

Background: AD and prion diseases both involve conformational changes and deposition of insoluble proteins; similar to anti-Aβ autoantibodies, anti-PrP autoantibodies may be present in healthy individuals.
Results: PrP autoantibodies (PrP-AA) purified from human IgG could significantly block PrP-(106–126) peptide fibril formation and PrP-induced neuronal death.
Conclusion: Human prion autoantibodies reduce prion peptide aggregation and associated neurotoxicity.
Significance: Purified PrP-AA may be a potential treatment for prion diseases.
Keywords: Antibodies, Brain, Neurons, Neurotoxin, Peptides, IVIg, Prion, Autoantibody, Fibril Formation, Neurotoxicity
Abstract
Prion diseases are a group of rare, fatal neurodegenerative disorders associated with a conformational transformation of the cellular prion protein (PrPC) into a self-replicating and proteinase K-resistant conformer, termed scrapie PrP (PrPSc). Aggregates of PrPSc deposited around neurons lead to neuropathological alterations. Currently, there is no effective treatment for these fatal illnesses; thus, the development of an effective therapy is a priority. PrP peptide-based ELISA assay methods were developed for detection and immunoaffinity chromatography capture was developed for purification of naturally occurring PrP peptide autoantibodies present in human CSF, individual donor serum, and commercial preparations of pooled intravenous immunoglobulin (IVIg). The ratio of anti-PrP autoantibodies (PrP-AA) to total IgG was ∼1:1200. The binding epitope of purified PrP-AA was mapped to an N-terminal region comprising the PrP amino acid sequence KTNMK. Purified PrP-AA potently blocked fibril formation by a toxic 21-amino acid fragment of the PrP peptide containing the amino acid alanine to valine substitution corresponding to position 117 of the full-length peptide (A117V). Furthermore, PrP-AA attenuated the neurotoxicity of PrP(A117V) and wild-type peptides in rat cerebellar granule neuron (CGN) cultures. In contrast, IgG preparations depleted of PrP-AA had little effect on PrP fibril formation or PrP neurotoxicity. The specificity of PrP-AA was demonstrated by immunoprecipitating PrP protein in brain tissues of transgenic mice expressing the human PrP(A117V) epitope and Sc237 hamster. Based on these intriguing findings, it is suggested that human PrP-AA may be useful for interfering with the pathogenic effects of pathogenic prion proteins and, thereby has the potential to be an effective means for preventing or attenuating human prion disease progression.
Introduction
Prion diseases, or transmissible spongiform encephalopathies (TSEs),2 are rapidly progressive neurodegenerative disorders with untreatable invariably fatal outcomes. Disease caused by altered forms of prion protein (PrP) include scrapie in sheep, bovine spongiform encephalopathy in cattle, as well as the human forms Kuru, Creutzfeldt-Jakob disease (CJD and vCJD), and the Gerstmann-Straussler-Scheinker (GSS) syndrome (1). These diseases are most likely caused by misfolding and aggregation of the normal host protein (PrPC) into a highly insoluble form PrPSc. In this process, a portion of the α-helix and random coil structure of PrPC, which is ubiquitously expressed in neurons and leukocytes, adopts the PrPSc β-pleated conformation, rendering the protein poorly soluble in water and resistant to protease digestion (1). Autopsy on the brains of prion disease patients has identified amyloid plaques comprised of insoluble PrPSc aggregates deposited around neurons in affected brain regions, which is thought to induce neuronal dysfunction and death, thus producing the clinical symptoms of infection (1–7). The primacy of a single protein causing disease across species by diverse mechanisms is unique in biology.
To date, there are no therapeutic treatments available for prion diseases. However, recent studies in cultured cells and mice indicate that immunotherapeutic strategies employing antibodies against the cellular form of PrPC can antagonize prion infectivity and disease development. Monoclonal antibodies (mAbs) or recombinant F(ab) fragments recognizing PrP effectively prevented prion infection of susceptible mouse neuroblastoma cells and abrogated de novo PrPSc formation in chronically infected cells (8–9). In addition, passive transfer of a PrP mAb into scrapie-infected mice suppressed peripheral prion replication and infectivity, and significantly delayed onset of the disease (10–12). Notably, no obvious adverse effects were observed in these studies. These findings suggest that immunotherapeutic strategies for human prion diseases are worth pursuing.
Recently, we and others (13–14) have suggested that an impaired or reduced ability to generate antibodies specific for beta amyloid (Aβ) peptides may be one mechanism contributing to Alzheimer disease (AD) pathogenesis. Intravenous immunoglobulin (IVIg) preparations containing natural levels of anti-Aβ antibodies or purified autoantibodies against Aβ have shown beneficial effects in trials with AD patients (13, 15–17). We have demonstrated that these autoantibodies prevent or disaggregate Aβ fibril formation and block their toxic effects in primary neurons (18).
Since the pathogenic mechanisms of AD and prion diseases both involve toxic conformational changes and deposition of insoluble protein aggregates (1, 19–23) and given the early successes with natural Aβ autoantibodies for treatment of AD, we hypothesized that anti-PrP autoantibodies (PrP-AA) may also be present in blood products derived from healthy individuals. The potential for efficacy of PrP-AA is also based on results demonstrating the ability of mouse mAbs to prevent fibril formation, disaggregate already formed fibrils, and inhibit the neurotoxic effect of PrPSc (24). A benefit of purified human PrP-AA over humanized mouse mAbs is a reduced potential for neutralizing host responses to residual mouse sequences in the chimeric antibody.
A peptide fragment spanning human PrP sequences 106–126 (PrP106–126) possesses several chemicophysical characteristics of PrPSc, including the propensity to form β-sheet-rich, insoluble, and protease-resistant fibrils similar to those found in prion-diseased brains (25–26). This peptide has been widely used in an in vitro model to study PrPSc-induced neurotoxicity (27–32). A mutation in the prion protein gene (PRNP) leading to a substitution of valine for alanine at peptide position 117 (A117V) is associated with GSS syndrome, an inherited prion disease (33–35) that is characterized by multi-centric amyloid plaques in the cerebellum and cortex (36). The A117V mutation lies within the PrP106–126 region. The finding that a modification of PrP106–126(A117V) alters the toxic mechanism in vitro suggests that there may be heterogeneity in the mechanism of neurotoxicity of PrPSc. The mechanism underlying the neurotoxic effects of PrP106–126(A117V) includes at least two components: The first is similar to that of PrPSc, which requires the presence of microglia and neuronal PrPC expression; while the second is independent of neuronal PrPC expression or presence of microglia (36).
In this study, we have found evidence that PrP-AA are present in human CSF and serum. These autoantibodies could be successfully purified from IVIg by using affinity chromatography columns conjugated with PrP106–126(A117V) peptide. Additionally, we identified a five amino acid binding epitope for PrP-AA. Furthermore, we demonstrated that purified PrP-AA effectively protects cultured cerebral granule neurons (CGN) against wild type and mutant PrP106–126 induced neurotoxicity.
EXPERIMENTAL PROCEDURES
Purification of PrP-AA and Autoantibodies against Aβ
The protocol was adapted from a previously described method (13). Disposable chromatography columns were packed with CNBr-activated Sepharose 4B (Amersham Biosciences, Piscataway, NJ). PrP106–126(A117V) (Bachem) and Aβ1–40 (Invitrogen) were conjugated to Sepharose beads (0.6 mg/ml drained Sepharose) according to the manufacturer's instructions. The labeled Sepharose columns were equilibrated and washed with PBS (pH 7.4). After passing individual donor or commercial pooled human IgG (Baxter or Octapharm) through the columns and collecting the unbound (i.e. pass-through) fractions, bound IgG fractions were released by passing elution buffer (50 mm glycine at pH 2.5) through the column. The pH-neutralized fractions were collected and tested by ELISA.
Epitope Mapping of Purified PrP-AA
An array of 11 amino acid peptides, which were sequentially frame shifted by one residue or had single amino acid replacements, were synthesized on a cellulose membrane (Department of Biochemistry, Schulich School of Medicine and Dentistry, University of Western Ontario) using the spot method of multiple peptide synthesis (37–38). During the mapping study, membranes bound with peptides were prepared by washing with 100% ethanol and PBS, three times each, followed by blocking with 5% no-fat milk in PBS overnight at 4 °C. The membrane was then washed with PBS once more before adding 0.2 μg/ml purified PrP-AA and incubating overnight at 4 °C. After incubating with anti-human IgG HRP antibody (1:2000), the blots were visualized with the Super Signal chemiluminescence substrate (Pierce).
ELISA
The ELISA assay for PrP-AA was modified from a previously described method (13). 96-well ELISA plates were coated with PrP106–126 (A117V) that was dissolved in a coating buffer (1.7 mm NaH2PO4, 98 mm Na2HPO4, 0.05% sodium azide, pH 7.4).
Determination of PrP-AA Isotype
The IgG subclasses of purified antibody samples were determined using a Quantibody human Ig isotype array (Raybiotech, INC, cat QAH-ISO-1-1).
Immunoprecipitation of PrP and PrPSc by Purified PrP-AA
Reaction mixtures of homogenates in buffer containing 100 mm NaCl and 25 mm Tris/HCl (pH 7.4) were prepared from the cerebellum of a PrP(A117V) transgenic mouse and the brain of a hamster inoculated with Hamster Scrapie Strain Sc237 (10% v/v, InPro Biotechnology, South San Francisco, CA) (39). After centrifuging at 11,000 × g for 30 min at 4 °C, the mouse or hamster brain homogenates (2.5 or 100 mg/ml, respectively) were incubated with or without 100 μg/ml proteinase K (PK) at 37 °C for 2 h. PK digestion was terminated with 10 mm phenylmethylsulfonyl fluoride and heated at 100 °C 5 min. Cooled reaction mixtures were incubated overnight at 4 °C with 1 μg of purified human PrP-AA or purified human autoantibodies against Aβ. Protein A-agarose was added, and a second overnight incubation was performed, followed by centrifuging and washing three times with PBS. Immunoprecipitates were loaded into 4–12% NuPage Bis-Tris gel (Invitrogen NP0321) for Western blotting with diluted (1/2000) commercial anti-PrP monoclonal antibodies (3F4, Chemicon, AB1562; and 6D11, Santa Cruz Biotechnology, sc-58581) followed by horseradish-peroxidase-conjugated goat anti mouse IgG. Binding was visualized by enhanced chemiluminescence (Thermo Scientific, 34095). The 3F4 monoclonal antibody was raised against amino acids 109–112 of human PrP. According to the manufacturer, 3F4 recognizes both protease sensitive and resistant forms of human and hamster PrP, but not mouse PrP after denaturing. Monoclonal antibody clone 6D11 was raised against amino acids 93–109 of human PrP. According to the manufacturer this antibody recognizes PrPc as well as PrPSc of human, mouse, and hamster origin.
Fluorometric Experiments
Fluorometry has been previously described (18, 40). Synthetic PrP106–126 was incubated with or without purified PrP-AA in PBS buffer at 37 °C overnight. Samples were added to 50 mmol/liter glycine pH 9.2, 2 μmol/liter thioflavin T (Sigma) in a final volume of 2 ml. Fluorescence was measured spectrophotometrically at excitation with emission wavelengths of 435 nm and 485 nm, respectively. Samples were run in triplicate and were plotted with the mean ± S.D.
Electron Microscopy
Synthetic PrP106–126 was incubated with or without purified PrP-AA in PBS buffer at 37 °C overnight. 2 μl of each sample were dropped onto 300 mesh carbon/formvar-coated grids and allowed to absorb for 3 min. A drop of the negative stain (NanoVan, Nanoprobes, Inc. Yaphank, NY) was placed on the grid for 8–10 s and then wicked off for drying. Images were taken using a Tecnai G12 BioTwin transmission electron microscope (FEI, Hillsboro, OR) with an AMT CCD camera (Advanced Microscopy Techniques, Danvers, MA).
Mass Spectrometry
Electrospray ionization mass spectrometry (ESI-MS, API 4000, Applied Biosystems) was used to detect the monomer of PrP. The instrument was equipped with a Z-spray ionization source. Both nebulizer and desolvation gases were nitrogen and the collision gas was argon. Mass spectrometric parameters were set as follows: collision gas (CAD) 8, curtain gas (CUR) 10, ion source gas 1 (GS1) 15, ion source gas 2 (GS2) 35, electrospray voltage 5000 in positive ion scan mode, and dry temperature at 500 °C. The mixture of methanol, water, and formic acid (90:10:0.1, v/v/v) were used as the mobile phase with a flow rate 0.2 ml/min. Synthetic PrP106–126 was incubated with or without purified PrP-AA in PBS buffer at 37 °C overnight. The samples were filtered and directly infused into the mass spectrometer (10 μl) through a LC system (Agilent 1100) with an auto sampler. All data were acquired at least in triplicate to confirm the reproducibility of the results.
Primary Rat Neuronal Culture and Neurotoxicity Assays
CGN were prepared from 7-day-old Sprague-Dawley rats as described previously (41). Briefly, rat CGN cells were prepared and seeded into 48-well poly-l-lysine-coated culture plates at a cell density of 2 × 105 cells/well in the BME medium with 10% fetal bovine serum and 25 mm KCl (Sigma). After incubating for 24 h, 10 μm cytosine arabino-furanoside (Sigma) was added to prevent glial proliferation. These cultures contain about 95% neurons (95% granule cells) with the remaining 5% of non-neuronal cells, mainly of astrocytic type (42–43). Treatments were performed after 14 days in vitro. PrP106–126 (A117V or 117A) or scrambled PrP106–126 (NGAKALMGGHGATKVMVGAAA) was pre-incubated in PBS, pH 7.2 at 37 °C for 48 h in the absence or presence of purified PrP-AA in vitro and was then added to cells. After the exposure of the cells to these incubates for 3 days, cell viability was determined by staining neurons with fluorescein diacetate/propidium iodide.
Glial Cell Culture
Primary cultures of rat cerebellar astroglial cells were prepared from the cerebellum of 7-day-old Sprague-Dawley rats as previously described (44–45). Cells dissociated from cerebella were plated at a density of 5 × 105/well on 24-well plates coated with poly-l-lysine and cultured in a complete medium containing 10% FBS. After 3 days, the medium was replaced with a fresh one containing 10% FBS, and the cells were cultured for additional 3–4 days before treatment until they were more than 90% confluent. As previous reports state, these cultures are composed of up to 90% of astrocytes positive for glial fibrillary acidic protein (44, 46).
Generation of Mice Heterozygous for the PRNPA117V Allele
The plasmid expression vector (pProPrpHGSal) (47), containing the proximal half of genomic mouse PNRP, including the promoter and coding sequences of exon 1, intron 1, and exon 2 fused to exon 3, was used to create the chimera. We inserted the hamster open reading frame (ORF) in place of the murine ORF. The hamster ORF sequence was amplified using PCR with hamster cDNA as the template and GCTATGTGGACTGATGTCGGC; CAGGGCCCACTAGTGCCAAG as the forward and reverse primers. The PCR fragment was cloned initially into pIRESneo. An A117V mutation (A117→V) was introduced by using the Quick Change Mutagenesis Kit (Stratagene). The mutation and absence of polymerase errors were verified by sequencing. The ApaI/PshA I insert was released and inserted in place of the murine ApaI/PshA I within the pProPrpHGSal vector, leading to a construct termed SHa-Mo PrP. An 11-kb DNA fragment containing the A117V mutant allele of the PNRP gene was excised from vector pProPrpHGSal by Not/SalI digestion and injected into the pronuclei of fertilized oocytes from PNRP knock-out mice (47). Genomic DNA, isolated from tail tissue of weanling animals, was screened for the presence of incorporated mouse/hamster chimeric PRNP transgene using PCR primers. The forward primer sequence (5′-CAA CCG AGC TGA AGC ATT CTG CCTT-3′) is in the mouse PrP region and the reverse primer sequence (5′-CAC GCG CTC CAT TAT CTT GAT G-3′) is in the hamster PrP region.
RESULTS
After identifying PrP-AA in all human CSF and serum samples from five normal individuals by using ELISA, we developed and used an affinity column coated with the mutant human PrP sequence encompassing residues 106–126 (KTNMKHMAGAAVAGAVVGGLG), which is termed PrP106–126(A117V), to isolate human PrP-AA from IVIg or serum from individual blood specimens. An intense signal was observed with antibody capture of PrP106–126(A117V) in an ELISA assay using bound PrP-AA (Fig. 1). The non-binding fraction (“pass-through” (PT)) was depleted of antibodies which bound PrP106–126(A117V) (Fig. 1). In contrast, purified PrP-AA could not be detected by ELISA coated with the unrelated Aβ1–40 peptide (data not shown).
FIGURE 1.
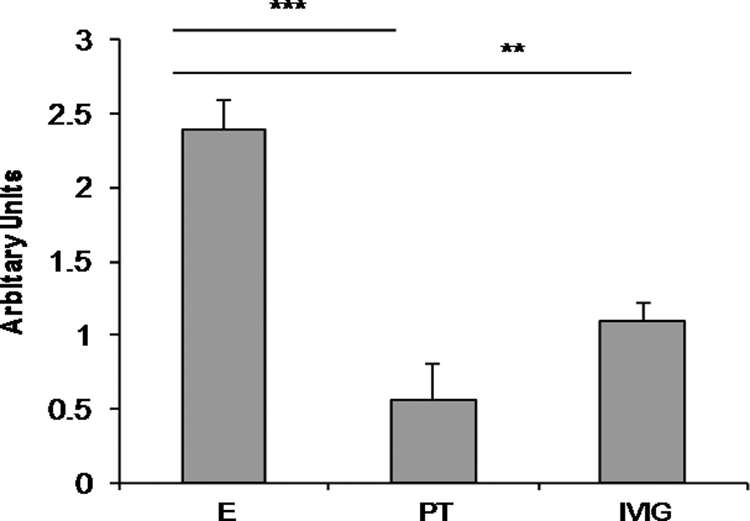
Analysis of PrP106–126(A117V) binding by purified PrP-AA in an ELISA assay. Purified PrP-AA, non-binding, pass-through IgG (PT) or original IVIg (all at 1 μg) were added to PrP106–126(A117V) peptide-coated wells. After washing, bound antibodies were detected with horseradish peroxidase-conjugated secondary anti-human IgG antibodies. Purified PrP-AA showed an enhanced signal compared with the original IVIg; whereas, the PT IgG was greatly diminished in binding capacity. E, PrP-AA; PT, pass-through IgG depleted of PrP-AA; IVIg, original IVIg used to purified PrP-AA; **, p < 0.01; ***, p < 0.001.
The specificity of PrP-AA was evaluated by immunoprecipitating PrP(A117V) from homogenates of brains from transgenic mice that express the human sequences encompassing residues 106–126. This was accomplished by knocking-in a hybrid mouse/hamster PNRP gene containing the A117V substitution, which has been used previously to investigate GSS (48). It has already been established that hamster PrP binds a human single chain PrP antibody (49), suggesting that brains expressing the coding region of the hamster protein could bind to human PrP antibodies. We confirmed the expression of PrP(A117V) in transgenic mouse brain homogenates using the commercially available mouse monoclonal antibody 3F4. This antibody recognized a protein of the correct mobility (∼29 kDa) in brain homogenates from transgenic PrP (A117V) mice, but not wild type or PNRP knock-out mice (Fig. 2A). Immunoprecipitation of ∼29 kDa proteins from brain homogenates of PrP(A117V) transgenic mice was accomplished with purified PrP-AA; whereas, no protein bands where observed after immunoprecipitation with PrP-AA-depleted IVIg (PT) (Fig. 2B). Western blotting of homogenates from brains of PrP(A117V) or PNRP knock-out mice demonstrated a major band corresponding to PrP only in the cortex and cerebella of the transgenic mice (Fig. 2C). Of note, although other minor protein species were evident upon detection of PrP-AA immunoprecipitates with the unrelated 3F4 antibody, PrP(A117V) was by far the predominant protein band observed (Fig. 2C). Taken together, these data indicate that PrP-AA binds PrP(A117V) with high specificity and affinity. Additionally, to examine whether PrP-AA could bind to protease-resistant PrPSc conformers, brain homogenates isolated from a Sc237 hamster pretreated with or without PK were immunoprecipitated by PrP-AA or autoantibodies against Aβ as a negative control. We clearly demonstrated that purified PrP-AA recognized both PrP and PK-resistant PrPSc (27–30 kDa) (Fig. 2D).
FIGURE 2.

Characterization of PrP-AA specificity for PrP. A, A117V transgenic mice, but not wild-type (WT) nor PNRP knock-out (KO) mice, were shown to express the PrP protein, which was detectable in brain homogenates using the murine monoclonal antibody 3F4. B, visualization of PrP(A117V) in brain homogenates (500 μg protein) of transgenic mice by immunoprecipitation with purified PrP-AA (E) or PT. Immunoprecipitated complexes were subjected to Western blot analysis with 3F4 antibody. C, purified PrP-AA recognized the PrP protein in Western blots of brain cortex and cerebellar (Cere) homogenates of A117V transgenic mice but not KO mice. Although, multiple bands were observed with overexposure, the strongest signal corresponded to the approximately band of 29 kDa PrP (A117V) observed in PrP(A117V) transgenic mice. D, Western blot analysis of immunoprecipitates from brain homogenates (1 mg transgenic mouse cerebellum and 10 mg Sc237 hamster brain) pretreated with or without proteinase K using PrP-AA or autoantibodies against Aβ. An anti-PrP antibody 6D11 which detects both mouse and hamster PrP, was used for detecting antibody. Numbers adjacent to horizontal lines indicate positions of molecular mass markers (kDa). 10 μl samples were loaded in each lane. Purified PrP-AA recognized both PrP and PK-resistant PrPSc (27–30kDa). Autoantibodies against Aβ did not recognize PrP nor PK-resistant PrPSc (27–30kDa). The photo was selected from a single representative experiment that was repeated three times with similar results. PT, pass-through IgG depleted of PrP-AA. Aβ-AA, autoantibodies against Aβ
The titer of PrP-AA in IVIg was determined to be 1:1200 within a total IgG concentration of 10 g/100 ml. The distribution of different IgG subclasses in the purified PrP-AA were as follow: IgG1 74.2%, IgG2 12%, IgG3 11.4%, and IgG4 2.4%. Thus, the IgG subclasses of purified PrP-AA are similar to the distribution of IgG subclasses in IVIg products and human serum. Furthermore, the PrP-AA binding epitope was determined using an array displaying a series of modified PrP106–126 peptides (Fig. 3, A and B). Binding of PrP-AA occurs at the extreme N terminus of PrP106–126, and requires, at a minimum residue KTNMK (106–110) as demonstrated in Fig. 3, C and D. Both lysines in this motif are critical for high affinity antibody binding since substitution or deletion of either completely abolished PrP-AA binding (Fig. 3).
FIGURE 3.

Mapping PrP-AA binding epitopes. Domain specificities of PrP-AA were determined using a peptide microarray. Sequences of either sequentially one amino acids shifted (A) or single amino acids deletions (C) peptides within region PrP106–126 which were synthesized and spotted on membranes are displayed in A and C. Membranes were then probed with PrP-AA (2 μg/ml) and then HRP conjugated anti-human-IgG antibody (triplicate membranes were probed). The sequence motif KTNMK appeared to be highly important since only peptide 1 is bound by PrP-AA, as shown in panel B. Further validation came from experiments shown in panel D, which show strong binding only when residues 1–5 are present, implying the two lysines (KXXXK) are key elements for binding.
Next we investigated by electron microscopy, mass spectrometry, and fluorometric measurement using a Thioflavin T (ThT) reagent that binds specifically to fibrillar structures whether purified human PrP-AA could block PrP fibril formation as well as disaggregate preformed fibrils (Fig. 4). Dose-response and kinetic studies showed that pre-incubating PrP106–126 monomers or preformed peptide fibrils with purified human PrP-AA dose-dependently prevented fibril formation and disrupted preformed fibril structures in a time-dependent manner, as evidenced by a substantial decrease in ThT fluorescence (Fig. 4, A and B) compared with the control using PT. These findings were confirmed in independent experiments using various concentrations of PrP-AA and reaction time (Fig. 4, C and D).
FIGURE 4.

Effects of PrP-AA on PrP peptide's fibril formation. A, dose-response study of PrP106–126(A117V) fibril formation and PrP-AA effects. B, kinetic study of 50 μm PrP106–126(A117V) fibril formation and 0.07 μm PrP-AA effects. C, incubation of 50 μm PrP106–126(A117V) peptides with or without purified PrP-AA in PBS. Purified PrP-AA significantly inhibited PrP106–126(A117V) fibril formation. D, incubation of preformed fibrils from 50 μm PrP106–126(A117V) peptides with purified PrP-AA (E, 0.07 μm) or pass-through IgG (PT, 0.07 μm) in PBS for 48 h. Purified PrP-AA significantly disaggregated preformed PrP106–126(A117V) fibrils as measured by ThT staining. Samples were run in triplicate and plotted as the mean ± S.D. (***, p < 0.001; **, p < 0.01; *, p < 0.05 compared with PrP only, one-way ANOVA). Representative data from triplicate mass spectra of the PrP106–126(A117V) monomer with (E) or without (F) PrP-AA were inserted to E and F. Electron micrographs of the products from experiments are shown in E and F (scale bar = 500 nm). E, PrP-AA; PT, pass-through IgG depleted of PrP-AA.
To confirm findings obtained from the ThT fluorescence assay and to exclude interference of ThT bound with PrP fibrils by antibodies, fibrils, and monomers were visualized by electron microscopy and measured by mass spectrometry. The mass spectra of PrP monomers incubated with (Fig. 4D) or without (Fig. 4E) PrP-AA revealed that PrP-AA treatments significantly increased the well-resolved PrP monomer peak, indicating that PrP-AA blocked PrP fibril formation. Electron microscopic examination of these reactions confirmed the data from mass spectrometry (Fig. 4, E and F).
We next assessed whether PrP-AA could protect cultured primary rat neurons from toxicity of predominantly fibrillar PrP106–126, as determined by measuring viability using FDA/PI stains (Figs. 5 to 7). The addition of PrP106–126 was allowed to form fibrils before addition induced neuronal death in a dose-dependent fashion (Fig. 5A). In contrast, a control peptide with a scrambled PrP106–126 sequence showed no neurotoxicity compared with PrP106–126 (Fig. 5B). These data demonstrate the specific toxicity to CGN of PrP106–126, which had been allowed to form fibers before addition.
FIGURE 5.

Neurotoxicity of PrP peptides on CGN. Dose-dependence of PrP106–126(A117V) fibril neurotoxicity was examined in CGN. The neurons were exposed to different dosages of PrP106–126(A117V) (5 μm to 100 μm) (A) or PrP106–126(A117V) (100 μm) and scrambled control peptide (100 μm) (B) for 3 days. Cell viability was determined by staining neurons with fluorescein diacetate/propidium iodide. Values are expressed as percentages (%) of control (untreated). The data represent the mean ± S.D. (bars) values of triplicate determinations from a single but representative experiment, which has been repeated three times with similar results (**, p < 0.01; ***, p < 0.001 by one-way ANOVA).
FIGURE 7.
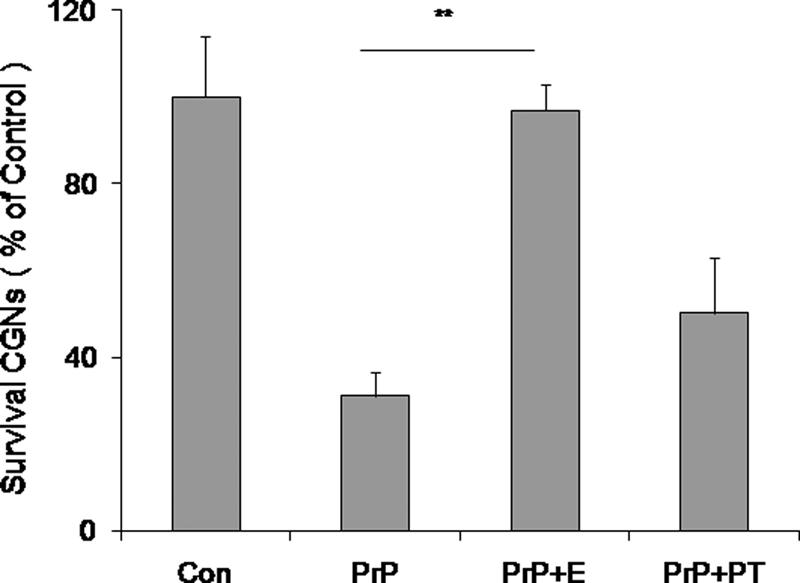
Analysis of PrP-AA in the culture system. The PrP-AA prevented PrP106–126(A117V) induced neurotoxicity in a neuron-glia co-culture system. Purified PrP-AA significantly blocked PrP106–126(A117V) fibril-induced neuronal death in the co-cultured system. CGN-glia were treated with 50 μm PrP106–126(A117V) fibril only and PrP106–126(A117V) fibril that had been preincubated with 0.07 μm PrP-AA for 24 h. Cell viability was determined by staining neurons with fluorescein diacetate/propidium iodide. Values are expressed as percentages (%) of control (untreated). The data represent the mean ± S.D. (bars) values of triplicate determinations from a single but representative experiment, which has been repeated three times with similar results (**, p < 0.01, by one-way ANOVA). PrP, PrP106–126(A117V) peptides; E, PrP-AA; PT, pass-through IgG depleted of PrP-AA.
Next we tested whether neurotoxicity of PrP106–126 monomers or fibers could be blocked by pre-incubating with purified PrP-AA before adding to cultures of CGN. Human PrP-AA almost completely prevented neurotoxicity of the mutated PrP106–126(A117V) added as monomers (Fig. 6A) or preformed fibrils (Fig. 6B). In addition, PrP-AA also potently blocked wild-type PrP106–126-induced neurotoxicity (Fig. 6C). Conversely, PrP-AA-depleted fraction of IVIg failed to protect against neurotoxicity produced by either peptide.
FIGURE 6.

Effects of PrP-AA on wild type or mutant PrP106–126 induced neurotoxicity. Exposure of rat CGN to 50 μm PrP106–126(A117V or wild type) fibril resulted in a reduction of neuronal survival during a 3 day incubation period. Purified PrP-AA (0.07 μm) significantly attenuated PrP106–126(A117V) fibril-induced neuronal death. A, PrP106–126(A117V) peptides (50 μm) were incubated with PrP-AA (0.07 μm) before being exposed to neurons. B, preformed PrP106–126(A117V) fibrils were incubated with PrP-AA (0.07 μm) before being exposed to neurons. C, preformed wild-type PrP106–126(117A) fibrils were incubated with PrP-AA (0.07 μm) before exposed to neurons. Cell viability was determined by staining neurons with fluorescein diacetate/propidium iodide. The data represent the mean ± S.D. of triplicate determinations from a representative experiment repeated at least three times with similar results (*, p < 0.05; ***, p < 0.001, compared with PrP106–126 only, one-way ANOVA). Con, untreated cultures; PrP, PrP106–126 (A117V or wild type) peptides; E, PrP-AA; PT, pass-through IgG depleted of PrP-AA.
Previous studies reported that, unlike the wild-type peptide, PrP106–126(A117V) fibrils induce inflammation-mediated neurotoxicity (36). To confirm that purified human PrP-AA protected against inflammation related neurotoxicity of PrP106–126(A117V), we applied co-culture system of CGN combined with glia cells. Consistent with the previous report, treatment of these mixed cultures with preformed PrP106–126(A117V) fibrils led to markedly greater CGN death (Fig. 7) compared with treatment of CGN monocultures (Fig. 6). This toxicity was greatly reduced with PrP-AA pretreatment of fibrils (Fig. 7). In contrast, PT demonstrated no neuroprotective effects.
DISCUSSION
We have identified specific prion protein-binding antibodies in both sera and CSF from normal individuals and have demonstrated neutralization of PrP toxicity in primary cerebellar neurons. This is the first identification and isolation of PrP antibodies from subjects with no documented exposure to prion antigens. Both immunoprecipitation and Western blot data suggest that PrP-AA strongly binds to the PrP monomer and PrPSc. We speculate that these autoantibodies may have normal physiological functions of immune-mediated PrP replication control or clearance, similar to what we have previously postulated for circulating Aβ antibodies (13, 15). Our results demonstrate that human PrP-AA can be isolated from currently marketed IVIg; thus, the potential for producing a consistent product to test in the clinic is enhanced.
It has been previously suggested that PrP antibodies may be an effective immunotherapy for prion diseases (50). Interestingly, even though TSE is a CNS disease, PrPSc accumulates in lymphoid tissues before CNS involvement. Accordingly, lymphoid PrPSc represents an early primary target for therapeutic strategies, given the greater accessibility of peripheral tissues compared with privileged CNS system which significantly impedes penetration of the antibodies through the blood brain barrier. Possible immunotherapies are active immunization with a PrP antigen or passive immunization with selective antibodies. Development of an active immunization therapy may be problematic since prion infections do not elicit a classical immune response and there likely would be great reticence to immunize asymptomatic or uninfected individuals given the known infectivity of this peptide (50). In addition, a phase II clinical trial in AD patients testing active immunization with the Aβ epitode, AN1792, failed due to severe side effects. Passive immunization, on the other hand, may represent a better approach given the lack of issues cited above.
Our present finding of fairly abundant levels of PrP-AA in normal human sera and concentrated pooled IgG, which can be purified and concentrated, represents a new opportunity for rapidly developing an effective and relatively safer immunotherapy for prion diseases. Alternatively, a humanized monoclonal antibody targeting the PrP epitope could be developed based on the binding sequence of PrP-AA. Although monoclonal antibodies may be viewed as more optimal than purified polyclonal antibodies from the standpoint of consistency of preparation, there is still concern that chronic dosing with humanized antibodies may generate anti-idiotypic responses directed to the residual mouse CDR sequences.
We demonstrate that purified PrP-AA dramatically inhibit PrP fibril formation and disrupt preformed PrP fibrils, as reported in previous studies using mouse PrP antibodies (8–9). The epitope for human PrP-AA is a unique, within the human genome, five-amino acid sequence located at the N terminus of the PrP106–126 peptide, which is conserved between humans and hamster PrP. Human PrP-AA recognizes the full-length hybrid hamster/mouse prion protein containing the A117V mutation when expressed in a transgenic mouse line. Interestingly enough, human PrP-AA also directly and strongly binds to a well known hamster protease-resistant PrPSc protein, SC237, indicating human IgG, somehow, may be involved in protecting humans to resist prion infections at a certain degree. The finding that PrP-AA binding is disrupted by mutating a small stretch of amino acids exclusively, suggests that the pool of purified IgG is comprised of only a small number of antibody clones. Furthermore, it identifies a discrete region within the full-length peptide that is crucial for fibril formation and neurotoxicity. Since binding occurs at a region of the PrP protein (e.g. 106–110) without known mutations, this purified PrP-AA should be effective for treatment of all prion diseases. Indeed, we have demonstrated prevention of both wild type and PrP106–126(A117V) fibril formation and peptide-induced neurotoxicity. In addition, the different pathways of neuronal death induced by these two peptides suggest that PrP-AA may have a broad function to treat prion diseases besides GSS. Additionally, since PrP-AA could interact with PrPSc, it is necessary to perform a future study to show whether the human PrP-AA can interfere with human PrPSc formation, replication, and PrPSc-induced neurotoxicity in the brain. Additionally, it is also important in future studies to test the effect of the PrP-AA on aggregation of full-length PrP or the N-terminal domain of wild-type PrP. Experiments are currently underway in transgenic models expressing various forms of the full-length protein to test this prediction.
This study provides strong evidence that PrP-AA is found in normal human blood and CSF and can be easily purified from pooled IgG. The similar features of PrP-AA to autoanti-Aβ antibodies suggests treatment of prion diseases with PrP-AA is highly feasible, especially since whole IVIg clinical trials for AD are currently ongoing and have demonstrated some efficacy (51). Thus, administration of purified human PrP-AA or IVIg may be used some day to prevent or slow down prion disease progression.
Footnotes
- TSE
- transmissible spongiform encephalopathies
- PrP
- prion protein
- CJD
- Creutzfeldt-Jakob disease
- AD
- Alzheimer disease
- IVIg
- intravenous immunoglobulin
- PrP-AA
- anti-PrP autoantibodies
- CGN
- cerebellar granule neuron
- Aβ
- beta amyloid.
REFERENCES
- 1. Prusiner S. B. (1991) Molecular biology of prion diseases. Science 252, 1515–1522 [DOI] [PubMed] [Google Scholar]
- 2. Bugiani O., Giaccone G., Piccardo P., Morbin M., Tagliavini F., Ghetti B. (2000) Neuropathology of Gerstmann-Sträussler-Scheinker disease. Microsc. Res. Tech. 50, 10–15 [DOI] [PubMed] [Google Scholar]
- 3. Ghetti B., Piccardo P., Frangione B., Bugiani O., Giaccone G., Young K., Prelli F., Farlow M. R., Dlouhy S. R., Tagliavini F. (1996) Prion protein amyloidosis. Brain Pathol. 6, 127–145 [DOI] [PubMed] [Google Scholar]
- 4. Giaccone G., Verga L., Bugiani O., Frangione B., Serban D., Prusiner S. B., Farlow M. R., Ghetti B., Tagliavini F. (1992) Prion protein preamyloid and amyloid deposits in Gerstmann-Sträussler-Scheinker disease, Indiana kindred. Proc. Natl. Acad. Sci. U.S.A. 89, 9349–9353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kitamoto T., Tateishi J., Tashima T., Takeshita I., Barry R. A., DeArmond S. J., Prusiner S. B. (1986) Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Ann. Neurol. 20, 204–208 [DOI] [PubMed] [Google Scholar]
- 6. Tateishi J., Kitamoto T., Hashiguchi H., Shii H. (1988) Gerstmann-Sträussler-Scheinker disease: immunohistological and experimental studies. Ann. Neurol. 24, 35–40 [DOI] [PubMed] [Google Scholar]
- 7. Ghetti B., Tagliavini F., Masters C. L., Beyreuther K., Giaccone G., Verga L., Farlow M. R., Conneally P. M., Dlouhy S. R., Azzarelli B. (1989) Gerstmann-Sträussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family. Neurology 39, 1453–1461 [DOI] [PubMed] [Google Scholar]
- 8. Peretz D., Williamson R. A., Kaneko K., Vergara J., Leclerc E., Schmitt-Ulms G., Mehlhorn I. R., Legname G., Wormald M. R., Rudd P. M., Dwek R. A., Burton D. R., Prusiner S. B. (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739–743 [DOI] [PubMed] [Google Scholar]
- 9. Enari M., Flechsig E., Weissmann C. (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. U.S.A. 98, 9295–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heppner F. L., Musahl C., Arrighi I., Klein M. A., Rülicke T., Oesch B., Zinkernagel R. M., Kalinke U., Aguzzi A. (2001) Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 294, 178–182 [DOI] [PubMed] [Google Scholar]
- 11. White A. R., Enever P., Tayebi M., Mushens R., Linehan J., Brandner S., Anstee D., Collinge J., Hawke S. (2003) Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422, 80–83 [DOI] [PubMed] [Google Scholar]
- 12. Sigurdsson E. M., Brown D. R., Daniels M., Kascsak R. J., Kascsak R., Carp R., Meeker H. C., Frangione B., Wisniewski T. (2002) Immunization delays the onset of prion disease in mice. Am. J. Pathol. 161, 13–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du Y., Dodel R., Hampel H., Buerger K., Lin S., Eastwood B., Bales K., Gao F., Moeller H. J., Oertel W., Farlow M., Paul S. (2001) Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology 57, 801–805 [DOI] [PubMed] [Google Scholar]
- 14. Weksler M. E., Relkin N., Turkenich R., LaRusse S., Zhou L., Szabo P. (2002) Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontol. 37, 943–948 [DOI] [PubMed] [Google Scholar]
- 15. Dodel R., Hampel H., Depboylu C., Lin S., Gao F., Schock S., Jäckel S., Wei X., Buerger K., Höft C., Hemmer B., Möller H. J., Farlow M., Oertel W. H., Sommer N., Du Y. (2002) Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann. Neurol. 52, 253–256 [DOI] [PubMed] [Google Scholar]
- 16. Dodel R. C., Du Y., Depboylu C., Hampel H., Frölich L., Haag A., Hemmeter U., Paulsen S., Teipel S. J., Brettschneider S., Spottke A., Nölker C., Möller H. J., Wei X., Farlow M., Sommer N., Oertel W. H. (2004) Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 75, 1472–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dodel R., Neff F., Noelker C., Pul R., Du Y., Bacher M., Oertel W. (2010) Drugs 70, 513–528 [DOI] [PubMed] [Google Scholar]
- 18. Du Y., Wei X., Dodel R., Sommer N., Hampel H., Gao F., Ma Z., Zhao L., Oertel W. H., Farlow M. (2003) Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain 126, 1935–1939 [DOI] [PubMed] [Google Scholar]
- 19. Beekes M. (2007) Prions and prion diseases. FEBS J. 274, 575. [DOI] [PubMed] [Google Scholar]
- 20. DeArmond S. J., Yang S. L., Lee A., Bowler R., Taraboulos A., Groth D., Prusiner S. B. (1993) Three scrapie prion isolates exhibit different accumulation patterns of the prion protein scrapie isoform. Proc. Natl. Acad. Sci. U.S.A. 90, 6449–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wisniewski T. (2001) Henry M. Wisniewski M.D. Ph.D. J. Alzheimers Dis. 3, 7–22 [DOI] [PubMed] [Google Scholar]
- 22. Wisniewski T., Sigurdsson E. M. (2007) Therapeutic approaches for prion and Alzheimer's diseases. FEBS. J. 274, 3784–3798 [DOI] [PubMed] [Google Scholar]
- 23. Brundin P., Melki R., Kopito R. (2010) Nat. Rev. Mol. Cell Biol. 11, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanan E., Goren O., Eshkenazy M., Solomon B. (2001) Immunomodulation of the human prion peptide 106–126 aggregation. Biochem. Biophys. Res. Commun. 280, 115–120 [DOI] [PubMed] [Google Scholar]
- 25. Fioriti L., Quaglio E., Massignan T., Colombo L., Stewart R. S., Salmona M., Harris D. A., Forloni G., Chiesa R. (2005) The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol. Cell Neurosci. 28, 165–176 [DOI] [PubMed] [Google Scholar]
- 26. Tagliavini F., Forloni G., D'Ursi P., Bugiani O., Salmona M. (2001) Studies on peptide fragments of prion proteins. Adv. Protein Chem. 57, 171–201 [DOI] [PubMed] [Google Scholar]
- 27. Forloni G., Angeretti N., Chiesa R., Monzani E., Salmona M., Bugiani O., Tagliavini F. (1993) Neurotoxicity of a prion protein fragment. Nature 362, 543–546 [DOI] [PubMed] [Google Scholar]
- 28. Forloni G., Del Bo R., Angeretti N., Chiesa R., Smiroldo S., Doni R., Ghibaudi E., Salmona M., Porro M., Verga L. (1994) A neurotoxic prion protein fragment induces rat astroglial proliferation and hypertrophy. Eur. J. Neurosci. 6, 1415–1422 [DOI] [PubMed] [Google Scholar]
- 29. Brown D. R., Herms J., Kretzschmar H. A. (1994) Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. Neuroreport 5, 2057–2060 [DOI] [PubMed] [Google Scholar]
- 30. Brown D. R., Schmidt B., Kretzschmar H. A. (1996) A neurotoxic prion protein fragment enhances proliferation of microglia but not astrocytes in culture. Glia 18, 59–67 [DOI] [PubMed] [Google Scholar]
- 31. Brown D. R., Schmidt B., Kretzschmar H. A. (1996) Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 380, 345–347 [DOI] [PubMed] [Google Scholar]
- 32. Brown D. R. (1998) Prion protein-overexpressing cells show altered response to a neurotoxic prion protein peptide. J. Neurosci. Res. 54, 331–340 [DOI] [PubMed] [Google Scholar]
- 33. Mastrianni J. A., Curtis M. T., Oberholtzer J. C., Da Costa M. M., DeArmond S., Prusiner S. B., Garbern J. Y. (1995) Prion disease (PrP-A117V) presenting with ataxia instead of dementia. Neurology 45, 2042–2050 [DOI] [PubMed] [Google Scholar]
- 34. Tateishi J. (1990) Slow virus, prion and nervous system Tanpakushitsu Kakusan Koso 35, 1327–1331 [PubMed] [Google Scholar]
- 35. Hsiao K. K., Cass C., Schellenberg G. D., Bird T., Devine-Gage E., Wisniewski H., Prusiner S. B. (1991) A prion protein variant in a family with the telencephalic form of Gerstmann-Sträussler-Scheinker syndrome. Neurology 41, 681–684 [DOI] [PubMed] [Google Scholar]
- 36. Brown D. R. (2000) Altered toxicity of the prion protein peptide PrP106–126 carrying the Ala(117)→Val mutation. Biochem. J. 346, 785–791 [PMC free article] [PubMed] [Google Scholar]
- 37. Molina F., Laune D., Gougat C., Pau B., Granier C. (1996) Improved performances of spot multiple peptide synthesis. Pept. Res. 9, 151–155 [PubMed] [Google Scholar]
- 38. Morel N., Simon S., Frobert Y., Volland H., Mourton-Gilles C., Negro A., Sorgato M. C., Créminon C., Grassi J. (2004) Selective and efficient immunoprecipitation of the disease-associated form of the prion protein can be mediated by nonspecific interactions between monoclonal antibodies and scrapie-associated fibrils. J. Biol. Chem. 279, 30143–30149 [DOI] [PubMed] [Google Scholar]
- 39. Yokoyama T., Kimura K. M., Ushiki Y., Yamada S., Morooka A., Nakashiba T., Sassa T., Itohara S. (2001) In vivo conversion of cellular prion protein to pathogenic isoforms, as monitored by conformation-specific antibodies. J. Biol. Chem. 276, 11265–11271 [DOI] [PubMed] [Google Scholar]
- 40. Du Y., Bales K. R., Dodel R. C., Liu X., Glinn M. A., Horn J. W., Little S. P., Paul S. M. (1998) a2-Macroglobulin attenuates Aβ(1–40) fibril formation and associated neutrotoxicity of cultured fetal rat cortical neurons. J. Neurochem. 70, 1182–1188 [DOI] [PubMed] [Google Scholar]
- 41. Du Y., Dodel R., Bales K., Hamilton-Byrd E., Paul S. M. (1997) Involvement of a Caspase-3-like cysteine protease in 1-methyl-4-phenylpyridinium (MPP+) mediated apoptosis of cultured cerebellar granule neurons. J. Neurochem. 69, 1382–1388 [DOI] [PubMed] [Google Scholar]
- 42. Resink A., Hack N., Boer G. J., Balázs R. (1994) Growth conditions differentially modulate the vulnerability of developing cerebellar granule cells to excitatory amino acids. Brain Res. 655, 222–232 [DOI] [PubMed] [Google Scholar]
- 43. Du Y., Bales K. R., Dodel R. C., Hamilton-Byrd E., Horn J. W., Czilli D. L., Simmons L. K., Ni B., Paul S. M. (1997) Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 11657–11662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ciani E., Paulsen R. E. (1995) Activation of a reporter gene responsive to NGFI-B in cultured neurons and astrocytes. J. Mol. Neurosci. 6, 131–139 [DOI] [PubMed] [Google Scholar]
- 45. Sparapani M., Dall'Olio R., Gandolfi O., Ciani E., Contestabile A. (1997) Neurotoxicity of polyamines and pharmacological neuroprotection in cultures of rat cerebellar granule cells. Exp. Neurol. 148, 157–166 [DOI] [PubMed] [Google Scholar]
- 46. Kiedrowski L., Costa E., Wroblewski J. T. (1992) In vitro interaction between cerebellar astrocytes and granule cells: a putative role for nitric oxide. Neurosci. Lett. 135, 59–61 [DOI] [PubMed] [Google Scholar]
- 47. Weissmann C., Flechsig E. (2003) PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 66, 43–60 [DOI] [PubMed] [Google Scholar]
- 48. Hegde R. S., Mastrianni J. A., Scott M. R., DeFea K. A., Tremblay P., Torchia M., DeArmond S. J., Prusiner S. B., Lingappa V. R. (1998) A transmembrane form of the prion protein in neurodegenerative disease. Science 279, 827–834 [DOI] [PubMed] [Google Scholar]
- 49. Flego M., Ascione A., Zamboni S., Dupuis M. L., Imperiale V., Cianfriglia M. (2007) Generation of human scFvs antibodies recognizing a prion protein epitope expressed on the surface of human lymphoblastoid cells. BMC Biotechnol. 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weissmann C., Raeber A. J., Montrasio F., Hegyi I., Frigg R., Klein M. A., Aguzzi A. (2001) Prions and the lymphoreticular system. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 356, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Relkin N. R., Szabo P., Adamiak B., Burgut T., Monthe C., Lent R. W., Younkin S., Younkin L., Schiff R., Weksler M. E. (2009) 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol. Aging 30, 1728–1736 [DOI] [PubMed] [Google Scholar]