

Background: Neutrophil-specific membrane-type 6 matrix metalloproteinase (MT6-MMP)/leukolysin has seven known substrates.
Results: We identified 72 new MT6-MMP substrates by proteomics and family-wide chemokine screens. Cell membrane-bound vimentin chemoattracts macrophages, whereas MT6-MMP-cleaved vimentin is an “eat-me” signal greatly increasing phagocytosis.
Conclusion: MT6-MMP substrates indicate a role for clearance of apoptotic neutrophils.
Significance: MT6-MMP cleaves many bioactive proteins important in innate immunity.
Keywords: Chemokines, Inflammation, Matrix Metalloproteinase (MMP), Phagocytosis, Proteolytic Enzymes, Proteomics
Abstract
The neutrophil-specific protease membrane-type 6 matrix metalloproteinase (MT6-MMP)/MMP-25/leukolysin is implicated in multiple sclerosis and cancer yet remains poorly characterized. To characterize the biological roles of MT6-MMP, it is critical to identify its substrates for which only seven are currently known. Here, we biochemically characterized MT6-MMP, profiled its tissue inhibitor of metalloproteinase inhibitory spectrum, performed degradomics analyses, and screened 26 chemokines for cleavage using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. MT6-MMP processes seven each of the CXC and CC chemokine subfamilies. Notably, cleavage of the neutrophil chemoattractant CXCL5 activates the chemokine, thereby increasing its agonist activity, indicating a feed-forward mechanism for neutrophil recruitment. Likewise, cleavage also activated CCL15 and CCL23 to increase monocyte recruitment. Utilizing the proteomics approach proteomic identification of cleavage site specificity (PICS), we identified 286 peptidic cleavage sites spanning from P6 to P6′ from which an unusual glutamate preference in P1 was identified. The degradomics screen terminal amine isotopic labeling of substrates (TAILS), which enriches for neo-N-terminal peptides of cleaved substrates, was used to identify 58 new native substrates in fibroblast secretomes after incubation with MT6-MMP. Vimentin, cystatin C, galectin-1, IGFBP-7, and secreted protein, acidic and rich in cysteine (SPARC) were among those substrates we biochemically confirmed. An extracellular “moonlighting” form of vimentin is a chemoattractant for THP-1 cells, but MT6-MMP cleavage abolished monocyte recruitment. Unexpectedly, the MT6-MMP-cleaved vimentin potently stimulated phagocytosis, which was not a property of the full-length protein. Hence, MT6-MMP regulates neutrophil and monocyte chemotaxis and by generating “eat-me” signals upon vimentin cleavage potentially increases phagocytic removal of neutrophils to resolve inflammation.
Introduction
As a critical cell of the innate immune system, neutrophils store components that are required for transendothelial migration and antibacterial and proinflammatory activities (1). In inflammation, neutrophils release proteases, including neutrophil-specific membrane-type 6 MMP3 (MT6-MMP; MMP25; leukolysin) and MMP8 and antimicrobial peptides, they phagocytose pathogens and release soluble mediators, including proinflammatory cytokines and chemokines, to propagate the inflammatory response (2). Short lived, neutrophils die by apoptosis in an MMP8-regulated process (3). Upon display of “eat-me” proteins on the apoptotic cell membrane, phagocytosis by macrophages is promoted (4, 5) as a key event prior to tissue remodeling and resolution of inflammation. However, the mechanisms that target neutrophils for phagocytic clearance are not well understood nor is it clear how eat-me signals are generated from normal proteins.
Cellular recruitment of neutrophils and monocytes is dependent in part upon specific chemoattracting cytokines, termed chemokines, which are produced and released from resident and recruited cells. Of the two main chemokine subfamilies, CXC chemokines primarily recruit neutrophils, whereas CC chemokines are more important in the recruitment of monocytes. Proteolysis of chemokine termini results in significant functional changes (6) with MMP cleavage (7) having importance in modifying cell recruitment and inflammation in vivo (6).
The neutrophil chemoattractants human CXCL8 and CXCL5 and murine CXCL5/LIX are potently activated by stromal MMPs and neutrophil-specific MMP8 (8–10), whereas human CXCL1, -2, -3, -5, -6, -7, and -8 are inactivated by MMP1, -9, and macrophage-specific MMP12 (8). This leukocyte-MMP-directed regulation of neutrophil and monocyte chemokines led us to address the role of the poorly understood neutrophil-specific cell membrane MT6-MMP in processing chemokines for which just seven substrates, mainly extracellular matrix proteins, have been identified in the past 13 years since cloning (11, 12).
MT6-MMP is membrane-associated through a glycosylphosphatidylinositol anchor and contains a furin cleavage sequence for intracellular activation in the Golgi (11, 12). Enzymatic activity of MT6-MMP is regulated by the abundant serum protein clusterin (13) and by the tissue inhibitors of metalloproteinases (TIMPs) 1, 2, and 3 (14–16); notably the role of TIMP4 is unknown, but it is frequently associated with vascular tissue. MT6-MMP is localized primarily in neutrophil gelatinase granules but is also found in specific granules, secretory vesicles, and lipid rafts on the plasma membrane of resting cells (15, 17). Stimulation of neutrophils by CXCL8 and interferon-γ induces MT6-MMP release, whereas stimulation and induction of apoptosis by phorbol 12-myristate 13-acetate relocates MT6-MMP to the neutrophil surface (15, 17), suggesting that the enzyme functions differently at multiple stages of the inflammatory process. MT6-MMP function is implicated in development and disease by increased expression (18), but its few known substrates are limited to the usual ones tested for MMP activity, type IV collagen, gelatin, fibronectin, fibrin, α-1 proteinase inhibitor, urokinase plasminogen activator receptor, and myelin basic protein (14, 19–21), revealing little to distinguish it from other MMPs in its potential in vivo roles.
Identification of the substrate repertoire of a protease (the substrate degradome (22)) is critical to deciphering the biological role of proteases. We recently developed a proteomics approach termed terminal amine isotopic labeling of substrates (TAILS) to specifically enrich for the new N termini (termed neo-N termini) of cleaved substrates from a protease-treated proteome (23). The use of isobaric mass tags for relative and absolute quantification (iTRAQ) enables highly controlled experiments by multiplex mass spectrometry analyses (24). TAILS has enabled identification of many new substrates for proteases (23–25).
To explore the biological roles of MT6-MMP, we expressed and purified a soluble form of MT6-MMP. First, we evaluated the ability of MT6-MMP to cleave both neutrophil and monocyte chemoattractants in a hypothesis-directed approach. Using the human lung fibroblast secretome as a relevant proteome that might be encountered by migrating neutrophils, we then applied TAILS to identify MT6-MMP substrates in a hypothesis-generating proteomics screen. In total, 72 substrates were identified, and 19 new substrates were biochemically confirmed. The results of this research provide insight into the role of MT6-MMP in the potentiation and resolution of inflammation.
EXPERIMENTAL PROCEDURES
Proteins
Recombinant human MMP1, -2, -3, -8, -9, -12, -13, and soluble MMP14 and recombinant murine TIMP1, -2, and -4 were expressed and purified from mammalian systems (26). Recombinant MMP7 (United States Biochemical Corp.), human vimentin and galectin (R&D Systems), and DQ gelatin (Molecular Probes) were purchased. Chemokines and the small molecule MMP inhibitor marimastat were chemically synthesized, purified, and validated for activity as described (27, 28). Insulin-like growth factor-binding protein (IGFBP)-7 protein and antibody, and cystatin C were kindly provided by Drs. Kaoru Miyazaki (Yokohama City University, Japan) and Magnus Abrahamson (University of Lund, Lund, Sweden), respectively.
Recombinant Human MT6-MMP Protein Expression and Purification
We expressed and purified different soluble (s) forms of MT6-MMP (Fig. 1A), all with a FLAG tag, that are catalytically active upon purification (sMT6-MMP) and could be activated by 1 mm 4-aminophenylmercuric acetate (sMT6-MMPΔF) because the furin site had been replaced or that remain catalytically inactive (sMT6-MMP(E234A)). To do so, a HindIII site was introduced at the 5′-end of full-length MT6-MMP cDNA (kindly provided by Dr D. Pei, Guangzhou Institutes of Biomedicine and Health, China) using the forward primer 5′-CCGAAGCTTATGCGGCTGCGGCTCCGG-3′. A FLAG tag, EcoRI site, and stop sequence were exchanged with glycosylphosphatidylinositol anchor region residues starting after Gly514 to create sMT6-MMP using the reverse primer 5′-CGGGAATTCCTACTTGTCATCGTCGTCCTTGTAGTCACCAGAGCTCGGGGCGGG-3′. After PCR (35 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 60 s), the gel-purified product was digested with HindIII and EcoRI, gel-purified and A-tailed, and then ligated into pGEM-T.
FIGURE 1.

Preparation of MT6-MMP. A, domain structures of MT6-MMP proteins generated here by site-directed mutagenesis indicating the signal peptide (S; yellow), propeptide (Pro; blue) with a furin cleavage site (navy), catalytic domain (green), flexible linker (gray), hemopexin domain (red), and stalk and hydrophobic tail for glycosylphosphatidylinositol anchoring (ST; orange). The stalk was truncated, and a FLAG tag (black) was appended to create an sMT6-MMP construct. An additional mutation to sMT6-MMP was made to remove the furin cleavage site (sMT6-MMPΔF). Alternatively, a further mutation to sMT6-MMP was made to exchange the catalytic Glu residue to Ala, resulting in the catalytically inactive sMT6-MMP(E234A) protein. B, purified sMT6-MMP, sMT6-MMP(E234A), and sMT6-MMPΔF proteins were visualized under non-reducing (−DTT) or reducing conditions (+DTT) on silver-stained 15% SDS-PAGE. The differences in protein migration in these two conditions are due to differences arising from the effects of disulfide bonding under non-reduced compared with reduced conditions. In comparing these conditions in the wild type and inactive mutants, the different electrophoretic migrations of protein in the two preparations relate to the presence or absence of autolytic cleavage within the hemopexin domain. Thus, in the absence of catalytic activity in the sMT6-MMP(E234A) recombinant form, no autocatalytic cleavage occurs in the hemopexin domain, resulting in electrophoretic migration under nonreducing conditions consistent with a disulfide bond-constrained domain that migrates faster than when reduced or when cleaved and non-reduced in the wild type sMT6-MMP form. Edman sequencing of sMT6-MMP(E234A) showed the N terminus to be Tyr108 (arrow). Sequencing showed the N terminus of sMT6-MMPΔF to be Leu101 (double arrows) with a cleavage product occurring within the hemopexin domain at Leu348 (double gray arrows). Western blot analysis of purified mTIMP2 (T2), sMT6-MMP(E234A) (E234A), and sMT6-MMPΔF (ΔF) with a primary antibody to TIMP2 indicates the presence of TIMP2 in the sMT6-MMP(E234A) but not sMT6-MMPΔF preparation. C, schematic of sMT6-MMPΔF indicating the seven amino acids remaining at the N terminus upstream of the catalytic domain and cleavage point at Leu348 within the hemopexin domain.
The furin activation site of sMT6-MMP was mutated at 103RRRRR107 to 103GAGAG107, resulting in sMT6-MMPΔFurin (sMT6-MMPΔF) using the primers 5′-GGGGCTGGTCGGTGCCGGTGCCGTTACGCTGTCAG-3′ and 3′-CCCCGACCAGCCACGGCCACGGCCAATGCGAGACTC-5′ (loop-in region is indicated in bold) in 18 PCR cycles (95 °C for 30 s, 55 °C for 60 s, and 68 °C for 13.5 min). Separately, a catalytically inactive mutant (Glu234 to Ala) was generated using forward 5′-GGCTGTCCATGCGTTTGGCCACGCC-5′ and reverse 3′-CCGACAGGTACGCAAACCGGTGCGG-5′ primers, producing sMT6-MMP(E234A). All PCR products were confirmed by DNA sequencing.
After ligation into pGW1GH vector (generously provided by J. M. Clements, British Biotech Pharmaceuticals, Oxford, UK), the plasmids were electroporated into CHO-K1 cells. Clonal selection was with mycophenolic acid in DMEM supplemented with 10% Cosmic calf serum, hypoxanthine-thymidine supplement (Invitrogen), and xanthine. Positive expression in clones was screened for in 24-h conditioned medium by Western blots using the M2 αFLAG primary antibody (Sigma).
We devised a protein purification procedure for MT6-MMP that was similar for each construct. After stably transfected CHO-K1 cells reached 90% confluence in mycophenolic acid-containing medium the cells were washed with PBS three times, and the medium was replaced with CHO serum-free medium. Conditioned medium was then collected every 24 h for 5–10 days, clarified by centrifugation at 1500 × g for 10 min, and filtered using a 0.2-μm filter. A green Sepharose column (Sigma) in water was used as a first purification step; following washing, proteins were eluted with a 0.5–1.5 m NaCl gradient. After dialysis against Tris-buffered saline (pH 7.4), samples were loaded onto an αFLAG-agarose column and eluted with 0.1 mm αFLAG peptide (Sigma). Protein purity was confirmed by silver-stained 15% SDS-PAGE and by Western blotting using both M2 αFLAG and rabbit αMT6-MMP (ab39031, Abcam). Proteins were quantified by Bradford assay, and the activity of both sMT6 and sMT6-MMPΔF was quantified by active site titration using recombinant TIMP1 and a FRET-quenched fluorescent (QF) methylcoumarin (Mca) and 2,4-dinitrophenyl (Dpa) substrate, QF24 (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2), with excitation/emission at 320/405 nm.
Kinetic Evaluation of Fluorescent Substrates
Enzyme kinetics of sMT6-MMP, sMT6-MMPΔF, and sMT6-MMP(E234A) were determined using QF24 and QF35 (Mca-Pro-Leu-Ala-Nva-Dpa-Ala-Arg-NH2), and activity was quantified relative to the fluorescence of a standard curve of Mca. Increasing amounts of active site-titrated enzyme were added to the substrate, and the reaction kinetics were evaluated at an excitation/emission of 320/405 nm at 37 °C on a Polarstar Optima 96-well fluorometer (BMG Labtech). The initial velocity (Vi) was calculated by (ΔRFU/t) × [Mca])/RFUMca where RFU is the relative fluorescent units due to enzyme activity and t is time. This value was entered into the equation kcat/Km = Vi/([E][S]) to solve for kcat/Km.
Peptide-based Active Site Evaluation of MT6-MMP by Proteomics
Proteomic identification of cleavage site specificity (PICS) (29, 30) was performed using 200 μg of a proteome-derived database-searchable peptide library (prepared from K562 cell (ATCC CCL-243) lysate by tryptic digestion followed by trypsin inactivation and blocking of all primary amines by dimethylation). After incubation with active sMT6-MMPΔF (0.8 μg; corresponding to a 1:250 (w/w) ratio in a total reaction volume <400 μl) in 50 mm HEPES, 10 mm CaCl2, 200 mm NaCl, pH 7.8 for 16 h at 37 °C, the PICS assay was halted by heat inactivation (70 °C for 30 min). Cleaved prime-side peptide products were biotinylated at the newly generated primary amine with 0.5 mm sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (Pierce) for 2 h at 22 °C. Biotin-labeled peptides were then bound to streptavidin-Sepharose (GE Healthcare) equilibrated in 50 mm HEPES, 150 mm NaCl, pH 7.4 for 16 h at 22 °C. Unbound peptides were removed by buffer washing under centrifugation (1,000 × g for 1 min) in spin columns (Pierce). Bound peptides were eluted with 40 mm DTT in 50 mm HEPES. Impurities were removed with C18 Sep-Pak cartridges, eluting with 80% acetonitrile, and the volume was decreased under vacuum centrifugation. Cleaved peptides were analyzed by LC-MS/MS on a QStar XL Hybrid electrospray ionization mass spectrometer (Applied Biosystems). Wiff files were searched with both Mascot and X! Tandem, the union of the peptides was analyzed by WebPICS (31), and IceLogos defining specificity were then generated (32).
Inhibition of MT6-MMP
Enzyme was incubated in the presence of increasing concentrations of recombinant murine TIMP1, TIMP2, TIMP4, or marimastat for 2 h at 37 °C before the addition of QF24. The kinetics of the reactions were assessed at 37 °C on a Polarstar Optima 96-well fluorometer. Data were imported, and Ki values were calculated in Prism (GraphPad).
Cleavage Assays
In vitro native substrate cleavage assays performed at enzyme to substrate ratios of 1:10 (w/w) of chemokines, vimentin, IGFBP-7, cystatin C, galectin-1, and secreted protein, acidic and rich in cysteine (SPARC) by sMT6-MMPΔF were performed in a 10-μl reaction containing 50 mm HEPES, 200 mm NaCl, 5 mm CaCl2, pH 7.4 for 16 h at 37 °C. Chemokine cleavage assay products were analyzed by MALDI-TOF mass spectrometry (MS) on a Voyager-DE STR (Applied Biosystems) using sinapinic acid matrix (33) and confirmed by silver-stained 15% Tris-Tricine SDS-PAGE. Chemokine cleavage was defined to be positive when the mass spectrometry spectra showed a cleavage product with >20% ion intensity of the full-length chemokine. Cystatin C processing was confirmed by silver-stained 15% Tris-Tricine SDS-PAGE. Vimentin, IGFBP-7, galectin-1, and SPARC cleavages were confirmed by silver-stained 15 or 7.5% Tris-glycine SDS-PAGE. Gelatin and casein processing were evaluated by zymography of gels polymerized with 0.2 mg/ml proteins. MMP processing of 25 μg/ml DQ gelatin was evaluated by an increase in fluorescence at an excitation/emission of 495/515 nm on a Polarstar Optima 96-well fluorometer.
HFL-1 Secretome Preparation
Human fetal lung fibroblast-1 (HFL-1) cells (obtained from Dr. C Roberts, University of British Columbia, Vancouver, Canada) were grown to 90% confluence in DMEM containing 10% fetal calf serum. Cells were washed with PBS three times and then with serum-free medium two times before adding fresh serum-free DMEM. After 16 h, conditioned medium was clarified by centrifugation at 1,500 × g for 10 min and filtered with a 0.2-μm filter. Conditioned medium was concentrated and buffer-exchanged to 50 mm HEPES by ultracentrifugation using 3-kDa-cutoff membranes (Amicon). The protein concentration was measured by Bradford assay (Bio-Rad).
TAILS of MT6-MMP
For proteomics screening to discover MT6-MMP native substrates, we used TAILS (23). Active sMT6-MMPΔF or sMT6-MMP(E234A) as control was added to concentrated, serum-free HFL-1 secretome at a 1:250 (w/w) ratio in 50 mm HEPES, 150 mm NaCl, 5 mm CaCl2 in a 541-μl volume and incubated for 16 h at 37 °C. To enrich for full-length protein N-terminal and neo-N-terminal peptides, the resulting cleavage products were subjected to TAILS as a two-plex iTRAQ experiment (23, 24). Briefly, guanidine hydrochloride and HEPES were added to the HFL-1 cleavage assay products to a final concentration of 2.5 m and 250 mm, respectively, to denature proteins. Cysteines were reduced with 1 mm tris(2-carboxyethyl)phosphine at 65 °C and alkylated using 5 mm iodoacetamide. Whole protein iTRAQ labeling from the sMT6-MMPΔF or sMT6-MMP(E234A) digested proteomes was achieved in 50% DMSO with iTRAQ labels 114 and 115, respectively. After 30 min at 25 °C, the reaction was quenched with 100 mm ammonium bicarbonate. Labeled samples were combined and precipitated with 9 volumes of acetone/methanol (8:1, v/v) at −20 °C. The protein precipitate was pelleted by centrifugation at 2,500 × g for 30 min at 4 °C, washed with methanol, and resuspended in 50 mm HEPES for digestion with TrypsinGold (Promega) at a 1:100 enzyme/protein ratio for 16 h at 37 °C. Complete digestion was confirmed by silver-stained 15% SDS-PAGE. To enrich for N termini, peptides were incubated under acidic and reductive conditions at 37 °C for 16 h with a dendritic polyglycerol aldehyde polymer (Flintbox); internal and C-terminal peptides react to the aldehyde-derivatized polymer. N-terminally blocked peptides (due to iTRAQ labeling or natural acetylation or cyclization) were recovered from the unbound fraction by ultracentrifugation using 10,000-kDa-cutoff membranes (Amicon) as described in full (34).
The enriched N-terminome samples were fractionated by strong cation exchange-HPLC using a polysulfoethyl A column (100 × 4.6 mm, 5 μm, 300 Å; PolyLC Inc.) on a 1200 series HPLC (Agilent Technologies). Bound peptides were washed for 15 min in 10 mm potassium phosphate, 25% acetonitrile, pH 2.7. Peptides were eluted over a 22-min gradient to 0.3 m NaCl, then 6 min to 0.4 m NaCl, and 2 min to reach 1 m NaCl. 16 fractions, collected every 1.5 min, were concentrated under vacuum, desalted using C18 OMIX tips, and combined based on HPLC relative peak height. Peptide samples (eight fractions) were analyzed by nanospray LC-MS/MS using a C18 column interfaced with a QStar XL mass spectrometer.
TAILS Data Analysis
Acquired MS2 scans were searched by both Mascot (version 2.2.2, Matrix Science) and X! Tandem (July 1, 2007 release) against the human International Protein Index protein database (version 3.69). Search parameters were as follows: semi-Arg-C cleavage specificity with up to two missed cleavages; fixed modifications of cysteine carbamidomethylation and lysine iTRAQ; variable modifications of N-terminal iTRAQ, N-terminal acetylation, and methionine oxidation; peptide tolerance and MS/MS tolerance at 0.4 Da; and scoring scheme of ESI-QUAD-TOF. Search results were statistically modeled using Peptide Prophet and iProphet of the TransProteomic Pipeline (35) (version 4.3, revision 0, Build 200902191420) and Libra for quantification of iTRAQ reporter ion intensities. Each data set included only the peptides with an iProphet probability error rate ≤0.05. Using in-house software, CLIPPER (27), data sets were converted to a common format using ClipperConvert. Mascot and X! Tandem lists were combined for each experiment and analyzed as a single experiment or in tandem by CLIPPER. The data were normalized using a correction factor obtained by analysis of the average log2(ratio) natural N-terminal peptides. Candidate substrates were those having a ratio ≥2 standard deviations from the mean. The highest confidence substrates were proteins identified by the same peptide found in two biological replicate experiments. High confidence substrates were identified by multiple spectra of multiple forms of the peptide in one experiment, whereas candidate substrates requiring biochemical validation were identified by peptides identified by one unambiguous spectrum.
Transwell Migration Assays
The chemotactic potential of full-length and MMP-cleaved vimentin and chemokines for THP-1 cells (ATCC) was evaluated by Transwell migration (33). The relative number of cells migrating to the lower chamber was determined by CyQUANT reagent (Invitrogen), and fluorescence was evaluated by excitation/emission at 485/538 nm. The chemotactic index was calculated by the ratio of the relative fluorescence of samples from cells migrating in response to stimuli compared with medium as a control. Experiments were carried out in at least quadruplicate and repeated twice. Statistical significance of cleaved versus full-length vimentin was evaluated by t test.
Phagocytosis Assay
Fluoresbrite® microparticles (Polysciences, Inc., Warrington, PA) were incubated with test stimuli in the presence of normal human serum for 30 min at 37 °C and then washed twice with Hanks' balanced saline solution. THP-1 cells were differentiated with 200 ng/ml phorbol 12-myristate 13-acetate for 24 h. Cells were combined with the coated microparticles for 1 h. Phagocytosis was stopped by the addition of ice-cold Hanks' balanced saline solution, and the cells were washed free of coated microparticles twice with Hanks' balanced saline solution, trypsinized, and washed. Phagocytosis of the coated microparticles was analyzed by FACS on a BD FACSCanto II with BD FACSDiva software (BD Biosciences). Statistical significance of cleaved versus full-length vimentin was evaluated by t test.
RESULTS
Expression of Recombinant sMT6-MMP, sMT6-MMPΔF, and sMT6-MMP(E234A)
On SDS-PAGE analysis, the main recombinant sMT6-MMP band electrophoresed at the expected position with some minor bands resulting from autoactivation (Fig. 1B) that, as expected, were absent in the preparation of the inactive sMT6-MMP(E234A). The amino acid sequence 108YALSG at the start of the 57-kDa (+DTT) sMT6-MMP(E234A) (Fig. 1B) is the correct start of the catalytic domain, indicating that furin cleavage and removal of the prodomain occurs normally. The lower band copurifying with sMT6-MMP(E234A) in equimolar concentrations was identified by Western blot to be TIMP2 (Fig. 1B), consistent with previous studies that identified TIMP2 complexed with mature MT6-MMP (16).
We also generated sMT6-MMPΔF that has the furin activation site deleted to have a form that can be activated by 1 mm 4-aminophenylmercuric acetate and for increased stability during purification and storage. sMT6-MMPΔF migrated as an ∼57-kDa doublet (−DTT) likely representing glycosylation variants (36) and at ∼37 and 30 kDa when reduced (Fig. 1B). In the absence of the furin cleavage site, the prodomain was still removed intracellularly at 100G↓L (Fig. 1A). Under reducing conditions, the bands at ∼30 kDa have the N-terminal sequence 348LVSPR352, representing a cleavage within the hemopexin domain (Fig. 1, B and C) as observed previously (13). Thus, the disulfide-bonded (Cys317-Cys508) hemopexin domain of sMT6-MMPΔF is cleaved with the C-terminal fragment being retained to the catalytic domain by the disulfide bond (Fig. 1C). The lack of hemopexin domain cleavage in sMT6-MMP(E234A) suggests that this is an autocatalytic event.
Conventional Substrate and Peptide Activities
Using the ΔFurin form of the enzyme, we found that the gelatinolytic activity of sMT6-MMP is virtually absent as shown by zymography (Fig. 2A) and by using DQ gelatin substrate (Fig. 2B). Similar weak activity in casein zymograms was also found (Fig. 2C).
FIGURE 2.

Characterization of MT6-MMP protein. A, gelatinase activity of sMT6-MMPΔF was evaluated by gelatin zymography. 10 ng of MMP-9 resulted in a clear band (arrow), whereas 100 and 1,000 ng of sMT6-MMPΔF resulted in only a slight clearance (arrowhead), indicating weak gelatinolytic activity (n = 2). B, quenched fluorescein-conjugated gelatin processing by sMT6-MMPΔF compared with MMP-2 processing was monitored at an excitation/emission of 485/530 nm (n = 2). C, casein processing by sMT6-MMPΔF was evaluated by zymography. 10 ng of MMP-7 resulted in a clear band (arrow), whereas 100 and 1,000 ng of sMT6-MMPΔF resulted in only a slight clearance (arrowhead), indicating that casein is not a good substrate for MT6-MMP (n = 2). D, the rate of cleavage of QF24 substrate by increasing concentrations of MMPs was compared by measurement of the increase in fluorescence at an excitation/emission of 320/405 nm (n = 2). E, heat map of the amino acid occurrence at positions P6 to P6′ of the 286 peptides analyzed and identified by MS/MS following PICS analysis of sMT6-MMPΔF cleavage of a tryptic library. F, IceLogo analysis of the PICS data, adjusting for relative abundance of the amino acids in the Homo sapiens proteome, indicating the relative occurrence of amino acids at positions P6 to P6′ from the 286 peptides analyzed and identified. G, inhibition of 3 nm sMT6-MMPΔF processing of 1.0 μm QF24 by TIMP1, -2, and -4 and by marimastat was evaluated by measurement of fluorescence at an excitation/emission of 320/405 nm, and the Ki was calculated in Prism (GraphPad) (n = 3). RFU, relative fluorescent units.
Using quenched fluorescent substrates, the kcat/Km of sMT6-MMPΔF was calculated to be 171 m−1 s−1 for QF24 and 36 m−1 s−1 for QF35; both values are considerably lower than that for sMT1-MMP (results not shown). As expected, sMT6-MMP(E234A) had no detectable activity (not shown). Cleavage rates of these FRET peptides by sMT6-MMPΔF were less than that of sMT6-MMP (Fig. 2D) possibly due to the extra seven amino acid residues at the N terminus. This may lead to an absence of a stabilizing potential salt bridge between the N-terminal primary amine and the carboxylate group of the conserved Asp232 in helix C as shown for MMP-8 (37). The sMT6-MMPΔF protein showed the expected improved stability and activity following storage at −80 °C as compared with sMT6-MMP and so was used in most experiments.
Proteomic Identification of Cleavage Site Specificity
The low activity of MT6-MMP against the conventional substrates commonly used to profile MMPs indicated that these were neither optimal nor natural substrates. Therefore, using a K562 cell proteome tryptic library, we used PICS, which enables the simultaneous detection of prime- and nonprime-side cleavage residues by mass spectrometry (29, 30). In total, 286 cleavage sites were identified from P6 to P6′ that showed a strong preference for Leu at P1′ and Val at P2′ and preferences for Pro/Val at P3 and Ala/Glu at P1 (Fig. 2E). After adjusting for the natural amino acid abundance, the IceLogo of the PICS data confirmed this and also revealed preferences for Asn in P1 and P2 (Fig. 2F). Notably, MT6-MMP has a preference for glutamate at P1, an unusual preference for MMPs. Thus, the reduced activity of MT6-MMP for standard MMP QF substrates is consistent with the different preferences observed by PICS analysis, specifically the preference for Gly at P1 by several MMPs that is not preferred for MT6-MMP but is present in QF24.
TIMP Inhibition
English et al. (14) found TIMP2 and TIMP3 to be much stronger inhibitors than TIMP1 of an MT6-MMP form consisting of the catalytic domain alone. However, other groups observed similar inhibition by TIMP1 and TIMP2 of both the catalytic domain and sMT6-MMP forms (13, 16). We found that TIMP1 and the previously untested inhibitor TIMP4 were more potent inhibitors of sMT6-MMPΔF than TIMP2, and all were stronger than the small molecule inhibitor marimastat (Fig. 2G).
Chemokine Processing by MT6-MMP
To evaluate the role of the neutrophil-specific MT6-MMP in modulating inflammatory cell recruitment, we evaluated its cleavage specificity for 26 chemokines. Substrate selectivity was observed in that cleavage sites were identified in seven of each of the subfamilies of CXC and CC chemokines (Fig. 3, A and B), whereas 12 chemokines were not processed by MT6-MMP. Cleavage of human but not murine (m) CXCL2 resulted in a product lacking the four N-terminal residues (Fig. 3C) as is common for many MMP-cleaved chemokines (38). Notably the N-terminal sequence of murine CXCL2, AVVASELR, differs significantly from human. Cleavage was also not observed in the related chemokines CXCL1 and CXCL3, which have Ser-Val residues in place of Pro-Leu at P3-P2 of CXCL2, consistent with PICS data (Fig. 2, E and F). Both human and murine CXCL5 were processed by MT6-MMP at the N terminus. Although mCXCL5(10–92) had not been previously identified, the products hCXCL5(8–78) and mCXCL5(5–92) were shown previously to have increased agonist activity (9, 10, 39, 40).
FIGURE 3.

MT6-MMP selectively processes both CXC and CC chemokines. Human or mouse chemokines (1 μg) were incubated at 37 °C for 16 h with recombinant sMT6-MMPΔF in a 10-μl reaction at an enzyme to substrate ratio of 1:10 (w/w). Cleavage assay products of CXCL (A) or CCL (B) chemokines were visualized on silver-stained 15% Tris-Tricine gels. Cleavages of the chemokines listed were not detectable by MALDI-TOF following incubation at a 1:20 molar ratio with sMT6-MMPΔF at 37 °C for 16 h (n > 2). C, cleavage products were assigned by MALDI-TOF mass spectrometry by comparison of measured with predicted mass to charge ratios (m/z) with +1 charge ionization ([M + H]+). Cleavage sites are indicated by an arrow in the corresponding sequence.
CXCL6 was cleaved to a product that lacks the first 28 residues; cleavage by MMPs beyond the conserved cysteine residues is rarely observed outside of degradation, yet this product was observed at all enzyme to chemokine ratios and was the only product also found with eight other MMPs (results not shown). The MT6-MMP C-terminal truncation product CXCL9(1–90) was previously observed following MMP7, -9, and -12 activity (10, 41). Processing of CXCL12α by MT6-MMP showed results consistent with those of other MMPs, removing four N-terminal residues to result in a loss of CXCL12 activity on CXCR4, a switch in receptors from CXCR4 to CXCR3, and increased neurotoxicity (42, 43).
Seven CC chemokines were processed by MT6-MMP (Fig. 3B). The removal of four N-terminal residues from CCL2, CCL7, and CCL13 by MT6-MMP is consistent with the previous findings of processing by multiple MMPs; these products are potent receptor antagonists (7, 8, 38). CCL4(1–69) was cleaved to the product CCL4(7–69), which is known to reduce dimerization (44). CCL15 and CCL23 were cleaved at multiple sites by MT6-MMP in their extended N termini, thereby activating these chemokines (45). Truncation of the first four residues of CCL16 has not been reported previously for any MMP.
TAILS Proteomics Analysis to Identify MT6-MMP Cellular Substrates
Secretomes from HFL-1 cells were incubated either with sMT6-MMPΔF (and labeled with iTRAQ 114) or with the catalytically inactive sMT6-MMP(E234A) as control (and labeled with iTRAQ 115) to identify substrates for MT6-MMP. Samples from two biological replicates (Experiments 1 and 2) were processed by TAILS to purify their N-terminomes (Fig. 4A). From TAILS1, 312 and 224 unique peptides at >95% confidence were identified by Mascot and X! Tandem, respectively, after statistical modeling using the TransProteomic Pipeline. The union yielded 407 unique and high confidence N-terminal and neo-N-terminal peptides (Fig. 4B). From TAILS2, Mascot and X! Tandem searches identified 224 and 166 unique peptides with >95% confidence, respectively, for a combined total of 300 unique identifications (Fig. 4B). 89 peptides were common to both experiments (Fig. 4C, supplemental Fig. S1, and supplemental Table I). Separate CLIPPER analysis, which has extremely stringent requirements for the quality of the spectra used to assign peptides, identified an additional 219 and 112 peptides in the TAILS1 and TAILS2 data sets that were not common to TAILS1 and TAILS2 (Fig. 4C and supplemental Tables II and III).
FIGURE 4.

Peptide and substrate identification by TAILS analysis. A, schematic overview of the method of the TAILS analysis of conditioned medium from cells treated with sMT6-MMPΔF or sMT6-MMP(E234A). B, four-way Venn diagram of unique peptide identifications with ≥95% confidence by Mascot and X !Tandem in TAILS1 and TAILS2. C, two-way Venn diagram of unique peptides that have reached the experimental cutoff for that data set identified in TAILS1, TAILS2, or both. SCX, strong cation exchange.
As expected, the abundance ratios of N-terminal semitryptic peptides derived from the natural N terminus of full-length proteins (±N-terminal methionine or ±signal peptide) are unchanged and equally distributed in both the sMT6-MMPΔF- and sMT6-MMP(E234A)-treated secretomes (supplemental Fig. S1). The distribution of unblocked (now labeled by iTRAQ) and naturally acetylated termini were similar, ∼50% (supplemental Fig. S2), as found previously by TAILS of secretomes (23, 24).
MT6-MMP substrate cleavage results in an iTRAQ ratio >1 (114(sMT6-MMPΔF)/115(sMT6-MMP(E234A)), whereas a ratio <1 indicates cleavage within and loss of the N terminus. For increased confidence in the substrates identified, the value of the cutoff ratio for each experiment was set at ≥2 standard deviations from the mean. Ultimately, 171 unique peptides from 58 proteins met all criteria for inclusion. Proteins that were identified in biological replicate experiments were considered to be the highest confidence candidate substrates of MT6-MMP (Table 1), and those identified by ≥2 different spectra were high confidence substrates (Table 2), whereas those identified by one form of a peptide were considered as candidate substrates of MT6-MMP (Table 3) requiring biochemical validation. Notably, where partially overlapping sequences from multiple cleavages are observed, the site of MT6-MMP processing may be obscured by subsequent aminopeptidase activity as is a limitation of proteomics experiments that evaluate a proteome in the native state.
TABLE 1.
Highest confidence substrates of MT6-MMP
Substrates were identified by the same peptide in biological replicates following HFL secretome treatment with sMT6-MMPΔF (114 label) or sMT6-MMP(E234A) (115 label) having a 114/115 ratio of <0.71 or >1.4 based upon a standard deviation of 0.24. Thus, these peptides represent the C-terminal cleavage product after cleavage by MT6-MMP.
| Protein | Ratio 114/115 | Peptide |
|---|---|---|
| Actin | 0.64 | 20GFAGDDAPR |
| 2.22 | 29AVFPSIVGR | |
| 11.14 | 105LTEAPLNPKANR | |
| 4.41 | 298VLSGGTTMYPGIADR | |
| 4.53 | 6LVVDNGSGMCKAGFAGDDAPRa | |
| 26.19 | 43VMVGMGQKDSYVGDEAQSKRb | |
| 21.62 | 52SYVGDEAQSKRb | |
| Collagen type I α-1 chain | 10.29 | 1001GPPGLAGPPGESGR |
| 31.73 | 977GPSGEPGKQGPSGASGERb | |
| 10.98 | 1004GLAGPPGESGRb | |
| 36.17 | 1005LAGPPGESGRb | |
| 42.57 | 1006AGPPGESGRb | |
| 12.67 | 1050APGAPGPVGPAGKSGDRb | |
| 12.27 | 1052GAPGPVGPAGKSGDRb | |
| 0.36 | 1075AGPVGPVGARb | |
| 6.55 | 1237SLSQQIENIRb | |
| 23.76 | 1239SQQIENIRb | |
| Collagen type I α-2 chain | 1.42 | 868GAPGILGLPGSR |
| 2.33 | 333VGAAGATGAR | |
| 4.68 | 1141SLNNQIETLLTPEGSR | |
| 17.94 | 113FQGPAGEPGEPGQTGPAGARb | |
| 14.84 | 115GPAGEPGEPGQTGPAGARb | |
| 0.65 | 121GEPGQTGPAGARa | |
| 20.15 | 331GPVGAAGATGARb | |
| 48.93 | 370GPPGPSGEEGKRb | |
| 1.48 | 385GEAGSAGPPGPPGLRa | |
| 43.99 | 559GPSGPAGEVGKPGERb | |
| 7.94 | 784GMTGFPGAAGRb | |
| 23.30 | 808GPPGPAGKEGLRb | |
| 3.10 | 868GAPGILGLPGSRb | |
| 24.51 | 967GPVGPAGKHGNRb | |
| 38.25 | 1103VSGGGYDFGYDGDFYRb | |
| 10.94 | 1109DFGYDGDFYRb | |
| 14.89 | 1110FGYDGDFYRb | |
| 29.73 | 1111GYDGDFYRb | |
| 27.86 | 1112YDGDFYRb | |
| 20.83 | 1144NQIETLLTPEGSRb | |
| 39.22 | 1142LNNQIETLLTPEGSRb | |
| 25.98 | 1143NNQIETLLTPEGSRb | |
| 42.37 | 1146IETLLTPEGSRb | |
| 22.07 | 1149LLTPEGSRb | |
| Cystatin C | 5.54 | 35LVGGPMDASVEEEGVRR |
| 26.71 | 35LVGGPMDASVEEEGVR | |
| 26.35 | 36VGGPMDASVEEEGVRb | |
| 7.48 | 37GGPMDASVEEEGVRb | |
| 31.22 | 42ASVEEEGVRb | |
| IGFBP-5 | 0.63 | 164KFVGGAENTAHPR |
| 2.97 | 213AVYLPNCDR | |
| 0.18 | 165FVGGAENTAHPRb | |
| 0.29 | 166VGGAENTAHPRa | |
| IGFBP-7 | 1.46 | 98AGAAAGGPGVSGVCVCKSR |
| 2.42 | 98AGAAAGGPGVSGVCVCKSRb | |
| 6.72 | 99GAAAGGPGVSGVCVCKSRb | |
| 32.03 | 100AAAGGPGVSGVCVCKSRb | |
| MT6-MMP | 8.38 | 318EGNFDAIANIR |
| 36.59 | 29VSLGVDWLTRa | |
| 8.67 | 254FYQGPVGDPDKYRa | |
| 4.41 | 255YQGPVGDPDKYRa | |
| 23.62 | 359FWEGLPAQVRb | |
| 8.73 | 386SGPQFWVFQDRa | |
| 17.25 | 473GDTYFFKGAHYWRa | |
| Transgelin | 0.66 | 2ANKGPSYGMSR |
| Tropomyosin α-4 chain | 3.56 | 133LVILEGELER |
| 0.68 | 16ALQQQADEAEDRa | |
| 19.62 | 115AKHIAEEADRb | |
| 15.24 | 134VILEGELERb | |
| Vimentin | 0.58 | 55SSPGGVYATR |
| 2.05 | 87SLADAINTEFKNTR | |
| 1.55 | 88LADAINTEFKNTR | |
| 0.30 | 54ASSPGGVYATRb | |
| 0.65 | 91AINTEFKNTRa | |
| 35.77 | 114FANYIDKVRb | |
| 0.39 | 258VDVSKPDLTAALRb | |
| 8.83 | 295FADLSEAANRb | |
| 2.69 | 296ADLSEAANRb | |
| 4.21 | 330VDALKGTNESLERb | |
| YIPF3 | 0.71 | 321DIPAMLPAAR |
| 26 S protease regulatory subunit 8 | 1.82 | 2ALDGPEQMELEEGKAGSGLR |
a Additional peptide identification in TAILS1.
b Additional peptide identification in TAILS2.
TABLE 2.
High confidence substrates of MT6-MMP
Substrates were identified by ≥2 spectra, including multiple charge states of modifications of the peptide, following HFL secretome treatment with sMT6-MMPΔF (114 label) or sMT6-MMP(E234A) (115 label) having a 114/115 ratio of <0.68 or >1.46 based upon a standard deviation of 0.27.
| Protein | Ratio 114/115 | Peptide | No. of spectra |
|---|---|---|---|
| α-Actinin-1 | 24.56 | 13MQPEEDWDRa | 2 |
| Collagen type II α-1 chain | 43.76 | 576GPAGPAGERa | 2 |
| 8.59 | 825GATGFPGAAGRa | 2 | |
| Collagen type IV α-2 chain | 6.84 | 76GLQGFPGLQGRa | 1 |
| 20.66 | 461FPGLPGSPGARa | 1 | |
| 32.24 | 651GPAGTPGQIDCDTDVKRa | 1 | |
| 5.81 | 655TPGQIDCDTDVKRa | 1 | |
| Collagen type VI α-1 chain | 1.71 | 443GDPGEAGPQGDQGRb | 1 |
| 16.85 | 497GPPGDPGLMGERa | 2 | |
| 7.81 | 500GDPGLMGERa | 1 | |
| Collagen type VI α-3 chain | 2.96 | 3106LTETDICKLPKDEGTCRa | 1 |
| Dickkopf-related protein 3 | 33.89 | 131TVITSVGDEEGRa | 2 |
| 15.46 | 54VEELMEDTQHKLRa | 1 | |
| 51.66 | 252ITWELEPDGALDRa | 1 | |
| Extracellular matrix protein 1 | 0.15 | 20ASEGGFTATGQRa | 3 |
| Fibrillin-1 | 0.37 | 25ADANLEAGNVKETRa | 2 |
| Latent transforming growth factor β-binding protein 2 | 1.48 | 1729FEGLQAEECGILNGCENGRb | 1 |
| 5.36 | 249SSAAGEGTLARa | 1 | |
| MMP-1 | 4.98 | 328LEAAYEFADRb | 2 |
| 7.30 | 364IYSSFGFPRb | 1 | |
| Peptidyl-prolyl cis-trans isomerase A | 7.88 | 9DIAVDGEPLGRa | 3 |
| 8.52 | 8FDIAVDGEPLGRa | 1 | |
| Procollagen C-endopeptidase enhancer 1 | 37.52 | 309SPSAPDAPTCPKQCRa | 2 |
| Protein-lysine 6-oxidase | 0.61 | 59SLGSQYQPQRb | 2 |
| 4.75 | 57LLSLGSQYQPQRb | 2 | |
| 0.39 | 22APPAAGQQQPPRa | 1 | |
| 2.12 | 58LSLGSQYQPQRb | 1 | |
| Serpin H1 | 0.23 | 139SVSFADDFVRb | 2 |
| Sulfhydryl oxidase 1 | 42.01 | 565AMGALELESRa | 7 |
| 32.69 | 566MGALELESRa | 2 | |
| Stromal cell-derived factor 4 | 12.27 | 84GKDLGGFDEDAEPRa | 1 |
| 7.94 | 88GGFDEDAEPRa | 1 | |
| Syndecan-4 | 7.00 | 24TEVIDPQDLLEGRa | 2 |
| 7.49 | 26VIDPQDLLEGRa | 1 | |
| Tropomyosin 3 | 6.84 | 170LVIIEGDLERa | 1 |
| 38.06 | 171VIIEGDLERa | 2 | |
| Tropomyosin β chain | 41.62 | 22AEQAEADKKQAEDRa | 2 |
| 48.86 | 24QAEADKKQAEDRa | 1 | |
| 17.45 | 74AEKKATDAEADVASLNRa | 1 | |
| 23.20 | 151AKHIAEDSDRa | 1 |
a Additional peptide identification in TAILS2.
b Additional peptide identification in TAILS1.
TABLE 3.
Candidate substrates of MT6-MMP
Substrates were identified by one spectrum following HFL secretome treatment with sMT6-MMPΔF (114 label) or sMT6-MMP(E234A) (115 label) having a 114/115 ratio of <0.39 or >2.4 based upon a standard deviation of 0.6.
| Protein | Ratio 114/115 | Peptide |
|---|---|---|
| α2-Macroglobulin | 8.24 | 708YESDVMGRa |
| Calumenin precursor | 10.54 | 53LGAEEAKTFDQLTPEESKERb |
| Cellular nucleic acid-binding protein | 50.29 | 45FVSSSLPDICYRb |
| Dihydrolipoyllysine-residue succinyltransferase | 0.66 | 51DDLVTVKTPAFAESVTEGDVRa |
| Elongation factor 1-δ | 2.58 | 60SLAGSSGPGASSGTSGDHGELVVRb |
| Fibulin-1 | 36.94 | 170EQEDPYLNDRb |
| Filamin-C | 4.27 | 2304FTVGPLGEGGAHKVRb |
| Galectin-1 | 3.22 | 35LGKDSNNLCLHFNPRb |
| Galectin-3-binding protein | 9.49 | 447FQAPSDYRb |
| Gelsolin precursor | 1.47 | 404GLGLSYLSSHIANVERa |
| Glyceraldehyde-3-phosphate dehydrogenase | 6.84 | 4VKVGVNGFGRa |
| Nestin | 5.54 | 1397LLDPAAWDRa |
| Polymerase I and transcript release factor | 11.70 | 344VGADDDEGGAERb |
| Protein-disulfide isomerase A6 precursor | 0.45 | 72LYSSSDDVIELTPSNFNRa |
| Protein-lysine 6-oxidase | 0.25 | 56SLLSLGSQYQPQRb |
| Sodium-dependent phosphate transport protein | 51.66 | 319ISSVLQANLRb |
| SPARC | 0.63 | 156LDSELTEFPLRa |
| Spondin-2 | 25.78 | 251FIPPAPVLPSRb |
| Sulfhydryl oxidase 1 precursor | 51.66 | 34ALYSPSDPLTLLQADTVRb |
| Transcription elongation factor SPT4 | 0.65 | 2ALETVPKDLRa |
| Tubulin α-4A chain | 6.46 | 92LITGKEDAANNYARa |
| VEGF C | 1.52 | 112AHYNTEILKSIDNEWRa |
| V-type proton ATPase subunit G 1 | 2.03 | 2ASQSQGIQQLLQAEKRa |
| WD repeat-containing protein 1 | 6.84 | 8VFASLPQVERb |
| 40 S ribosomal protein SA | 4.33 | 90FAAATGATPIAGRb |
| 5,6-Dihydroxyindole-2-carboxylic acid oxidase | 21.06 | 425FPLENAPIGHNRb |
| 60 S ribosomal protein L26-like 1 | 0.57 | 1MKFNPFVTSDRa |
a Peptide identification in TAILS1.
b Peptide identification in TAILS2.
Validation of TAILS Candidate Substrates
In total, 58 substrates were identified by TAILS, and we biochemically confirmed five of these, namely cystatin C, IGFBP-7, galectin-1, vimentin, and SPARC. By SDS-PAGE, the shift of cystatin C following MT6-MMP processing is evident and complete (Fig. 5A). From the TAILS data, cystatin C cleavage occurs at the same site, 34R↓L, as for MMP2, which we showed reduces its cathepsin inhibitory activity (46). MT6-MMP cleavage of IGFBP-7 was also confirmed (Fig. 5B). IGFBP-7 was previously identified as an MMP-2 substrate by iTRAQ degradomics (47) but was not biochemically confirmed. Related to IGFBP-7, IGFBP-1, -2, -3, -4, and -6 are known MMP substrates (24, 46, 48).
FIGURE 5.
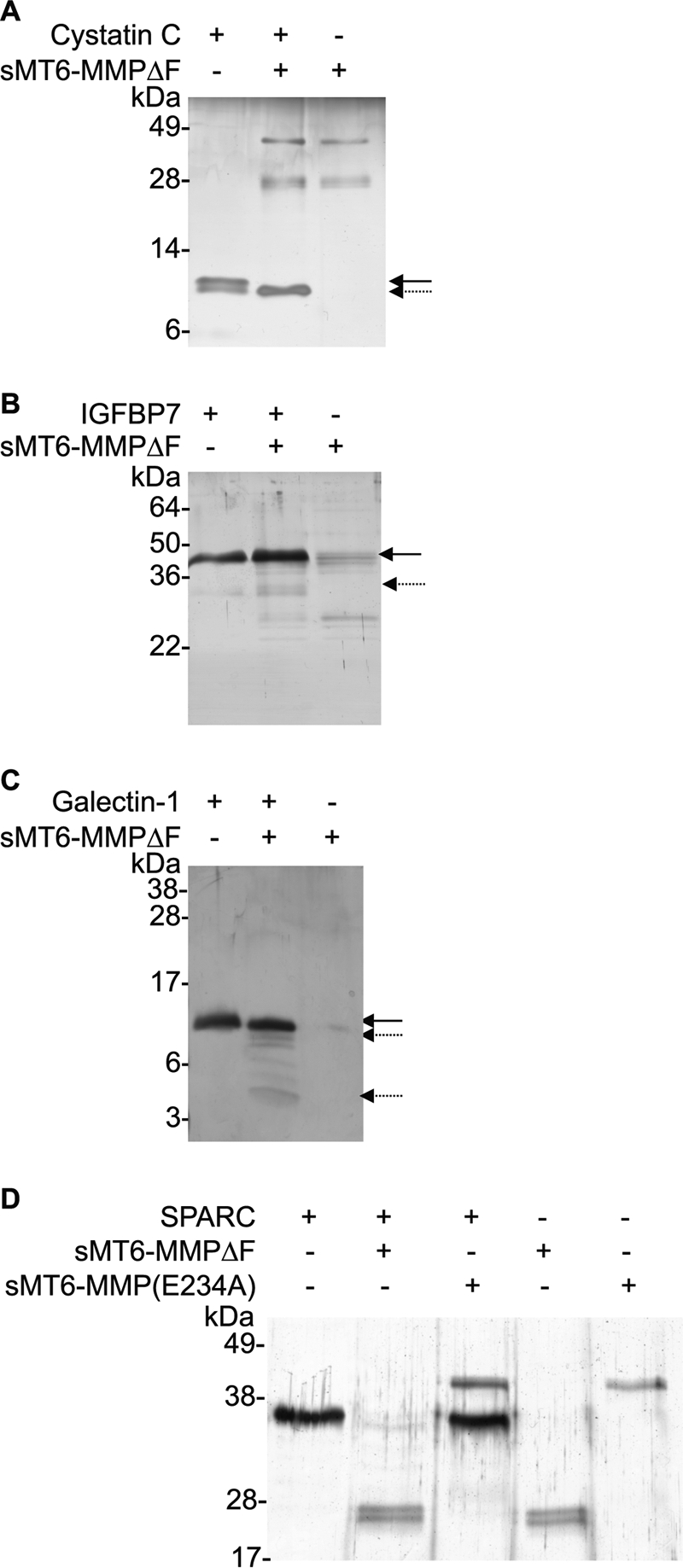
MT6-MMP cleaves cystatin C, IGFBP-7, galectin-1, and SPARC. Candidate substrates (1 μg) cystatin C (A), IGFBP-7 (B), galectin-1 (C), and SPARC (D) were incubated in a 10-μl reaction at an enzyme to substrate of 1:10 (w/w) with sMT6-MMPΔF for 16 h at 37 °C. Cleavage assay products were visualized under reducing conditions by silver-stained SDS-PAGE. The arrow indicates full-length protein; the broken arrow indicates cleavage product in the presence of MT6-MMP.
Because galectin-1 is known to be a substrate of MMP2 and -14 (46, 48), we considered it likely that it was also a substrate for MT6-MMP even though it was identified by only one high confidence peptide (Table 3). This was the case with SDS-PAGE analysis of incubations of MT6-MMP and galectin-1, confirming cleavage (Fig. 5C). SPARC was identified previously as a substrate of MMP2, -3, -7, -9, and -13 (48), and we now confirm the same for MT6-MMP (Fig. 5D). Unlike the other substrates validated in vitro, the one peptide identified for SPARC was only present in the control digests; that is, the experimental iTRAQ ratio decreased, thereby indicating degradation. Indeed, our analyses revealed complete loss of the full-length SPARC band, confirming degradation rather than cleavage to specific fragments. These analyses again confirm that TAILS is highly predictive for substrate identification even from single peptides. Thus, we expect that the majority if not all of the candidates are bona fide substrates.
Vimentin Functions as a Chemoattractant and upon MMP Cleavage as a Eat-me Signal for Monocytes
We identified vimentin as one of the highest confidence substrates of MT6-MMP (Table 1) by 10 different peptides shown on the vimentin sequence in Fig. 6A; three were common to both experiments. We characterized this substrate further. By in vitro cleavage assay, we confirmed that vimentin is cleaved not only by MT6-MMP (Fig. 6B) but also by seven additional MMPs (Fig. 6C). Interestingly, vimentin is displayed on the surface of apoptotic neutrophils (49), and so we reasoned it may act as an eat-me signal (4).
FIGURE 6.

MMP-processed vimentin has increased chemotactic and phagocytic potential for THP-1 cells. A, protein sequence of vimentin. Underlined or bold text indicates peptides identified with increased or decreased iTRAQ-ratio by TAILS analysis, respectively. B, vimentin (1 μg) was incubated with recombinant sMT6-MMPΔF at an enzyme to substrate ratio of 1:10 (w/w) in a 10-μl reaction volume at 37 °C for 16 h. Cleavage assay products were visualized by silver-stained 15% SDS-PAGE. The arrow indicates full-length vimentin; the broken arrow indicates cleavage product in the presence of MT6-MMP (n = 2). Comparable preparations were used for migration and phagocytosis assays. C, vimentin was incubated in the presence of MMP1, -2, -7, -8, -9, -12, -13, and -14 for 16 h at 37 °C. Cleavage assay products were visualized by silver-stained 15% SDS-PAGE. D, Transwell migration of THP-1 cells for 90 min toward full-length or sMT6-MMPΔF-cleaved vimentin or an equivalent concentration of sMT6-MMPΔF through a 5-μm pore-sized filter. Migrated cells were quantified by CyQUANT assay and are displayed as chemotactic index, defined as the ratio of cells migrating in response to stimulus compared with buffer control. Results shown are the mean ± S.E. of two replicates of experiments completed in quadruplicate. E, vimentin was incubated in the presence of sMT6-MMPΔF (Cleaved Vimentin), sMT6-MMP(E234A), or buffer to allow for processing prior to incubation and coating of Fluoresbrite microbeads. Fluorescence of THP-1 cells was evaluated by FACS following incubation of cells with Fluoresbrite microbeads previously coated with full-length or MMP-processed vimentin. Increased fluorescence indicates increased phagocytosis of coated microparticles. Results shown are the mean ± S.D. of three replicates of experiments completed in duplicate. ** and ***, statistical significance of cleaved versus full-length vimentin was evaluated by t test as indicated in the figure.
To explore this potential role of vimentin and MT6-MMP in neutrophil function, we showed in Transwell migration assays that full-length vimentin is a chemoattractant for THP-1 monocytic cells, whereas vimentin cleaved by MT6-MMP (Fig. 6D) or MMP12 (not shown) loses chemoattractant activity. The peak chemotactic response was at 450 nm, which is comparable with that of other intracellular proteins having chemoattractant activity (50). In contrast, phagocytosis by differentiated THP-1 cells of microparticles coated with cleaved vimentin cut by MT6-MMP (Fig. 6E) or MMP12 (not shown) was increased more than 2-fold. Notably, full-length vimentin showed no activity over controls. Hence, macrophage chemoattraction and phagocytosis of apoptotic neutrophils may be important functions of cell surface-displayed and then neutrophil MT6-MMP-cleaved vimentin.
DISCUSSION
With just seven substrates reported in the past 13 years since the discovery of MT6-MMP, its function has remained enigmatic. Given the neutrophil-specific expression, we hypothesized that this cell surface MMP would be involved in the proteolytic regulation of specific inflammatory molecules involved in innate immune processes. For the first time, we found that MT6-MMP is inhibited by the predominantly vascularly expressed TIMP4, and by using chemokine family-wide screens and proteomics analyses, we profiled the active site specificity of MT6-MMP. In identifying 286 cleavage sites by PICS, the active site substrate recognition cleft was mapped to extend from P3 to P6′. With 72 new substrates for MT6-MMP discovered, including 14 chemokines that recruit neutrophils and monocytes, the substrate degradome of this MMP has been greatly expanded. Notably, the precise cleavage of the CC and CXC chemokines by MT6-MMP leads to their inactivation or activation, consistent with a role in the regulation of innate immune cellular responses. Furthermore, several of the 58 new substrates identified by TAILS are known to contribute to inflammation through actions on leukocyte migration. Indeed, with only 20% of the new substrates identified as being extracellular matrix proteins, rather than being primarily an extracellular matrix degrader, MT6-MMP appears to be mainly involved in bioactive molecule cleavage. Notably, vimentin on the cell surface is a chemoattractant for monocytes, and we found that MMP-cleaved vimentin potently increases phagocytosis, suggesting that this contributes to marking apoptotic neutrophils for phagocytic clearance by macrophages.
Neutrophils are among the first cells recruited in inflammation. In this early phase of innate immunity, the predominant neutrophil chemoattractant CXCL8 is produced by macrophages, monocytes, and neutrophils and is activated particularly by neutrophil-specific MMP8 in a feed-forward mechanism (9) and also by MMP9, -12, -13, and -14 (8–10, 26) but strangely not by MT6-MMP. Similarly, a murine functional orthologue, mCXCL5/LIX, is activated by MMP8 and to a lesser extent by MMP9 (9, 10), which only poorly compensates for the lack of MMP8 in the Mmp8−/− mouse (9). Following neutrophil activation by CXCL8 and interferon-γ, polymorphonuclear neutrophils express MT6-MMP on the surface and shed the enzyme in an active, soluble form (15, 17) that corresponds to the sMT6-MMP construct. We identified that MT6-MMP processes and activates the neutrophil chemoattractants CXCL2 and human and murine CXCL5 to the products CXCL2(5–73), CXCL5(8–78), and mCXCL5(5–92, 10–92), which we and others previously showed have enhanced agonist and chemotactic potential over their full-length counterparts (9, 10, 39, 40, 51). These results suggest a discrete role for MT6-MMP in promoting a different phase of neutrophil migration. By acting in a positive feedback manner, MT6-MMP activation of human and murine CXCL5 and human CXCL2 has potential to amplify the initial neutrophil recruitment driven by MMP-8 activation of CXCL8. Pleiotropic effects are also likely because the MT6-MMP cleavage of CXCL6, which was also observed for MMP1, -2, -3, -7, -8, -9, -12, -13, and -14 (not shown), is predicted to inactivate the chemokine by destabilization of the protein core as truncation occurs C-terminally to both partners of the two disulfide bridges at CXC.
In addition to neutrophil recruitment, MT6-MMP can promote the recruitment of monocytes. Once recruited to tissue, monocyte receptor expression changes from CCR1high (binding CCL15 and CCL23) to CCR2high (CCL2). Both CCL15 and CCL23 are produced by monocytes and macrophages but are relatively weak CCR1 receptor agonists in their full-length forms. Processing of CCL15 and CCL23 by MT6-MMP results in N terminus-truncated forms that we have recently shown to exhibit increased agonist activity with increased chemotactic potential (45). This suggests a role for neutrophils, through MT6-MMP, in a positive feed-forward mechanism for the progression of the inflammatory response through increased recruitment of monocytes expressing CCR1. In contrast, CXCL12 and the chemokines CCL2, CCL7, and CCL13 lose agonist activity or become receptor antagonists following N terminus truncation (7, 8, 38, 42) as observed by MT6-MMP proteolysis. Overall, this differential processing of chemokines by MT6-MMP may then halt recruited cells, signaling that they have reached the target site, or terminate continued recruitment as one aspect in the dampening of the inflammatory response.
Neutrophil apoptosis is a final critical event in the innate immune response, important for chemotaxis and activation of macrophages (4, 5, 52, 53), during which there is also enhanced cell surface expression of MT6-MMP (15). One of the highest confidence substrates we identified was vimentin, which recent studies have shown has a bona fide extracellular role as a “moonlighting” protein (54). Vimentin is actively secreted by macrophages following TNF-α stimulation (55) and is displayed on the surface of apoptotic neutrophils (49). We found that upon loss of chemoattractant activity MMP-cleaved vimentin potently promotes phagocytosis by differentiated THP-1 cells. Hence, vimentin displayed on the cell surface of apoptotic neutrophils and cleaved by MT6-MMP may function as an eat-me signal for macrophage phagocytosis.
A number of cleaved proteins identified by TAILS are components of cell proliferation and collagen production pathways and so may be involved in the early phase of wound healing. These include procollagen C-proteinase enhancer, peptidyl-prolyl cis-trans isomerase A, and IGFBP-5 and -7, the latter of which was biochemically confirmed as a novel substrate. IGFBPs are critical regulators of IGF, sequestering and inhibiting the activity of ∼99% of circulating IGF (56, 57); cleavage and inactivation of IGFBPs promote IGF activity. Unbound IGF binds surface-expressed IGF receptor to promote cell growth (58, 59). Hence, the specific cleavage of IGFBP-5 and -7 shown here for the first time by any MMP suggests a role for MT6-MMP in unmasking IGF that may promote healing.
Finally, a number of extracellular matrix-associated proteins were identified as substrates, including fibrillin-1, extracellular matrix protein 1, syndecan-4, and type I, II, IV, and VI collagens; galectin-1 and SPARC were also biochemically validated as new MT6-MMP substrates. The latter two are matricellular proteins with multifunctional roles, including proinflammatory functions and wound healing (60–62). This highlights the usefulness of unbiased proteomics screens using TAILS to identify protease substrates from even one prime-side-cleaved peptide.
Thus, through hypothesis-directed and hypothesis-generating approaches, we have discovered a large number of new MT6-MMP substrates that shed light on the in vivo role of this enzyme and neutrophil function. Notably, the two polymorphonuclear neutrophil-specific proteases MMP8 and MT6-MMP act in a temporally distinct manner on discrete neutrophil CXC chemokines. This implicates MT6-MMP in the later recruitment of neutrophils through the activation of CXCL2 and CXCL5 and in the progression of the acute inflammatory response toward monocyte recruitment by activation of CCL15 and CCL23. Interstitial roles for MT6-MMP are also suggested wherein monocyte chemoattractants, including vimentin, are cleaved, halting haptotaxis and then stimulating phagocytosis of apoptotic neutrophils. In view of the biological activities of these new MT6-MMP substrates and cleavage products, MT6-MMP can potentially contribute to the complex regulation needed in inflammation and wound healing that is being explored in ongoing studies.
Supplementary Material
This work was supported in part by a Canadian Institutes of Health Research grant, an infrastructure grant from the Michael Smith Research Foundation (University of British Columbia Centre for Blood Research), and the British Columbia Proteomics Network.

This article contains supplemental Figs. S1 and S2 and Tables I, II, and III.
- MMP
- matrix metalloproteinase
- HFL
- human fetal lung fibroblast
- IGFBP
- insulin-like growth factor-binding protein
- iTRAQ
- isobaric tags for relative and absolute quantitation
- MT6-MMP
- membrane-type 6 MMP
- PICS
- proteomic identification of cleavage site specificity
- SPARC
- secreted protein, acidic and rich in cysteine
- TAILS
- terminal amine isotopic labeling of substrates
- TIMP
- tissue inhibitor of metalloproteinases
- sMT6
- soluble MT6
- QF
- FRET-quenched fluorescent
- Mca
- methylcoumarin
- Dpa
- 2,4-dinitrophenyl
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- m
- murine
- h
- human
- IFG
- insulin-like growth factor.
REFERENCES
- 1. Borregaard N., Cowland J. B. (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89, 3503–3521 [PubMed] [Google Scholar]
- 2. Serhan C. N., Savill J. (2005) Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 3. Cox J. H., Starr A. E., Kappelhoff R., Yan R., Roberts C. R., Overall C. M. (2010) Matrix metalloproteinase 8 deficiency in mice exacerbates inflammatory arthritis through delayed neutrophil apoptosis and reduced caspase 11 expression. Arthritis Rheum. 62, 3645–3655 [DOI] [PubMed] [Google Scholar]
- 4. Guzik K., Potempa J. (2008) Friendly fire against neutrophils: proteolytic enzymes confuse the recognition of apoptotic cells by macrophages. Biochimie 90, 405–415 [DOI] [PubMed] [Google Scholar]
- 5. Kennedy A. D., DeLeo F. R. (2009) Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61 [DOI] [PubMed] [Google Scholar]
- 6. Mortier A., Gouwy M., Van Damme J., Proost P. (2011) Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 317, 642–654 [DOI] [PubMed] [Google Scholar]
- 7. McQuibban G. A., Gong J. H., Tam E. M., McCulloch C. A., Clark-Lewis I., Overall C. M. (2000) Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 [DOI] [PubMed] [Google Scholar]
- 8. Dean R. A., Cox J. H., Bellac C. L., Doucet A., Starr A. E., Overall C. M. (2008) Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464 [DOI] [PubMed] [Google Scholar]
- 9. Tester A. M., Cox J. H., Connor A. R., Starr A. E., Dean R. A., Puente X. S., López-Otín C., Overall C. M. (2007) LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One 2, e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Den Steen P. E., Wuyts A., Husson S. J., Proost P., Van Damme J., Opdenakker G. (2003) Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur. J. Biochem. 270, 3739–3749 [DOI] [PubMed] [Google Scholar]
- 11. Kojima S., Itoh Y., Matsumoto S., Masuho Y., Seiki M. (2000) Membrane-type 6 matrix metalloproteinase (MT6-MMP, MMP-25) is the second glycosyl-phosphatidyl inositol (GPI)-anchored MMP. FEBS Lett. 480, 142–146 [DOI] [PubMed] [Google Scholar]
- 12. Pei D. (1999) Leukolysin/MMP25/MT6-MMP: a novel matrix metalloproteinase specifically expressed in the leukocyte lineage. Cell Res. 9, 291–303 [DOI] [PubMed] [Google Scholar]
- 13. Matsuda A., Itoh Y., Koshikawa N., Akizawa T., Yana I., Seiki M. (2003) Clusterin, an abundant serum factor, is a possible negative regulator of MT6-MMP/MMP-25 produced by neutrophils. J. Biol. Chem. 278, 36350–36357 [DOI] [PubMed] [Google Scholar]
- 14. English W. R., Velasco G., Stracke J. O., Knäuper V., Murphy G. (2001) Catalytic activities of membrane-type 6 matrix metalloproteinase (MMP25). FEBS Lett. 491, 137–142 [DOI] [PubMed] [Google Scholar]
- 15. Fortin C. F., Sohail A., Sun Q., McDonald P. P., Fridman R., Fülöp T. (2010) MT6-MMP is present in lipid rafts and faces inward in living human PMNs but translocates to the cell surface during neutrophil apoptosis. Int. Immunol. 22, 637–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radichev I. A., Remacle A. G., Shiryaev S. A., Purves A. N., Johnson S. L., Pellecchia M., Strongin A. Y. (2010) Biochemical characterization of the cellular glycosylphosphatidylinositol-linked membrane type-6 matrix metalloproteinase. J. Biol. Chem. 285, 16076–16086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang T., Yi J., Guo A., Wang X., Overall C. M., Jiang W., Elde R., Borregaard N., Pei D. (2001) Subcellular distribution and cytokine- and chemokine-regulated secretion of leukolysin/MT6-MMP/MMP-25 in neutrophils. J. Biol. Chem. 276, 21960–21968 [DOI] [PubMed] [Google Scholar]
- 18. Sohail A., Sun Q., Zhao H., Bernardo M. M., Cho J. A., Fridman R. (2008) MT4-(MMP17) and MT6-MMP (MMP25), A unique set of membrane-anchored matrix metalloproteinases: properties and expression in cancer. Cancer Metastasis Rev. 27, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andolfo A., English W. R., Resnati M., Murphy G., Blasi F., Sidenius N. (2002) Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1-D2 linker region and expose epitopes not present in the intact soluble receptor. Thromb. Haemost. 88, 298–306 [PubMed] [Google Scholar]
- 20. Nie J., Pei D. (2004) Rapid inactivation of α-1-proteinase inhibitor by neutrophil specific leukolysin/membrane-type matrix metalloproteinase 6. Exp. Cell Res. 296, 145–150 [DOI] [PubMed] [Google Scholar]
- 21. Shiryaev S. A., Savinov A. Y., Cieplak P., Ratnikov B. I., Motamedchaboki K., Smith J. W., Strongin A. Y. (2009) Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in autoimmune multiple sclerosis. PLoS One 4, e4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. López-Otín C., Overall C. M. (2002) Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 3, 509–519 [DOI] [PubMed] [Google Scholar]
- 23. Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 24. Prudova A., auf dem Keller U., Butler G. S., Overall C. M. (2010) Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics 9, 894–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Becker-Pauly C., Barre O., Schilling O., Auf dem Keller U., Ohler A., Broder C., Schutte A., Kappelhoff R., Stocker W., Overall C. M. (2011) Proteomic analyses reveal an acidic prime side specificity for the astacin metalloprotease family reflected by physiological substrates. Mol. Cell. Proteomics 10, M111.009233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butler G. S., Tam E. M., Overall C. M. (2004) The canonical methionine 392 of matrix metalloproteinase 2 (gelatinase A) is not required for catalytic efficiency or structural integrity: probing the role of the methionine-turn in the metzincin metalloprotease superfamily. J. Biol. Chem. 279, 15615–15620 [DOI] [PubMed] [Google Scholar]
- 27. auf dem Keller U., Prudova A., Gioia M., Butler G. S., Overall C. M. (2010) A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol. Cell. Proteomics 9, 912–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clark-Lewis I., Vo L., Owen P., Anderson J. (1997) Chemical synthesis, purification, and folding of C-X-C and C-C chemokines. Methods Enzymol. 287, 233–250 [DOI] [PubMed] [Google Scholar]
- 29. Schilling O., Huesgen P. F., Barré O., Auf dem Keller U., Overall C. M. (2011) Characterization of the prime and non-prime active site specificities of proteases by proteome-derived peptide libraries and tandem mass spectrometry. Nat. Protoc. 6, 111–120 [DOI] [PubMed] [Google Scholar]
- 30. Schilling O., Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 [DOI] [PubMed] [Google Scholar]
- 31. Schilling O., auf dem Keller U., Overall C. M. (2011) Factor Xa subsite mapping by proteome-derived peptide libraries improved using WebPICS, a resource for proteomic identification of cleavage sites. Biol. Chem. 392, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 32. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 33. Starr A. E., Overall C. M. (2009) Chapter 13. Characterizing proteolytic processing of chemokines by mass spectrometry, biochemistry, neo-epitope antibodies and functional assays. Methods Enzymol. 461, 281–307 [DOI] [PubMed] [Google Scholar]
- 34. Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J. N., Overall C. M. (2011) Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 6, 1578–1611 [DOI] [PubMed] [Google Scholar]
- 35. Pedrioli P. G. (2010) Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol. Biol. 604, 213–238 [DOI] [PubMed] [Google Scholar]
- 36. Zhao H., Sohail A., Sun Q., Shi Q., Kim S., Mobashery S., Fridman R. (2008) Identification and role of the homodimerization interface of the glycosylphosphatidylinositol-anchored membrane type 6 matrix metalloproteinase '(MMP25). J. Biol. Chem. 283, 35023–35032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reinemer P., Grams F., Huber R., Kleine T., Schnierer S., Piper M., Tschesche H., Bode W. (1994) Structural implications for the role of the N terminus in the 'superactivation' of collagenases. A crystallographic study. FEBS Lett. 338, 227–233 [DOI] [PubMed] [Google Scholar]
- 38. McQuibban G. A., Gong J. H., Wong J. P., Wallace J. L., Clark-Lewis I., Overall C. M. (2002) Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 100, 1160–1167 [PubMed] [Google Scholar]
- 39. Nufer O., Corbett M., Walz A. (1999) Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry 38, 636–642 [DOI] [PubMed] [Google Scholar]
- 40. Wuyts A., D'Haese A., Cremers V., Menten P., Lenaerts J. P., De Loof A., Heremans H., Proost P., Van Damme J. (1999) NH2- and COOH-terminal truncations of murine granulocyte chemotactic protein-2 augment the in vitro and in vivo neutrophil chemotactic potency. J. Immunol. 163, 6155–6163 [PubMed] [Google Scholar]
- 41. Cox J. H., Dean R. A., Roberts C. R., Overall C. M. (2008) Matrix metalloproteinase processing of CXCL11/I-TAC results in loss of chemoattractant activity and altered glycosaminoglycan binding. J. Biol. Chem. 283, 19389–19399 [DOI] [PubMed] [Google Scholar]
- 42. McQuibban G. A., Butler G. S., Gong J. H., Bendall L., Power C., Clark-Lewis I., Overall C. M. (2001) Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 276, 43503–43508 [DOI] [PubMed] [Google Scholar]
- 43. Zhang K., McQuibban G. A., Silva C., Butler G. S., Johnston J. B., Holden J., Clark-Lewis I., Overall C. M., Power C. (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 6, 1064–1071 [DOI] [PubMed] [Google Scholar]
- 44. Laurence J. S., LiWang A. C., LiWang P. J. (1998) Effect of N-terminal truncation and solution conditions on chemokine dimer stability: nuclear magnetic resonance structural analysis of macrophage inflammatory protein 1β mutants. Biochemistry 37, 9346–9354 [DOI] [PubMed] [Google Scholar]
- 45. Starr A. E., Dufour A., Maier J., Overall C. M. (2012) Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 287, 5848–5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dean R. A., Overall C. M. (2007) Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611–623 [DOI] [PubMed] [Google Scholar]
- 47. Dean R. A., Butler G. S., Hamma-Kourbali Y., Delbé J., Brigstock D. R., Courty J., Overall C. M. (2007) Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell. Biol. 27, 8454–8465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Butler G. S., Overall C. M. (2009) Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry 48, 10830–10845 [DOI] [PubMed] [Google Scholar]
- 49. Moisan E., Girard D. (2006) Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J. Leukoc. Biol. 79, 489–498 [DOI] [PubMed] [Google Scholar]
- 50. Oppenheim J. J., Dong H. F., Plotz P., Caspi R. R., Dykstra M., Pierce S., Martin R., Carlos C., Finn O., Koul O., Howard O. M. (2005) Autoantigens act as tissue-specific chemoattractants. J. Leukoc. Biol. 77, 854–861 [DOI] [PubMed] [Google Scholar]
- 51. King A. G., Johanson K., Frey C. L., DeMarsh P. L., White J. R., McDevitt P., McNulty D., Balcarek J., Jonak Z. L., Bhatnagar P. K., Pelus L. M. (2000) Identification of unique truncated KC/GROβ chemokines with potent hematopoietic and anti-infective activities. J. Immunol. 164, 3774–3782 [DOI] [PubMed] [Google Scholar]
- 52. Guzik K., Bzowska M., Smagur J., Krupa O., Sieprawska M., Travis J., Potempa J. (2007) A new insight into phagocytosis of apoptotic cells: proteolytic enzymes divert the recognition and clearance of polymorphonuclear leukocytes by macrophages. Cell Death Differ. 14, 171–182 [DOI] [PubMed] [Google Scholar]
- 53. Parente L., Solito E. (2004) Annexin 1: more than an anti-phospholipase protein. Inflamm. Res. 53, 125–132 [DOI] [PubMed] [Google Scholar]
- 54. Butler G. S., Overall C. M. (2009) Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 8, 935–948 [DOI] [PubMed] [Google Scholar]
- 55. Mor-Vaknin N., Punturieri A., Sitwala K., Markovitz D. M. (2003) Vimentin is secreted by activated macrophages. Nat. Cell Biol. 5, 59–63 [DOI] [PubMed] [Google Scholar]
- 56. Firth S. M., Baxter R. C. (2002) Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 23, 824–854 [DOI] [PubMed] [Google Scholar]
- 57. Pollak M. (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 [DOI] [PubMed] [Google Scholar]
- 58. Clemmons D. R., Sleevi M., Allan G., Sommer A. (2007) Effects of combined recombinant insulin-like growth factor (IGF)-I and IGF binding protein-3 in type 2 diabetic patients on glycemic control and distribution of IGF-I and IGF-II among serum binding protein complexes. J. Clin Endocrinol. Metab. 92, 2652–2658 [DOI] [PubMed] [Google Scholar]
- 59. Liu X. J., Malkowski M., Guo Y., Erickson G. F., Shimasaki S., Ling N. (1993) Development of specific antibodies to rat insulin-like growth factor-binding proteins (IGFBP-2 to -6): analysis of IGFBP production by rat granulosa cells. Endocrinology 132, 1176–1183 [DOI] [PubMed] [Google Scholar]
- 60. Almkvist J., Karlsson A. (2004) Galectins as inflammatory mediators. Glycoconj. J. 19, 575–581 [DOI] [PubMed] [Google Scholar]
- 61. Chlenski A., Cohn S. L. (2010) Modulation of matrix remodeling by SPARC in neoplastic progression. Semin. Cell Dev. Biol. 21, 55–65 [DOI] [PubMed] [Google Scholar]
- 62. Malik R. K., Ghurye R. R., Lawrence-Watt D. J., Stewart H. J. (2009) Galectin-1 stimulates monocyte chemotaxis via the p44/42 MAP kinase pathway and a pertussis toxin-sensitive pathway. Glycobiology 19, 1402–1407 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.