGE and Coomassie Blue stain; only samples with estimated purity higher than 90% were used. Collected proteins were concentrated in 25 mm HEPES, pH 7.4, 150 mm NaCl, 0.1 mm EGTA, 6.25% glycerol, 1 mm DTT (dithiothreitol), 0.03% Brij-35 buffer using Amicon Ultra-15 centrifugal filter units, aliquoted, and stored at −80 °C.
JNK1β1 Activation
Active MKK4 and MKK7 (Millipore) were used for in vitro activation of JNK1β1 following the method of Lisnock et al. (30) with slight modifications: the final concentration of JNK1β1 was 500 nm, whereas MKK7 was added at 100 nm 1 h prior to the addition of 1 nm MKK4. Total reaction incubation time was 2 h at 30 °C. Activation achieved ranged from 70 to 90% based on MS analysis.
JNK1β1 Kinetic Assays
JNK activity was determined as incorporation of 33P from [γ-33P]ATP into recombinant proteins B-F-ATF2 or H-N-cJun. Reactions were carried out in a volume of 120 μl containing 25 mm HEPES, pH 7.4, 10 mm MgCl2, 1 mm DTT, 20 mm β-glycerophosphate, 0.1 mm Na3VO4, 0.5 mg/ml of bovine serum albumin (BSA), 1.5 μCi of [33P]ATP (3000 Ci/mmol), 0.1–32 μm ATP, 0.05–4 μm recombinant substrate, and 2 nm active JNK1β1. Reactions were carried out at 30 °C in 96-well plates and stopped by adding an equal volume of 100 mm H3PO4 after 45–60 min. Fifty microliters of each reaction were transferred to pre-wet Immobilon-P Multiscreen HTS IP plates (Millipore) in triplicate or quadruplicate. Vacuum was applied to filter samples and the wells were washed exhaustively with 10 mm HEPES, pH 7.4, 100 mm NaCl, 25 mm EDTA. Finally, a wash with 50% ethanol was done to minimize drying time prior to the addition of 100 μl of Ultima Gold XR liquid scintillation counting mixture (PerkinElmer Life Sciences). Plates were read in a TopCount microplate scintillation counter (Packard Instrument Co.). Initial velocities of the reactions were fitted with two-substrate equations using GraFit version 5.0.13 (31) as published before (27, 28).
JNK1β1 Inhibition Assays
Inhibition of JNK1β1 was done with the non-hydrolyzable ATP analog, AMP-PCP (5–400 μm), and the JIP1 δ-domain peptide 153RPKRPTTLNLF163, JIP1 pep (32) (5–400 nm, NeoPeptide). These assays were performed keeping the concentration of one JNK substrate constant, whereas the concentration of the other substrate and the inhibitor were varied. When ATP was kept constant the concentration was 10–20 μm, 1 μm was chosen for ATF2 and H-N-cJun. Data were analyzed as previously published (27, 28).
c-Jun in Vitro Biotinylation
H-N-cJun (500 μg/ml) was biotinylated using the EZ-Link NHS-LC-LC-biotin in 1× PBS using a 5:1 molar ratio of biotin reagent:protein for 30 min at RT following the fortéBIO suggested protocol. Biotinylated H-N-cJun was separated from the biotinylation reaction reagents by D-salt dextran desalting columns. Biotin incorporation was confirmed by Western blot analysis of samples using IRDye 680 streptavidin (Licor) to visualize the labeled protein.
Biolayer Interferometry (BLI)
A fortéBio Octet Red instrument was used to study kinetics of JNK1β1 binding to its substrates B-F-ATF2 and H-N-cJun. All the assays were performed with agitation set to 1000 rpm in fortéBIO 10× kinetic buffer to minimize nonspecific interactions. To this assay buffer, 10 mm MgCl2 and 2 mm DTT were added to stabilize the JNK-substrate interactions. The final volume for all the solutions was 200 μl/well. Assays were performed at 30 °C in solid black 96-well plates (Geiger Bio-One). 5–25 μg/ml of biotinylated ligand (B-F-ATF2 or H-N-cJun) in 10× kinetic buffer was used to load the ligand on the surface of streptavidin biosensors (SA) for 200–300 s. Typical capture levels were between 0.5 and 3 nm and variability within a row of eight tips did not exceed 0.1 nm. A 500–800 s biosensor washing step was applied prior to the analysis of the association of the ligand on the biosensor to the analyte in solution (0.15–10 μm JNK1β1 unphosphorylated or phosphorylated) for 100–1000 s. Finally, the dissociation of the interaction was followed for 100–1000 s. Dissociation wells were used only once to ensure buffer potency. Correction of any systematic baseline drift was done by subtracting the shift recorded for a sensor loaded with ligand but incubated with no analyte. Data analysis and curve fitting were done using Octet software version 7.0. Experimental data were fitted with the binding equations available for 1:1 interaction, 2:1 heterogeneous ligand (HL), 1:2 bivalent analyte, and mass transport binding model (Fig. 1, a–d, respectively). Global analyses of the complete data sets assuming binding was reversible (full dissociation) were done using nonlinear least squares fitting. Hence, a single set of binding parameters was obtained simultaneously for all the analyte concentrations in every experiment. Additionally, steady-state kinetic analyses were done for every data set to calculate the KD using the estimated response at equilibrium for each analyte concentration rather than the kon and koff values. All the experiments were repeated at least twice and the results obtained were similar. The data reported here correspond to the results of one representative experiment.
FIGURE 1.

Reaction equations for protein-protein interaction binding models. a, 1:1 binding model. b, 2:1 heterogeneous ligand. c, 1:2 bivalent analyte. d, mass transport. Where L is ligand, A is analyte, LA is the ligand-analyte complex, kon is the association rate constant, koff is the dissociation rate constant, Abulk is the analyte molecules in solution, Asurface corresponds to the analyte molecules close to the surface of the biosensor.
Statistical Analyses
All the enzyme kinetic and inhibition data were evaluated using F test of the Grafit version 5.0.13 software to compare the goodness of the different fitting analyses, χ2 values were below two in all the experiments. Goodness of fit for the interferometry data were assessed by evaluation of the residual plots, the χ2 and R2 values generated for all the fitting analyses were done.
RESULTS
High Level of c-Jun Fusion Protein Expression in E. coli
To date, no detailed kinetic characterization of any JNK isoform with c-Jun has been published and this may be because of the lack of reports of high level expression and purification of c-Jun. To address this shortcoming, we attempted the expression and purification of numerous c-Jun constructs in E. coli. High levels of expression and increased solubility of c-Jun(1–89) was possible by the construction of the N-terminal His-tagged-NusA recombinant fusion protein (Fig. 2a). The yield of purified protein typically was near 10 mg/liter and >90% purity as assessed by SDS-PAGE (Fig. 2B). It was decided not to proteolytically cleave the NusA solubility tag of c-Jun based on the fact that in our hands, numerous attempts to express high levels of soluble c-Jun or truncations of this protein with no tags have only been partly successful due to low yields, the formation of insoluble aggregates, or protein sticking to membranes during concentration.
FIGURE 2.
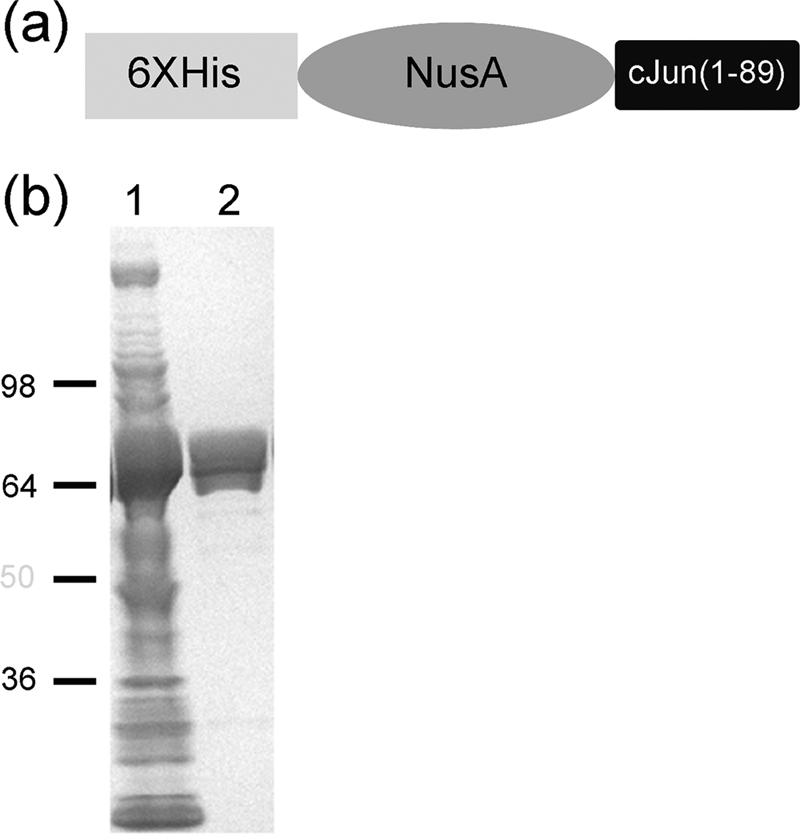
Expression and purification of H-N-cJun construct. a, schematic representation of the c-Jun(1–89) construct designed. b, Coomassie staining of SDS-PAGE, lane 1, soluble fraction of total cell lysate; lane 2, purified H-N-cJun(1–89).
Kinetic Analyses of JNK1β1 Activity
The kinetic mechanisms of JNK1β1 phosphorylation of B-F-ATF2 and H-N-cJun were assessed by varying the concentration of the recombinant substrates and ATP. Nonlinear regression analysis of the data using the two substrate equations presented in Ember et al. (27, 28) showed that the mechanism of phosphorylation of the substrates followed ternary complex formation with noninteracting substrate binding sites. This also suggested a sequential kinetic mechanism; i.e. all the substrates bind to the enzyme before products are formed. A representative example of the graphs showing the two-substrate profiles and double-reciprocal plots for B-F-ATF2 and H-N-cJun phosphorylation by JNK1β1 is given in Fig. 3, a–d. Table 1 presents the steady-state kinetic parameters for the substrate phosphorylation reactions studied.
FIGURE 3.

Two-substrate kinetic analysis of JNK1β1 activity. a, nonlinear least squares fitting (black lines) of the curves obtained for JNK1β1 phosphorylation of B-F-ATF2 two-substrate kinetic profile (symbols) using an equation for the ternary complex with noninteracting substrate binding sites. b, double-reciprocal plots and linear regression of the data shown in a. c, ternary complex with noninteracting binding site equation fitting to H-N-cJun phosphorylation data. d, double-reciprocal of data shown as in c and linear regression of the data. e, schematic representation of the two possible kinetic mechanisms for JNK1β1: random sequential Bi Bi (left) and random Bi ordered Bi (right).
TABLE 1.
Kinetic constants for active JNK1β1 from two-substrate kinetics
Results are the average of at least three independent experiments done in quadruplicate. Errors reported correspond to the mean ± S.E.
| Substrate | Km | kcata | kcat/Km |
|---|---|---|---|
| μm | min−1 | μm−1min−1 | |
| B-F-ATF2 | 1.1 ± 0.4 | 2.2 ± 0.7 | 3.2 ± 1.3 |
| ATP | 6.5 ± 1.3 | ||
| H-N-cJun | 2.8 ± 0.9 | 16.8 ± 5.1 | 6.2 ± 2.1 |
| ATP | 4.5 ± 0.1 |
a Enzyme catalytic constant was calculated using the Vmax determined from the two-substrate profile and the concentration of the enzyme used in the assay.
To determine whether the kinetic mechanism was ordered or random with respect to the substrates, the inhibition of B-F-ATF2 and H-N-cJun phosphorylation was studied using the ATP competitive inhibitor AMP-PCP and an 11-mer peptide consisting of the JIP1 δ-domain (32). Table 2 presents the inhibition parameters determined for the reactions studied and supplemental Figs. S1 and S2 show representative data for all the inhibition assays. As expected, AMP-PCP was a competitive inhibitor against ATP and a pure noncompetitive inhibitor against B-F-ATF2 and H-N-cJun. On the other hand, we found that JIP1 pep was a competitive inhibitor for B-F-ATF2 and H-N-cJun, and a pure noncompetitive inhibitor against ATP.
TABLE 2.
Inhibition constants and mechanisms of inhibition for JNK1β1
Results are the average of at least three separate experiments done in quadruplicate ± S.E.
| Inhibitor | Variable substrate | Type of inhibition | Ki |
|---|---|---|---|
| μm | |||
| AMP-PCP | ATP | Competitive | 29.2 ± 3.1 |
| B-F-ATF2 | Noncompetitive | 115.6 ± 66.8 | |
| JIP1 pep | ATP | Noncompetitive | 0.492 ± 0.196 |
| B-F-ATF2 | Competitive | 0.078 ± 0.015 | |
| AMP-PCP | ATP | Competitive | 91.5 ± 46.8 |
| H-N-cJun | Noncompetitive | 487.8 ± 129.1 | |
| JIP1 pep | ATP | Noncompetitive | 0.318 ± 0.074 |
| H-N-cJun | Competitive | 0.173 ± 0.039 |
Best fits for the experimental data were assessed by comparison of the mean ± S.E., nonlinear regression analysis, and F-tests for competitive, pure noncompetitive, mixed noncompetitive, and uncompetitive inhibition fits. The inhibition experiment results support the idea of a kinetic mechanism with random sequential addition of substrates and either random or ordered release of products (Fig. 3E). Studies of the enzymatic reaction in the reverse direction are not feasible for JNK and comprehensive product inhibition analysis could only be carried out if phosphorylated ATF2 and c-Jun were available. Nonetheless, we performed a partial characterization of product inhibition kinetics by analyzing the effects of ADP on the phosphorylation of B-F-ATF2 at constant or variable concentrations of ATP and we observed that ADP was competitive versus ATP as expected and had minimal inhibition of B-F-ATF2 also as expected suggesting that the release of the products could be either random or ordered.
To ensure that phosphorylation of the H-N-cJun construct was occurring in the c-Jun portion of the fusion protein, we performed a Western blot analysis of H-N-cJun samples treated with active JNK1β1 in the presence of ATP using anti-c-Jun phospho-Ser73 antibody (Cell Signaling). A band for phosphorylated c-Jun was only observed in those samples containing the protein substrate, active JNK1β1 and ATP (supplemental Fig. S3a). Additionally, we showed that His-tagged NusA (H-N), a construct lacking c-Jun(1–89), does not behave as a substrate for JNK1β1 in enzymatic assays (supplemental Fig. S3b). These experiments allowed us to confirm that H-N-cJun was effectively used as a substrate by JNK1β1. Interestingly, activity assays carried out with ATF2 or c-Jun concentrations above 2 μm showed a partial inhibition of JNK1β1 activity; consequently, it was not possible to obtain good quality data using substrate concentrations higher than 2 μm.
Rates of Interactions between Substrates and Active or Inactive JNK1β1
Kinetic analyses of the interactions between JNK1β1 and B-F-ATF2 or H-N-cJun were accomplished using BLI. The objective of these studies was to determine the impact of the ATP presence and the phosphorylation state of the kinase on the association/dissociation rate constants and the equilibrium dissociation constants of JNK1β1 interaction with B-F-ATF2 or H-N-cJun. All the binding curves in Figs. 4 and 5 showed initial very short and fast association/dissociation steps, followed by slower and much longer association/dissociation steps. Fig. 4 shows representative data for both association and dissociation phases of the curves obtained for JNK1β1 binding to immobilized B-F-ATF2 in the absence and presence of ATP for both the inactive and the active forms of the kinase. Similarly, Fig. 5 shows the same type of data for the interaction of JNK1β1 and immobilized H-N-cJun (experimental data are represented by black lines and curve fitting by gray lines).
FIGURE 4.

Interferometry studies of the interaction between immobilized B-F-ATF2 and JNK1β1: comparison of the effects of ATP and JNK1β1 phosphorylation state on the interaction. (i) experimental data for association and dissociation are represented by black and gray lines that show nonlinear least squares fitting. Concentrations of analyte (JNK1β1) are shown to the right side of each binding curve. (ii) secondary plots: steady-state analysis of the binding data to determine equilibrium dissociation constants (response at equilibrium plotted against analyte concentration). a, inactive (unphosphorylated) JNK1β1 interaction with B-F-ATF2. Steady-state analysis generated a KD = 2.60 ± 0.34 μm. b, inactive (unphosphorylated) JNK1β1 binding to immobilized B-F-ATF2 in the presence of 200 μm ATP-Mg2+. Steady-state analysis produced a KD = 0.80 ± 0.04 μm. c, active (phosphorylated) JNK1β1 interaction with B-F-ATF2. Steady-state analysis showed a KD = 11.0 ± 1.1 μm. d, active (phosphorylated) JNK1β1 interaction with immobilized B-F-ATF2 in the presence of 200 μm ATP-Mg2+. KD value was 1.70 ± 0.25 μm by steady-state analysis.
FIGURE 5.

Kinetics of the interaction between immobilized H-N-cJun and JNK1β1: comparison of the effects of ATP and JNK1β1 phosphorylation state on the interaction. (i) experimental data for association and dissociation are represented by black lines and nonlinear least squares fitting by gray lines. Concentrations of analyte are indicated on the right side of each binding curve. (ii) secondary plots: response at equilibrium against analyte concentration for steady-state analysis of the data to determine equilibrium dissociation constants. a, inactive (unphosphorylated) JNK1β1 and H-N-cJun interaction. Steady-state KD was 1.00 ± 0.35 μm. b, inactive (unphosphorylated) JNK1β1 and H-N-cJun interaction in the presence of 200 μm ATP-Mg2+. KD value obtained from steady-state analysis was 0.65 ± 0.07 μm. c, active (phosphorylated) JNK1β1 and H-N-cJun interaction. KD was 17.0 ± 7.5 μm, and d, active (phosphorylated) JNK1β1 and H-N-cJun interaction in the presence of 200 μm ATP-Mg2+. KD = 3.5 ± 0.95 μm.
Comparison of the interaction of B-F-ATF2 or H-N-cJun with inactive (unphosphorylated) JNK1β1 in the absence (Figs. 4a(i) and 5a(i)) and the presence of 200 μm ATP (Figs. 4b(i) and 5b(i)) showed that the association and dissociation were much more rapid in the presence of ATP and the interaction came to equilibrium much more quickly than in the absence of ATP. Phosphorylation of JNK1β1 seemed to have a similar effect to the presence of ATP based on the kinetics of the interaction between active (phosphorylated) JNK1β1 and B-F-ATF2 or H-N-cJun (Figs. 4c(i) and 5c(i)) as compared with the inactive (unphosphorylated) kinase (Figs. 4a(i) and 5a(i)). ATP-dependent effects were also observed in the binding of active JNK1β1 and B-F-ATF2 (Fig. 4d(i)) as compared with the binding of the same ligand-analyte pair in the absence of ATP (Fig. 4c(i)). On the other hand, the presence of ATP seemed to have little effect on the kinetics of the interaction between active JNK1β1 and H-N-cJun (Fig. 5, c(i) and d(i)). Interestingly, the signal intensity during the dissociation phase of the interaction of phosphorylated JNK1β1 and immobilized B-F-ATF2 or H-N-cJun approached baseline much more rapidly than in the case of the unphosphorylated kinase indicating an almost complete or full dissociation of the active kinase from the substrate during the experiment (compare Figs. 4a(i) and 5a(i) with 4c(i) and 5c(i)).
Nonlinear least squares fitting of the experimental data generated best fits using a 2:1 HL model, which assumes the existence of two independent ligand binding sites (Fig. 1B), hence the two values shown in Tables 3 and 4 for each kinetic parameter describing the interactions studied. Residual plots derived from curve fitting in Figs. 4 and 5, showed, in general, very small (less than 10% of the response) and random residuals, supporting the use of the 2:1 HL fitting model for the data (supplemental Figs. S4 and S5). Only a minor deviation from the experimental data were observed for the initial fast step of both the association and dissociation phases; however, R2 values were above 0.9 and χ2 values below 2.0 for all the fits.
TABLE 3.
Kinetics of interaction between inactive and active JNK1β1 with B-F-ATF2 in the absence or presence of ATP
Reported values are representative of a single experiment ± S.E. Similar results were obtained in replicate experiments.
| Ligand | Analyte JNK1β1 |
Rate constants |
KD | ||
|---|---|---|---|---|---|
| Inactive | Active | kon | koff | ||
| m−1s−1 | s−1 | μm | |||
| B-F-ATF2 | * | 5.85 ± 0.02 × 102a | 6.56 ± 0.02 × 10−4b | 1.12c | |
| 1.94 ± 0.03 × 104d | 5.29 ± 0.05 × 10−3e | 0.26f | |||
| 2.60 ± 0.34g | |||||
| B-F-ATF2 | * | 5.36 ± 0.11 × 102a | 2.5 ± 0.01 × 10−3b | 4.32c | |
| 2.19 ± 0.09 × 104d | 9.45 ± 0.12 × 10−2e | 4.66f | |||
| 11.0 ± 1.1g | |||||
| B-F-ATF2, ATP | * | 3.21 ± 0.03 × 102a | 7.05 ± 0.04 × 10−4b | 3.27c | |
| 2.38 ± 0.04 × 104d | 5.23 ± 0.05 × 10−2e | 2.42f | |||
| 0.80 ± 0.04g | |||||
| B-F-ATF2, ATP | * | 7.28 ± 0.16 × 103a | 3.54 ± 0.04 × 10−3b | 0.63c | |
| 1.39 ± 0.06 × 105d | 8.72 ± 0.15 × 10−2e | 0.49f | |||
| 1.70 ± 0.25g | |||||
a kon1, derived from the curve fitting of data with the 2:1 HL model.
b koff1, derived from the curve fitting of data with the 2:1 HL model.
c KD1, derived from the curve fitting of data with the 2:1 HL model.
d kon2, derived from the curve fitting of data with the 2:1 HL model.
e koff2, derived from the curve fitting of data with the 2:1 HL model.
f KD2, derived from the curve fitting of data with the 2:1 HL model.
g KD obtained from steady state analysis of the secondary plot Response at equilibrium versus concentration of analyte.
TABLE 4.
Kinetics of interaction between inactive and active JNK1β1 with H-N-cJun in the absence or presence of ATP
Reported values are representative of a single experiment ± S.E. Similar results were obtained in replicate experiments.
| Ligand | Analyte JNK1β1 |
Rate constants |
KD | ||
|---|---|---|---|---|---|
| Inactive | Active | kon | koff | ||
| m−1s−1 | s−1 | μm | |||
| H-N-cJun | * | 4.24 ± 0.22 × 102a | 3.27 ± 0.13 × 10−4b | 0.77c | |
| 1.10 ± 0.06 × 103d | 5.77 ± 0.32 × 10−4e | 0.52f | |||
| 1.00 ± 0.35g | |||||
| H-N-cJun | * | 8.38 ± 0.84 × 102a | 1.65 ± 0.01 × 10−3b | 4.80c | |
| 4.53 ± 0.13 × 104d | 2.17 ± 0.01 × 10−1e | 1.97f | |||
| 17.00 ± 7.50g | |||||
| H-N-cJun, ATP | * | 4.39 ± 0.01 × 103a | 7.20 ± 0.02 × 10−4b | 0.15c | |
| 1.86 ± 0.04 × 104d | 5.65 ± 0.07 × 10−3e | 0.30f | |||
| 0.65 ± 0.07g | |||||
| H-N-cJun, ATP | * | 2.00 ± 1.64 × 103a | 2.19 ± 0.07 × 10−3b | 1.27c | |
| 7.04 ± 1.05 × 104d | 8.95 ± 0.58 × 10−2e | 1.10f | |||
| 3.50 ± 0.95g | |||||
a kon1, derived from the curve fitting of data with the 2:1 HL model.
b koff1, derived from the curve fitting of data with the 2:1 HL model.
c KD1, derived from the curve fitting of data with the 2:1 HL model.
d kon2, derived from the curve fitting of data with the 2:1 HL model.
e koff2, derived from the curve fitting of data with the 2:1 HL model.
f KD2, derived from the curve fitting of data with the 2:1 HL model.
g KD obtained from steady state analysis of the secondary plot Response at equilibrium versus concentration of analyte.
Additionally, equilibrium dissociation constants were obtained from the steady-state analyses (response at equilibrium plotted as a function of analyte concentration) presented in Figs. 4, a(ii)–d(ii), and 5, a(ii)–d(ii). A single steady-state equilibrium dissociation constant, KD, is shown on Tables 3 and 4 for every JNK1β1 and B-F-ATF2 or H-N-cJun interaction studied. It is interesting to note that the steady-state KD for B-F-ATF2 was 4.2-fold higher for active JNK1β1 as compared with inactive JNK1β1 in the absence of ATP, whereas the steady-state KD for the active enzyme plus ATP was 6.5-fold lower than active JNK1β1 without ATP (Table 3). Showing a similar trend, the affinity for H-N-cJun was 17-fold lower for active JNK1β1 than inactive JNK1β1 in the absence of ATP, whereas the steady-state KD value for active JNK1β1 plus ATP was 4.8-fold lower than active JNK1β1 without ATP (Table 4).
To investigate if JNK1β1 bound specifically to the c-Jun portion of the fusion protein H-N-cJun, we biotinylated and immobilized His6-tagged NusA, lacking the c-Jun(1–89) fragment and analyzed the binding of inactive JNK1β1 (supplemental Fig. S3c). No binding was observed for His6-tagged NusA and inactive JNK1β1 up to 2.5 μm. Finally, because the active JNK1β1 used in these binding assays was prepared by treatment with active MKK4 and MKK7 and the two upstream activators were not separated from the reaction mixture prior to the binding studies, a control experiment using the same activation reaction conditions but in the absence of JNK1β1 was done. Serial dilutions of the active MKK4 and MKK7 solution showed no significant interaction with immobilized B-F-ATF2 (data not shown), indicating that the observed alterations of the binding curves upon activation of JNK1β1 are likely due to the phosphorylation state of the kinase and not to the presence of the upstream activators.
DISCUSSION
Enzyme Kinetics Characterization of JNK1β1
Both in vitro and in vivo studies have shown that JNK1 is associated with insulin resistance (12, 13), normal brain cytoarchitecture (14), TNFα-induced cell death (16), and T cell receptor-mediated TH cell proliferation, apoptosis, and differentiation (15). An exhaustive kinetic analysis of the substrate preference and activity of JNK1 would undoubtedly contribute to the understanding of the differences between JNK variants and contribute to the design of specific isoform-selective inhibitors of JNK, which ultimately is one of our goals.
To date, the only JNK variants that have been enzymatically characterized are JNK1α1 (27), JNK2α2 (29), and JNK3α1 (28). Here we present the first study of a β variant of JNK, which could contribute to establish differences and similarities between variants and the role of the different subdomains of the protein. The initial velocity studies reported here suggested a random sequential kinetic mechanism for JNK1β1, forming a ternary complex with its substrates, i.e. JNK1β1-ATF2-ATP or JNK1β1-cJun-ATP. This was deduced from the analysis of the converging lines obtained for the double-reciprocal Lineweaver-Burk plots of the two-substrate profiles (Fig. 3, b and d), which corresponded to a ternary and sequential rather than a ping-pong mechanism that is characterized by a plot with parallel lines (27, 33). Additionally, the values of Km and Kia were virtually identical for the reactions done varying either the concentration of ATP or the concentration of protein substrate (B-F-ATF2 or H-N-cJun). These results indicated similar affinities of JNK1β1 for one substrate when the second substrate was bound to its corresponding binding site or when it was absent (27, 33). Based on these results, it is possible to suggest that JNK1β1 has noninteracting substrate binding sites, which is in agreement with previously published data for JNK1α1 (27), JNK3α1 (28), and JNK2α2 (29).
We report here a Km value of 1.1 ± 0.4 μm for B-F-ATF2, which is about 4.6-fold higher than the value reported for JNK1α1 (0.24 ± 0.01 μm) (27), almost identical to the value reported for JNK2α2 (0.8 ± 0.3 μm) (29) and ∼6.9-fold higher than the Km value published for JNK3α1 (0.16 ± 0.01 μm) (28). Because Km roughly reflects the affinity of an enzyme for its substrate under certain conditions (33), it can be suggested that JNK1β1 binds slightly less tightly to ATF2 than JNK1α1 and JNK3α1, whereas it practically matches JNK2α2 affinity for the substrate. To date, only one report has been published comparing all JNK splice variant binding capacities to the transcription factors c-Jun, ATF2, and Elk-1 (10). Based on the results of an in vitro assay where 35S-labeled JNK binding to immobilized GST-tagged truncations of ATF2, c-Jun, or Elk-1 was estimated by autoradiography, the authors suggested that among JNK1α1, JNK1β1, JNK2α2, and JNK3α1, the highest ATF2 binding capacity corresponds to JNK2α2, closely followed by JNK1β1, whereas JNK3α1 and JNK1α1 showed the lowest binding to the substrate. Differences in the experimental procedures employed here and in the report in question could account for the divergence in the binding capacity for the different JNK splice variants, especially because the quantitation from Gupta et al. (10) was done from estimation of signal intensity in autoradiographs.
Furthermore, the kcat for the B-F-ATF2 phosphorylation reaction by JNK1β1 was 2.2 ± 0.7 min−1 (Table 1); which is slightly lower than the reported values for other JNK splice variants. JNK1β1 kcat was approximately half of what that parameter is for JNK3α1, 5.5 min−1 (28), around 3.5-fold lower than JNK1α1 (7.7 min−1) (27), and more than 13-fold lower than JNK2α2 (30.6 min−1) (29). This suggests that JNK1β1 has a slightly lower capacity to convert the substrate ATF2 into product phospho-ATF2 as compared with the other splice variants characterized so far (27, 28). Another possibility for the differences in kcat and Km for JNK1β1 compared with the other splice variants could be heterogeneous activation (i.e. less than 100% activation of our sample) in this study, or for the splice variants in the other studies. Lisnock et al. (30) showed that mono- and bisphosphorylated JNK3α1 had nearly identical activities so this possibility appears unlikely but cannot be strictly ruled out.
On the other hand, we report here a Km value for H-N-cJun(1–89) of 2.8 ± 0.9 μm (Table 1), which is similar to the value estimated by Kallunki et al. (8) based on immune complex kinase assays (2.5 ± 0.5 μm) and by Arthur et al. (34) who reported a Km value of 3.8 ± 0.6 μm for the GST-c-Jun (amino acids 1–135) phosphorylation reaction by JNK1 (no splice variant specified) by estimating the incorporation of 32P into GST-c-Jun upon SDS-PAGE separation and autoradiograph quantitation.
Although the kcat value obtained for H-N-cJun (16.8 ± 5.1 min−1) was within the range of the kcat values reported for the alternative JNK substrate ATF2, it is important to point out that the JNK1β1 turnover for c-Jun seemed to be about 7.5-fold higher than for ATF2. Additionally, a comparison of the catalytic efficiencies (kcat/Km) of JNK1β1 for B-F-ATF2 and H-N-cJun presented in Table 1 (3.2 ± 1.3 and 6.2 ± 2.1 μm−1 min−1, respectively) suggested no significant preference of the kinase for one substrate over the other. This corresponds to the first comprehensive characterization of the c-Jun kinetic mechanism for any splice variant of JNK.
Interestingly, if the concentration of the protein substrate ATF2 or c-Jun used in the kinetic assays exceeded 2 μm, JNK1β1 activity was partly inhibited. This does not seem to be the case for the other JNK splice variants studied thus far. We believe this could be due to either substrate (ATF2 or c-Jun) or product (phospho-ATF2 or phospho-c-Jun) inhibition. The latter possibility seems to be less likely because during the assays no more than 15% of substrate was converted to product, therefore presumably not enough product was generated to inhibit the enzyme activity. Because the inhibition in question was observed for both ATF2 and c-Jun, it did not seem to depend on the nature of the fusion protein used as substrate. It is possible that the nonproductive complexes formed at high substrate concentrations represent a splice variant-specific mechanism of enzyme activity regulation in vivo at physiological concentrations of the substrate. We are currently exploring this hypothesis.
Inhibition of JNK1β1 by ATP Analog and δ Domain of JIP1
To confirm the nature of the kinetic mechanism followed by JNK1β1 during the catalytic phosphorylation of substrates B-F-ATF2 and H-N-cJun, inhibition studies were conducted with AMP-PCP and JIP1 pep. Table 2 shows the kinetic parameters obtained for the inhibition of JNK1β1 substrate phosphorylation by AMP-PCP or JIP1 pep. The observed modes of inhibition are consistent with the random and sequential mechanism for the substrates, which bind to noninteracting binding sites (27, 33). Moreover, this kinetic mechanism coincides with the mechanisms reported by Ember et al. (27, 28), and Niu et al. (29) for JNK1α1, JNK3α1, and JNK2α2.
Interestingly, the inhibition of JNK1β1 activity by JIP1 pep when the concentration of ATP was varied and the concentration of protein substrate was held constant showed a pure noncompetitive inhibition mechanism rather than a mixed noncompetitive mechanism, which was the case for JNK1α1 and JNK3α1 (27, 28). Although the experimental data fitting to the equation for mixed noncompetitive inhibition was almost as good as the fitting done using the equation for pure noncompetitive inhibition; the similarity of the values obtained for Kis and Kii in the mixed noncompetitive fit (between 0.5- and 2-fold difference) did not allow us to justify the additional parameter included in the analysis of the inhibition.
JNK1β1 activity was inhibited by AMP-PCP in a manner similar to JNK1α1 and JNK3α1 (27, 28) based on the less than 2.5-fold difference between the Ki values for the JNK variants mentioned. A comparison of the Ki values for the inhibition of B-F-ATF2 (78 ± 15 nm) and H-N-cJun phosphorylation (173 ± 39 nm) by JIP1 pep (Table 2) suggested a similar ability of the peptide to block the phosphorylation of both substrates (only 2.2-fold difference in Ki values). Similar Ki values have been reported for the inhibition of B-F-ATF2 phosphorylation by JIP1 pep in the case of JNK1α1 (55 ± 4 nm) and JNK3α1 (25 ± 6 nm) (27, 28). On the other hand, Niu et al. (29) reported that JIP1 pep inhibited JNK2α2 phosphorylation of GST-ATF2 (amino acids 19–96) with a Ki of 1.1 μm, which could reflect kinetic differences in the substrate phosphorylation due to the nature of the fusion protein used in their studies.
Taken together these results suggest that the kinetic mechanism for the phosphorylation of ATF2 is the same for all the splice variants of JNK characterized so far. On the other hand, the kinetic parameters (Km and kcat) did show differences for JNK1β1 when compared with other variants. These differences could be due to the variability in primary structure (98% sequence identity for JNK1α1 and JNK1β1, 91% identity for JNK3α1 and JNK1β1, and 85% identity for JNK2α2 and JNK1β1). It is tempting to speculate that the differences in kinetic parameters are due to subtle but unique protein-protein interactions between JNK variants and substrates perhaps associated with features of JNK subdomains like the α and β segments of the splice variants.
The Kinetics of JNK1β1 Interaction with B-F-ATF2 or H-N-cJun Are Affected by ATP Presence or Phosphorylation of Kinase
BLI is a methodology that can be employed to study JNK interaction with substrates, activators, and scaffolding proteins; as long as the affinity of the interaction is within the detection limits of the instrument (KD 10−3–10−12 m for the Octet RED instrument used here) and the size of the analyte is large enough (>250 Da) to be detected upon binding to the immobilized ligand. To date, we have employed this technology to study the interactions of JNK1β1, JNK3α1, JNK1β2, and JNK1α1 with B-F-ATF2 and JNK3α1 with c-Jun.3
The real-time binding data shown in Figs. 4 and 5 for JNK1β1/B-F-ATF2 and JNK1β1/H-N-cJun, respectively, suggested that both association and dissociation processes were described by two steps: an initial quick event followed by a slower and much longer event. Both the presence of ATP and the phosphorylation of JNK1β1 seemed to increase the biphasic character of the association and dissociation phases of the binding curves, and as a consequence equilibrium was reached faster.
Inactive JNK1β1 bound to B-F-ATF2 with similar affinity to H-N-cJun (Tables 3 and 4). ATP caused a slight decrease in the KD of inactive JNK1β1 for B-F-ATF2 and H-N-cJun, whereas phosphorylation of the kinase increased the KD for both substrates. Finally, in the presence of ATP, active JNK1β1 showed higher affinity for B-F-ATF2 and H-N-cJun when compared with the active kinase in the absence of ATP (steady-state KD values in Tables 3 and 4). The lower affinity observed for the interaction of active JNK1β1 and B-F-ATF2 or H-N-cJun compared with the interaction of inactive JNK1β1 seemed to be mainly driven by an increase in the dissociation rate constants. Conversely, ATP-induced changes in KD were mainly driven by faster on rates (Tables 3 and 4).
Binding curve fitting was done using the 2:1 HL equations, which assume that there are at least two populations of immobilized ligand that differ in their ability to bind to the analyte and therefore the binding curves are described by two reactions with different rates. Based on the structural information and binding analysis done by other authors for several JNK variants (35–42), we expected Langmuirian kinetics with 1:1 stoichiometry for the studied interactions. Some of the factors that can cause deviations from pseudo-first order approximation of binding data include: mass transfer effects, immobilized ligand density, inhomogeneity of immobilized ligand or soluble analyte, immobilization chemistry, and rebinding of dissociated analyte (43).
To eliminate some of the possibilities that cause non-Langmuirian kinetics we attempted the following: 1) fitting data with equations for mass transfer; 2) immobilization of ligand at low density (between 0.5 and 3 nm signal shift during ligand loading step); 3) use of 10× kinetic buffer to minimize nonspecific interactions; 4) analysis of the interactions in reverse orientation; and 5) use of a “sink” (125 nm JIP1 pep in the dissociation solution) to prevent rebinding of dissociated JNK1β1 to the immobilized substrate and to achieve reliable kinetic data (44). None of these changes altered significantly the shape of the binding curves and more importantly, did not drastically improve the data fitting process with binding models different from the 2:1 HL model.
Hence, we believe that the observed mode of interaction between JNK1β1 and its substrates is both a combination of the actual kinetics of the protein-protein interaction in vitro and a certain degree of ligand heterogeneity due to the biotinylation process, in particular for H-N-cJun, which was biotinylated in an in vitro reaction that attaches biotin randomly to solvent-exposed primary amines (45, 46). The 2:1 binding model for the interaction of JNK1β1 and its substrates does not seem to be a unique feature of this particular splice variant based on the fact that we have seen similar behaviors for JNK1α1, JNK1β2, and JNK3α1 by BLI analysis.3
It is possible that both active and inactive JNK1β1 have the ability to form two different complexes with each substrate: a productive complex by binding an accessible or properly oriented ligand immobilized on the surface of the biosensor likely represented by kon2 and a nonproductive complex perhaps reflective of poorly oriented or less accessible substrate represented by kon1 (Fig. 1b and Tables 3 and 4). These complexes form both in the presence and absence of ATP. Based on our data we cannot completely rule out the possibility that substrate immobilization on the biosensor could have caused restricted conformational freedom for the substrate, steric hindrance, and/or limited number of appropriate substrate orientations for JNK1β1 binding (47), therefore giving rise to the complex interaction kinetics observed in our studies.
Because we expected the kon2 values shown in Tables 3 and 4 to more accurately represent the kinetics of JNK-substrate interaction, a comparison of this parameter for the active enzyme in the presence of ATP with the kcat/Km values from Table 1 would allow us to determine the validity of our kinetic analysis. For a given enzymatic reaction, kon cannot be greater than kcat/Km (48) because the reaction rate can only be as fast as the rate of substrate-enzyme association. In the present analysis, kcat/Km for B-F-ATF2 was ∼2.5-fold higher than the values of kon2 for the interaction of active JNK1β1 with B-F-ATF2 in the presence of ATP. This presents only a minor discrepancy based on what was stated before and we believe the observed differences could be due to the inherent differences in the technologies used to obtain the kinetics for the reaction and for the protein-protein interaction. Additionally, the kcat/Km is an apparent second-order rate constant defined by a complex equation that includes the microscopic rate constants in the reaction equation prior to an irreversible step (k1, k−1, k2, etc.). On the other hand, kon only represents the substrate-kinase binding step of the reaction. Therefore, the complexity of the studied interactions could contribute to the mentioned discrepancy.
Ngoei et al. (49) recently published an analysis of the interaction between JNK1α1 and JIP1 pep using surface plasmon resonance technology. The authors presented binding curves for inactive and active JNK1α1 interacting with JIP1 pep. Their data showed a remarkable similarity to the binding curves shown here. However, they suggested that the phosphorylation of JNK1α1 significantly increased its affinity for JIP1 pep. The differences in KD shifts upon JNK activation between our data and Ngoei et al. (49) could reflect: 1) distinct mechanisms of interaction between JNK and its substrates as compared with the interaction between the kinase and the δ-domain peptide of the scaffold protein JIP1; or 2) an inappropriate representation of the JNK-JIP1 interaction due to use of the small peptide rather than the full-length scaffold protein to study the kinetics of the interaction. Nonetheless, it is not unreasonable to expect a shift in the binding capabilities of an inactive versus an active enzyme. These changes could be caused by conformational alterations that allow JNK1β1 in the presence of ATP or phosphorylated JNK1β1 to bind differently to ATF2 and c-Jun altering its affinity for those ligands. Changes in affinity for interacting proteins and increase in enzymatic activity upon phosphorylation (activation) have been reported for other MAP kinases like ERK2 (reviewed by Rubinfeld and Seger (50)) and by Burkhard et al. (51) who demonstrated by surface plasmon resonance that phosphorylation of ERK2 affected interactions with Elk-1 and stathmin but not interactions with c-Fos and RSK-1.
Interestingly, activation of JNK1β1 seemed to be coupled to the almost complete dissociation of the kinase from the ligand (Figs. 4, c(i) and d(i), and 5, c(i) and d(i)). This observation suggests that the stability of the active kinase-substrate complex was lower than the stability of the complex between the inactive kinase and the substrates. This could be a regulatory mechanism to control the activity of the active enzyme in vivo. Additionally, in the presence of ATP, active JNK1β1 should be able to catalyze the phosphorylation of the immobilized substrate and once that reaction has occurred, rapid dissociation of the kinase from the substrate can be expected.
It is possible that the changes in the ATF2 and c-Jun KD values observed by BLI in the absence and presence of ATP were not large enough to cause a shift from hyperbolic to sigmoidal enzyme kinetics for JNK1β1, implying that the kinase is not an allosteric enzyme. However, it is interesting to speculate that changes within JNK could occur upon phosphorylation or ATP binding altering the dynamics of the protein. These ideas are currently being investigated in our laboratory by nuclear magnetic resonance of JNK in the presence and absence of ligands.
In summary, we were able to show that: 1) JNK1β1 followed a random and sequential enzymatic mechanism with noninteracting substrate binding sites; 2) JNK1β1 showed a slightly lower Km and kcat for ATF2 than other JNK splice variants characterized so far; 3) a higher turnover number for c-Jun when compared with ATF2; 4) no substrate preference between c-Jun and ATF2; and 5) inhibition of ATF2 phosphorylation activity by AMP-PCP and JIP1 pep remarkably similar to JNK1α1 and JNK3α1. We were also able to show that B-F-ATF2 and H-N-cJun had affinities similar to each other when binding to either inactive or active JNK1β1. Additionally, we were able to demonstrate that both activation of JNK1β1 and ATP presence induced alterations of the kinetics of the interaction with substrates ATF2 and c-Jun. Taken together these findings suggest that JNK1β1 in vitro enzymatic activity toward ATF2 was distinguishable from other splice variants studied so far and that inhibition studies for ATF2 phosphorylation by a small molecule nonhydrolyzable ATP analog (AMP-PCP) or by the substrate mimetic peptide, JIP1 pep, did not uncover distinctive features of JNK1β1 when compared with the other variants studied. Differences in the kinetics of the interactions with binding partners like substrates, scaffold proteins, activators, inhibitors, and phosphatases could be a mechanism for subtle regulation of binding and activity of JNK and therefore modulation of the function of this important stress-associated signaling pathway.
Supplementary Material
Acknowledgments
pEL124–544 vector was a kind gift from Dr. Dominic Esposito at SAIC-Frederick. We thank Lisa Cherry and Weimin Chen for developing JNK1β1 and B-F-ATF2 expression vectors, respectively. We also thank Jeremy Chambers and Lisa Cherry for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant NS057153 (to P. L.).

This article contains supplemental Figs. S1–S5.
M. Figuera-Losada and P. V. LoGrasso, unpublished data.
- MAPK
- mitogen-activated protein kinase
- ATF2
- activating transcription factor 2
- B
- bioease
- BLI
- biolayer interferometry
- HL
- heterogeneous ligand
- JIP1
- JNK interacting protein 1
- JNK
- c-Jun N-terminal kinase
- MKK4
- mitogen-activated protein kinase kinase 4
- MKK7
- mitogen-activated protein kinase kinase 7
- N
- NusA solubility tag
- NHS-LC-LC-biotin
- succinimidyl-6-[biotinamido]-6-hexanamidohexanoate
- AMP-PCP
- 5′-adenylyl(β,γ-methylene)diphosphonate.
REFERENCES
- 1. Bender K., Blattner C., Knebel A., Iordanov M., Herrlich P., Rahmsdorf H. J. (1997) UV-induced signal transduction. J. Photochem. Photobiol. B 37, 1–17 [DOI] [PubMed] [Google Scholar]
- 2. Dérijard B., Hibi M., Wu I. H., Barrett T., Su B., Deng T., Karin M., Davis R. J. (1994) JNK1, a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025–1037 [DOI] [PubMed] [Google Scholar]
- 3. Hara T., Namba H., Yang T. T., Nagayama Y., Fukata S., Kuma K., Ishikawa N., Ito K., Yamashita S. (1998) Ionizing radiation activates c-Jun NH2-terminal kinase (JNK/SAPK) via a PKC-dependent pathway in human thyroid cells. Biochem. Biophys. Res. Commun. 244, 41–44 [DOI] [PubMed] [Google Scholar]
- 4. Kyriakis J. M., Avruch J. (1996) Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 18, 567–577 [DOI] [PubMed] [Google Scholar]
- 5. Westwick J. K., Weitzel C., Minden A., Karin M., Brenner D. A. (1994) Tumor necrosis factor α stimulates AP-1 activity through prolonged activation of the c-Jun kinase. J. Biol. Chem. 269, 26396–26401 [PubMed] [Google Scholar]
- 6. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 [DOI] [PubMed] [Google Scholar]
- 7. Sluss H. K., Barrett T., Dérijard B., Davis R. J. (1994) Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell Biol. 14, 8376–8384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kallunki T., Su B., Tsigelny I., Sluss H. K., Dérijard B., Moore G., Davis R., Karin M. (1994) JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 8, 2996–3007 [DOI] [PubMed] [Google Scholar]
- 9. Mohit A. A., Martin J. H., Miller C. A. (1995) p493F12 kinase. A novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14, 67–78 [DOI] [PubMed] [Google Scholar]
- 10. Gupta S., Barrett T., Whitmarsh A. J., Cavanagh J., Sluss H. K., Dérijard B., Davis R. J. (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760–2770 [PMC free article] [PubMed] [Google Scholar]
- 11. Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G. S. (2006) Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 103, 10741–10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 13. Kaneto H., Nakatani Y., Miyatsuka T., Kawamori D., Matsuoka T. A., Matsuhisa M., Kajimoto Y., Ichijo H., Yamasaki Y., Hori M. (2004) Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 10, 1128–1132 [DOI] [PubMed] [Google Scholar]
- 14. Chang L., Jones Y., Ellisman M. H., Goldstein L. S., Karin M. (2003) JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 4, 521–533 [DOI] [PubMed] [Google Scholar]
- 15. Dong C., Yang D. D., Wysk M., Whitmarsh A. J., Davis R. J., Flavell R. A. (1998) Defective T cell differentiation in the absence of Jnk1. Science 282, 2092–2095 [DOI] [PubMed] [Google Scholar]
- 16. Liu J., Minemoto Y., Lin A. (2004) c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor α-induced c-Jun kinase activation and apoptosis. Mol. Cell. Biol. 24, 10844–10856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sabapathy K., Hochedlinger K., Nam S. Y., Bauer A., Karin M., Wagner E. F. (2004) Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 15, 713–725 [DOI] [PubMed] [Google Scholar]
- 18. Hunot S., Vila M., Teismann P., Davis R. J., Hirsch E. C., Przedborski S., Rakic P., Flavell R. A. (2004) JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang D. D., Kuan C. Y., Whitmarsh A. J., Rincón M., Zheng T. S., Davis R. J., Rakic P., Flavell R. A. (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389, 865–870 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi T., Sakai K., Sasaki C., Zhang W. R., Warita H., Abe K. (2000) c-Jun N-terminal kinase (JNK) and JNK interacting protein response in rat brain after transient middle cerebral artery occlusion. Neurosci. Lett. 284, 195–199 [DOI] [PubMed] [Google Scholar]
- 21. Bruckner S. R., Tammariello S. P., Kuan C. Y., Flavell R. A., Rakic P., Estus S. (2001) JNK3 contributes to c-Jun activation and apoptosis but not oxidative stress in nerve growth factor-deprived sympathetic neurons, J. Neurochem. 78, 298–303 [DOI] [PubMed] [Google Scholar]
- 22. Klintworth H., Newhouse K., Li T., Choi W. S., Faigle R., Xia Z. (2007) Activation of c-Jun N-terminal protein kinase is a common mechanism underlying paraquat- and rotenone-induced dopaminergic cell apoptosis. Toxicol. Sci. 97, 149–162 [DOI] [PubMed] [Google Scholar]
- 23. Morishima Y., Gotoh Y., Zieg J., Barrett T., Takano H., Flavell R., Davis R. J., Shirasaki Y., Greenberg M. E. (2001) β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 21, 7551–7560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dreskin S. C., Thomas G. W., Dale S. N., Heasley L. E. (2001) Isoforms of Jun kinase are differentially expressed and activated in human monocyte/macrophage (THP-1) cells. J. Immunol. 166, 5646–5653 [DOI] [PubMed] [Google Scholar]
- 25. Yang J. Y., Moulin N., van Bemmelen M. X., Dubuis G., Tawadros T., Haefliger J. A., Waeber G., Widmann C. (2007) Splice variant-specific stabilization of JNKs by IB1/JIP1. Cell Signal. 19, 2201–2207 [DOI] [PubMed] [Google Scholar]
- 26. Waeber G., Delplanque J., Bonny C., Mooser V., Steinmann M., Widmann C., Maillard A., Miklossy J., Dina C., Hani E. H., Vionnet N., Nicod P., Boutin P., Froguel P. (2000) The gene MAPK8IP1, encoding islet brain-1, is a candidate for type 2 diabetes, Nat. Genet. 24, 291–295 [DOI] [PubMed] [Google Scholar]
- 27. Ember B., LoGrasso P. (2008) Mechanistic characterization for c-Jun N-terminal kinase 1α1. Arch. Biochem. Biophys. 477, 324–329 [DOI] [PubMed] [Google Scholar]
- 28. Ember B., Kamenecka T., LoGrasso P. (2008) Kinetic mechanism and inhibitor characterization for c-Jun N-terminal kinase 3α1. Biochemistry 47, 3076–3084 [DOI] [PubMed] [Google Scholar]
- 29. Niu L., Chang K. C., Wilson S., Tran P., Zuo F., Swinney D. C. (2007) Kinetic characterization of human JNK2α2 reaction mechanism using substrate competitive inhibitors. Biochemistry 46, 4775–4784 [DOI] [PubMed] [Google Scholar]
- 30. Lisnock J., Griffin P., Calaycay J., Frantz B., Parsons J., O'Keefe S. J., LoGrasso P. (2000) Activation of JNK3α1 requires both MKK4 and MKK7. Kinetic characterization of in vitro phosphorylated JNK3α1. Biochemistry 39, 3141–3148 [DOI] [PubMed] [Google Scholar]
- 31. Leatherbarrow R. J. (2001) GraFit, 5 Ed., Erithacus Software Limited, Surrey, United Kingdom [Google Scholar]
- 32. Barr R. K., Kendrick T. S., Bogoyevitch M. A. (2002) Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 277, 10987–10997 [DOI] [PubMed] [Google Scholar]
- 33. Segel I. H. (1975) Enzyme Kinetics, Wiley-Interscience, New York [Google Scholar]
- 34. Arthur P. G., Matich G. P., Pang W. W., Yu D. Y., Bogoyevitch M. A. (2007) Necrotic death of neurons following an excitotoxic insult is prevented by a peptide inhibitor of c-Jun N-terminal kinase. J. Neurochem. 102, 65–76 [DOI] [PubMed] [Google Scholar]
- 35. Bowers S., Truong A. P., Neitz R. J., Neitzel M., Probst G. D., Hom R. K., Peterson B., Galemmo R. A., Jr., Konradi A. W., Sham H. L., Tóth G., Pan H., Yao N., Artis D. R., Brigham E. F., Quinn K. P., Sauer J. M., Powell K., Ruslim L., Ren Z., Bard F., Yednock T. A., Griswold-Prenner I. (2011) Design and synthesis of a novel, orally active, brain penetrant, tri-substituted thiophene-based JNK inhibitor. Bioorg. Med. Chem. Lett. 21, 1838–1843 [DOI] [PubMed] [Google Scholar]
- 36. Chamberlain S. D., Redman A. M., Wilson J. W., Deanda F., Shotwell J. B., Gerding R., Lei H., Yang B., Stevens K. L., Hassell A. M., Shewchuk L. M., Leesnitzer M. A., Smith J. L., Sabbatini P., Atkins C., Groy A., Rowand J. L., Kumar R., Mook R. A., Jr., Moorthy G., Patnaik S. (2009) Optimization of 4,6-bis-anilino-1H-pyrrolo[2,3-d]pyrimidine IGF-1R tyrosine kinase inhibitors towards JNK selectivity. Bioorg. Med. Chem. Lett. 19, 360–364 [DOI] [PubMed] [Google Scholar]
- 37. Heo Y. S., Kim S. K., Seo C. I., Kim Y. K., Sung B. J., Lee H. S., Lee J. I., Park S. Y., Kim J. H., Hwang K. Y., Hyun Y. L., Jeon Y. H., Ro S., Cho J. M., Lee T. G., Yang C. H. (2004) Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 23, 2185–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hom R. K., Bowers S., Sealy J. M., Truong A. P., Probst G. D., Neitzel M. L., Neitz R. J., Fang L., Brogley L., Wu J., Konradi A. W., Sham H. L., Tóth G., Pan H., Yao N., Artis D. R., Quinn K., Sauer J. M., Powell K., Ren Z., Bard F., Yednock T. A., Griswold-Prenner I. (2010) Design and synthesis of disubstituted thiophene and thiazole based inhibitors of JNK. Bioorg. Med. Chem. Lett. 20, 7303–7307 [DOI] [PubMed] [Google Scholar]
- 39. Kamenecka T., Jiang R., Song X., Duckett D., Chen W., Ling Y. Y., Habel J., Laughlin J. D., Chambers J., Figuera-Losada M., Cameron M. D., Lin L., Ruiz C. H., LoGrasso P. V. (2010) Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-Jun N-terminal kinase (JNK) inhibitors. J. Med. Chem. 53, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Probst G. D., Bowers S., Sealy J. M., Truong A. P., Hom R. K., Galemmo R. A., Jr., Konradi A. W., Sham H. L., Quincy D. A., Pan H., Yao N., Lin M., Tóth G., Artis D. R., Zmolek W., Wong K., Qin A., Lorentzen C., Nakamura D. F., Quinn K. P., Sauer J. M., Powell K., Ruslim L., Wright S., Chereau D., Ren Z., Anderson J. P., Bard F., Yednock T. A., Griswold-Prenner I. (2011) Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg. Med. Chem. Lett. 21, 315–319 [DOI] [PubMed] [Google Scholar]
- 41. Scapin G., Patel S. B., Lisnock J., Becker J. W., LoGrasso P. V. (2003) The structure of JNK3 in complex with small molecule inhibitors. Structural basis for potency and selectivity. Chem. Biol. 10, 705–712 [DOI] [PubMed] [Google Scholar]
- 42. Stebbins J. L., De S. K., Machleidt T., Becattini B., Vazquez J., Kuntzen C., Chen L. H., Cellitti J. F., Riel-Mehan M., Emdadi A., Solinas G., Karin M., Pellecchia M. (2008) Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. U.S.A. 105, 16809–16813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Shannessy D. J., Winzor D. J. (1996) Interpretation of deviations from pseudo-first order kinetic behavior in the characterization of ligand binding by biosensor technology. Anal. Biochem. 236, 275–283 [DOI] [PubMed] [Google Scholar]
- 44. Abdiche Y., Malashock D., Pinkerton A., Pons J. (2008) Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377, 209–217 [DOI] [PubMed] [Google Scholar]
- 45. Bayer E. A., Wilchek M. (1980) The use of the avidin-biotin complex as a tool in molecular biology. Methods Biochem. Anal. 26, 1–45 [DOI] [PubMed] [Google Scholar]
- 46. Green N. M. (1975) Avidin. Adv. Protein Chem. 29, 85–133 [DOI] [PubMed] [Google Scholar]
- 47. Edwards P. R., Gill A., Pollard-Knight D. V., Hoare M., Buckle P. E., Lowe P. A., Leatherbarrow R. J. (1995) Kinetics of protein-protein interactions at the surface of an optical biosensor. Anal. Biochem. 231, 210–217 [DOI] [PubMed] [Google Scholar]
- 48. Gilbert H. F. (2000) Basic Concepts in Biochemistry: A Students Survival Guide, 2nd Ed., McGraw-Hill, Health Professions Division, New York [Google Scholar]
- 49. Ngoei K. R., Catimel B., Church N., Lio D. S., Dogovski C., Perugini M. A., Watt P. M., Cheng H. C., Ng D. C., Bogoyevitch M. A. (2011) Characterization of a novel JNK (c-Jun N-terminal kinase) inhibitory peptide. Biochem. J. 434, 399–413 [DOI] [PubMed] [Google Scholar]
- 50. Rubinfeld H., Seger R. (2005) The ERK cascade. A prototype of MAPK signaling. Mol. Biotechnol. 31, 151–174 [DOI] [PubMed] [Google Scholar]
- 51. Burkhard K. A., Chen F., Shapiro P. (2011) Quantitative analysis of ERK2 interactions with substrate proteins. Roles for kinase docking domains and activity in determining binding affinity. J. Biol. Chem. 286, 2477–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.