

Background: Aptamers are single-stranded oligonucleotides folded into three-dimensional shapes that bind complementary molecular surfaces.
Results: This aptamer blocks factor IXa activity toward macromolecular substrates but only partially blocks small substrates.
Conclusion: The aptamer binds factor IXa so as to block the extended substrate-binding site.
Significance: These studies provide a mechanistic understanding of the function of an anticoagulant in late-stage clinical development.
Keywords: Aptamers, Blood Coagulation Factors, Enzyme Inhibitors, Proteolytic Enzymes, RNA, Antithrombotics, Coagulation Factor IX
Abstract
An aptamer targeting factor IXa has been evaluated in animal models and several clinical studies as a potential antithombotic therapy. We elucidate the molecular mechanism by which this aptamer acts as an anticoagulant. The aptamer binds tightly to factor IXa and prolongs the clotting time of human plasma. The aptamer completely blocks factor IXa activation of factor X regardless of the presence of factor VIIIa. However, the aptamer does not completely block small synthetic substrate cleavage, although it does slow the rate of cleavage. These data are consistent with the aptamer binding to the catalytic domain of factor IXa in such a way as to block an extended substrate-binding site. Therefore, unlike small molecule inhibitors, aptamers appear to be able to bind surfaces surrounding an active site and thereby sterically interfere with enzyme activity. Thus, aptamers may be useful agents to probe and block substrate-binding sites outside of the active site of an enzyme.
Introduction
Thrombosis represents a major risk of morbidity and mortality, particularly in the elderly (1). Currently, heparin is widely used as an antithrombotic; it is a potent anticoagulant that targets multiple activated factors in the coagulation cascade (2). However, heparin therapy can be associated with bleeding as well as complications such as heparin-induced thrombocytopenia (3). Heparin is not alone in this respect, and in general, current therapies represent a balance between the risk of thrombosis without treatment and the risk of bleeding due to treatment (4). One approach to reducing complications and improving antithrombotic therapy is to selectively target a specific coagulation protein (2).
Factor IX is an attractive target for antithrombotic therapy. It participates in the propagation phase of coagulation; after activation by either factor XIa or the factor VIIa-tissue factor complex, factor IXa binds to factor VIIIa on a platelet surface and activates factor X. Two studies have suggested that the primary effect of heparin on prolonging the activated partial thromboplastin time (aPTT)2 is through blocking factor IXa activation of factor X (5, 6). Studies in dogs and baboons showed that when active site-inhibited factor IXa is used as an anticoagulant in place of heparin, the extracorporeal circuit remains free of fibrin deposition, and intraoperative blood loss is significantly diminished relative to the standard heparin protocol (7, 8). In addition, an antibody directed against the amino-terminal region of factor IX acts as a potent antithrombotic agent in rat and guinea pig models of arterial thrombosis (9).
An emerging class of therapeutics is the folded nucleic acid structures called aptamers (10–12). Aptamers are small single-stranded oligonucleotides that fold into a specific three-dimensional shape that is assembled upon a secondary structure derived from multiple hydrogen-bonded base pairs. The surfaces on these molecular shapes can bind to complementary molecular surfaces on their target (13). This complementary shape binding is similar to the types of interactions observed between antibody-antigen pairs, and it allows aptamers to bind to proteins that do not naturally recognize nucleic acids (14). Because aptamer binding is very dependent on the internal hydrogen-bonded base pairs, the three-dimensional structure of the aptamer can be disrupted with an appropriately chosen oligonucleotide that can interrupt such base pairing. Application of this concept led us to develop the idea of therapeutic aptamers generated in concert with antidote oligonucleotides (15–21). Such complementary antidote oligonucleotides allow for complete neutralization of aptamer activity and create the possibility of fine-grained control of aptamer levels (18, 20, 21).
Applying the aptamer-antidote principles to the question of targeted antithrombotics, we have developed an aptamer (9.3t) targeted against factor IXa (15). The action of the aptamer can be controlled by an antidote oligonucleotide (15, 18). This aptamer can prolong the clotting time (aPTT) of normal human plasma; at saturating levels of aptamer, the aPTT was prolonged to the values seen in hemophilia B (factor IX-deficient) plasma. In a porcine model of cardiopulmonary bypass, this aptamer controlled thrombin generation better than heparin and was able to maintain patency in the bypass circuit for 30 min when used instead of heparin as an anticoagulant (17). Moreover, recently, an optimized version of this aptamer was able to replace heparin as the anticoagulant in a phase 2a clinical trial in patients with acute coronary syndrome undergoing percutaneous coronary intervention (22). Currently, a large ∼800-person phase 2b study is under way to further evaluate the aptamer and its matched antidote in this patient population. The studies presented here were initiated to identify the mechanisms by which this anti-factor IXa aptamer acts as an anticoagulant and antithrombotic.
EXPERIMENTAL PROCEDURES
Reagents
Factor Xa substrate (Pefachrome® FXa) was from Pentapharm (Basel, Switzerland), as was factor IXa substrate (Pefachrome® FIXa3960, H-d-Leu-phenylglycine-Arg-p-nitroanilide acetate). Cleavage of Pefachrome FIXa was not enhanced by ethylene glycol. Dansyl-Glu-Gly-Arg chloromethyl ketone (DEGR-CK) was purchased from Calbiochem. Anti-factor IX antibodies were purchased from Affinity Biologicals (Ancaster, Ontario, Canada). Unless indicated otherwise, all reactions were run in 20 mm HEPES (pH 7.4), 150 mm NaCl, 5 mm CaCl2, and 1 mg/ml ovalbumin.
Proteins
Factor IX was isolated from plasma by chromatography on DEAE-Sepharose eluted with NaCl, heparin-Sepharose eluted with NaCl, copper-loaded iminodiacetic acid (metal chelate)-Sepharose eluted with glycine, and HiTrap Q eluted with CaCl2 at a fixed NaCl concentration (pseudo-affinity). It was converted to factor IXa with a 1:200 molar ratio of factor XIa (Hematologic Technologies, Essex Junction, VT) as described previously and repurified by pseudo-affinity chromatography (23). Factor VIII (recombinant B-domainless) was the generous gift of the Harold R. Roberts Comprehensive Hemophilia Diagnostic and Treatment Center at the University of North Carolina at Chapel Hill. Factor X was isolated from plasma by chromatography on DEAE-Sepharose eluted with NaCl, heparin-Sepharose eluted with NaCl, hydroxylapatite eluted with phosphate, and HiTrap Q eluted with CaCl2 at a fixed NaCl concentration (pseudo-affinity). Antithrombin was isolated from plasma on heparin-Sepharose as described previously (24).
Lipid Vesicles
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Large unilamellar vesicles containing 15% phosphatidylserine, 41% phosphatidylcholine, 44% phosphatidylethanolamine were prepared by extrusion (25, 26).
Aptamer
Before all assays, the aptamer was refolded by heating to 65 °C for 5 min and cooling slowly to room temperature. The aptamer is designated 9.3t (15). The aptamer 9.3tM was used as a control; it differs from 9.3t by only 2 bases but does not bind factor IX (15). The aptamer was dephosphorylated using bacterial alkaline phosphatase and 5′-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP.
Clotting Assays
Citrated human plasma was purchased from King George Bio-Medical, Inc. (Overland Park, KS). aPTTs were performed in a Diagnostica Stago ST4 coagulometer using TriniCLOT aPTT S reagent (Trinity Biotech) with aptamer-treated plasma for 5 min before initiating clotting with TriniCLOT aPTT S (0.02 m CaCl2).
Factor IX Activation by Factor XIa
Factor IX (5 μm) was incubated with aptamer 9.3t (6 μm) for 30 min before addition of 25 nm factor XIa. At timed intervals, samples were removed into SDS buffer with 1% mercaptoethanol and heated to 95 °C for 3 min. Samples were run on a 10–15% PhastGel and stained with silver stain. Band intensity was determined using the NIH program ImageJ (rsb.info.nih.gov/ij/).
Factor IX Activation by Factor VIIa-Tissue Factor Complex
Factor IX (1 μm) was incubated with aptamer 9.3t (5 μm) for 30 min before addition of 2 nm factor VIIa with 1 nm relipidated recombinant human tissue factor. At timed intervals, samples were removed into SDS buffer (with or without 1% mercaptoethanol) and heated to 95 °C for 3 min. Samples were run on a 10–15% PhastGel, transferred to a nylon membrane, and blotted with rabbit polyclonal anti-factor IX antibodies. The presence of anti-factor IX antibodies was detected with IRDye 680-labeled anti-rabbit antibodies and visualized on a LI-COR ODYSSEY system. The fraction activated is the proportion of the staining in each lane found in the factor IXa band (unreduced) or the heavy chain (reduced) Western blots. The rate of factor IX activation (k) was determined by fitting the data to Equation 1.
Factor X Activation Assays
For assays with factor VIIIa, factor IXa (0.6 nm) was incubated with varied concentrations of 9.3t or 9.3tM. Factor VIIIa (1.4 nm) with 80 μm lipid was added, reducing the factor IXa concentration to 0.4 nm, and incubated for 3 min before addition of factor X and factor X substrate. Final concentrations in the assay were 0.2 nm factor IXa, 0.7 nm factor VIIIa, 40 μm lipid, 135 nm factor X, and 0.5 mm factor X substrate. Factor X activation was monitored by substrate cleavage, and data were analyzed to determine the initial rate as described previously (28).
For assays with preformed factor IXa-factor VIIIa complex, 0.4 nm factor IXa was incubated with 1.4 nm factor VIIIa and 80 μm lipid for 3 min before addition of aptamer for 1 min. Factor X and factor Xa substrate were added to give the same final concentrations as described above.
For assays without factor VIIIa, 6 or 12 nm factor IXa was incubated with aptamer 9.3t or 9.3tM for 15 min before being added to factor X, lipid, and factor Xa substrate. Final concentrations were 3 or 6 nm factor IXa, 150 μm factor X, 50 μm lipid, and 0.5 mm factor X substrate. Factor X activation was monitored as described above.
Measurement of Substrate Cleavage
Factor IXa (400 nm) was incubated with varied concentrations of aptamer 9.3t or 9.3tM before being added to H-d-Leu-phenylglycine-Arg-p-nitroanilide acetate (Pefachrome FIXa3960) to give final concentrations of 200 nm factor IXa, the indicated concentration of the aptamer, and 0.5 mm substrate. The substrate is a peptide sequence attached to a p-nitroanilide group. Cleavage of the substrate released a p-nitroaniline group; appearance was monitored at 405 nm. Substrate cleavage was monitored for 10 min, during which time the increase in absorbance at 405 nm was linear. Background hydrolysis of the substrate was measured by monitoring the absorbance at 405 nm in the absence of factor IXa but in the presence of aptamer.
Affinity Calculations
Factor IXa binding to aptamer 9.3t is an equilibrium reaction as shown in Equation 2.
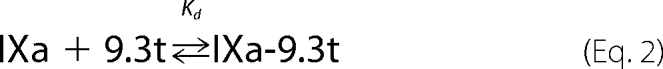 |
The equilibrium dissociation constant (Kd) is defined as shown in Equation 3.
 |
The terms for free factor IXa and free aptamer 9.3t are rewritten in terms of [IXa-9.3t], total factor IXa ([IXa]t), and total aptamer ([9.3t]t) as shown in Equation 4.
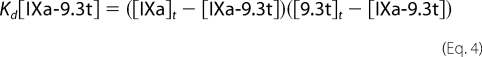 |
Because the concentration of the IXa-9.3t complex cannot be assumed to be small relative to the total concentration of the aptamer, the equation has to be evaluated without simplifying assumptions. Rearranging gives Equation 5.
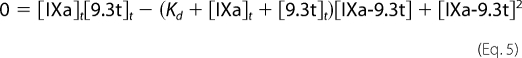 |
Solving for the concentration of the IXa-9.3t complex gives Equation 6.
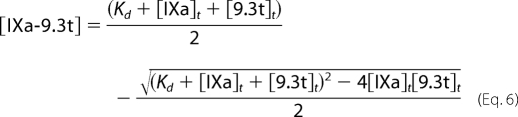 |
Because factor IXa in complex with the aptamer is inactive, the activity is proportional to the amount of free factor IXa as shown in Equation 7.
Activity expressed as the relative rate is given by Equation 8.
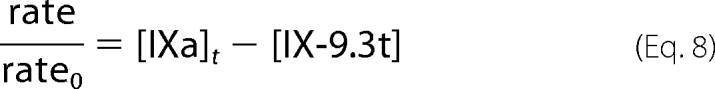 |
Substituting Equation 5 into Equation 7 gives Equation 9.
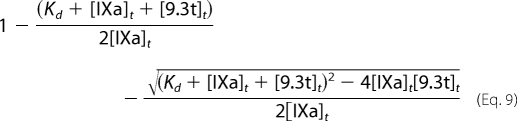 |
RESULTS
Aptamer Inhibition of Clotting Assays
As reported previously (15), the ability of aptamer 9.3t to prolong clotting was studied by adding varied concentrations of the aptamer to plasma and then measuring the aPTT. Aptamer 9.3t, but not a mutant version of the aptamer (9.3tM), dose-dependently prolonged the aPTT of plasma (Fig. 1). The aPTT reached values seen in hemophilic human plasma (29).
FIGURE 1.
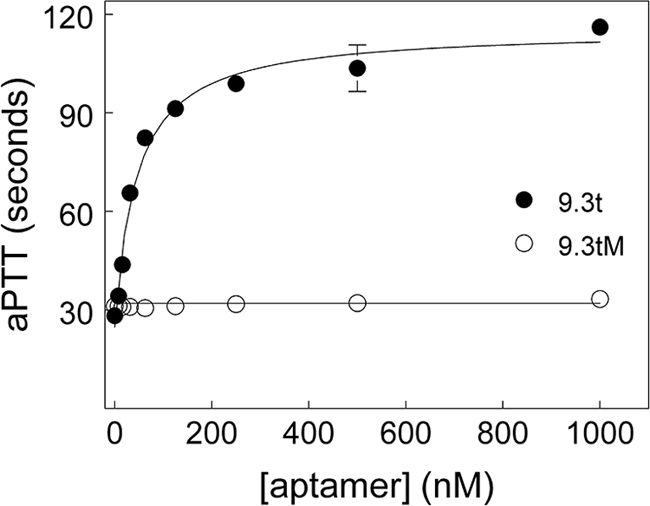
Aptamer prolongs clotting time of plasma. The indicated concentrations of aptamer 9.3t (●) or 9.3tM (○) were added to plasma. The clotting time is plotted against the concentration of the aptamer. With these reagents, hemophilia B plasma had a clotting time of ∼120 s.
Ligand Binding Assay Shows Aptamer Binding to Both Factors IX and IXa
Binding of aptamer 9.3t to protein was measured using a double-filter binding assay as described previously (30). A fixed concentration of radiolabeled aptamer 9.3t was incubated with varied protein concentrations and then added to membrane. Aptamer bound to protein was captured on the (top) nitrocellulose membrane. Unbound aptamer passed through and was captured by the (bottom) nylon membrane. Counts associated with protein were expressed as a percentage of the total counts (nitrocellulose capture + nylon capture). Aptamer 9.3t binding to factor IX was similar to that to factor IXa (Fig. 2); this is consistent with a previous study that used surface plasmon resonance to show that the aptamer binds with similar affinity to factors IX and IXa (31). Mutant aptamer 9.3tM did not bind to either factor IX or IXa.
FIGURE 2.

Aptamer binding to factors IX and IXa measured by double-filter binding. A fixed amount of radiolabeled aptamer was incubated with the indicated concentration of factor IX or IXa. Aptamer bound to factor IXa was captured on a nitrocellulose membrane, with unbound aptamer captured on nylon. Radioactivity bound to cellulose is plotted versus the concentration of factor IX (triangles) or factor IXa (circles). ● and ▴, binding of aptamer 9.3t; ○ and △, binding of mutant aptamer 9.3tM.
Aptamer Does Not Change Factor IX Activation by Factor XIa
Because aptamer 9.3t bound both factors IX and IXa, we first examined if the aptamer would inhibit activation of factor IX by factor XIa. The aptamer did not change factor XIa activation of factor IX (Fig. 3A).
FIGURE 3.

Aptamer effect on factor IX activation. A, factor IX was incubated with the aptamer (●) or a buffer control (○) before being activated with factor XIa in buffer without albumin. At the indicated times, samples were run on a 10–15% gradient PhastGel and silver-stained. Images were analyzed using ImageJ. The band intensity in the light chain (units of the band intensity from ImageJ scans) is plotted versus time. The insets show grayscale images of the gels. B, factor IX was incubated with the aptamer (●) or a buffer control (○) before being activated with factor VIIa-tissue factor. At the indicated times, samples were run on a 10–15% gradient PhastGel and transferred to a nylon membrane for Western blotting with anti-factor IX primary and fluorescent secondary antibodies. Fluorescence was detected with a LI-COR ODYSSEY system. Separate experiments examined reduced and unreduced samples, and all data are shown. The insets show representative Western blots.
Aptamer Slows Factor IX Activation by Factor VIIa-Tissue Factor Complex
We also examined factor IX activation by the factor VIIa-tissue factor complex. The aptamer decreased the rate of factor IX activation to ∼50% of the rate seen in its absence (Fig. 3B).
Aptamer Blocks Activity of Factor IXa-Factor VIIIa Complex
Because factor IXa activity was increased by >2000-fold when it associated with its cofactor, factor VIIIa, we evaluated if the aptamer could inhibit the activity of the factor IXa-factor VIIIa complex. Factor IXa was incubated with varied concentrations of aptamer 9.3t or mutant aptamer 9.3tM for 20 min at room temperature. Factor IXa activity was assessed by adding factor VIIIa with lipid and incubating for 3 min before adding factor X and factor Xa substrate. Aptamer 9.3t, but not mutant aptamer 9.3tM, inhibited factor X activation by the factor IXa-factor VIIIa complex in a dose-dependent fashion (Fig. 4A). Based upon this inhibition, the affinity of the aptamer for factor IXa was calculated to be 0.16 nm.
FIGURE 4.

Aptamer blocks factor IXa activation of factor X with or without factor VIIIa. A, the indicated concentrations of aptamer 9.3t were incubated with factor IXa (●) or preformed factor IXa-factor VIIIa complex (■), and factor X activation was measured. Mutant aptamer 9.3tM data for both data sets are shown (○). Factor Xa generation is expressed relative to the initial rate in the absence of the aptamer. The data for the aptamer incubated with factor IXa were fit to the quadratic equation (Equation 9) described under “Experimental Procedures” and gave a value of 0.16 ± 0.01 nm. The data fit for the aptamer added to the factor IXa-factor VIIIa complex gave a value of 0.07 ± 0.01 nm. B, the indicated concentrations of aptamer 9.3t were incubated with 6 nm (▴) and 3 nm (▾) factor IXa, and factor X activation on lipid vesicles was measured. Mutant aptamer 9.3tM data for 6 nm factor IXa are shown (○). The data for the aptamer incubated with factor IXa were fit to the quadratic equation (Equation 9) described under “Experimental Procedures” and gave a value of 0.9 nm (0.84 ± 0.06 and 0.93 ± 0.12 nm for 3 and 6 nm factor IXa, respectively).
To determine whether the aptamer was interfering with formation of the factor IXa-factor VIIIa complex, factor IXa was incubated with factor VIIIa and lipid for 3 min before addition of the aptamer for 1 min. Factor X and factor Xa substrate were then added to give the same final concentrations as before. Even though the incubation times were shorter, the aptamer was not less effective at inhibiting factor X activation (Fig. 4A) and may have been more effective (calculated affinity of 0.07 nm). This suggests that the aptamer is not competing with factor VIIIa for factor IXa binding.
Inhibition of Factor IXa Activation of Factor X
We next sought to determine whether the aptamer could inhibit factor IXa in the absence of factor VIIIa. Factor IXa activity in the absence of factor VIIIa was determined by activating factor X using lipid vesicles as the surface. Two concentrations of factor IXa were incubated with varied concentrations of either aptamer 9.3t or 9.3tM (Fig. 4B). A dose-dependent decrease in factor X activation was observed. Here, the apparent affinity of the aptamer for factor IXa was 0.9 nm in the absence of factor VIIIa.
Aptamer Blocks Antithrombin Inhibition of Factor IXa
We next wanted to determine whether the aptamer would interfere with the ability of antithrombin to inhibit factor IXa function. Factor IXa (1 μm) was incubated with 2 μm aptamer 9.3t or 9.3tM for 15 min. Antithrombin (20 μm) was added, and formation of the factor IXa-antithrombin complex was monitored by sampling into SDS buffer and heating to 95 °C for 3 min. Samples were run on a polyacrylamide gel and transferred to a nylon membrane for Western blotting. Aptamer 9.3t blocked formation of factor IXa-antithrombin complexes for at least 12 h (Fig. 5). Thus, the aptamer inhibits antithrombin from inactivating factor IXa.
FIGURE 5.

Aptamer blocks antithrombin inhibition of factor IXa. Factor IXa was incubated with aptamer 9.3t (●) or 9.3tM (○) before being added to antithrombin. At the indicated times, samples were run on a 10–15% gradient PhastGel, transferred to a nylon membrane, and Western-blotted for factor IXa. Images were analyzed using ImageJ. The proportion of the intensity in the factor IXa band is plotted versus time. The inset shows a grayscale image of the gel. Note that the time course is different for the left and right portions of the gel.
Aptamer Partially Inhibits Factor IXa Cleavage of Synthetic Substrate
To further elucidate the mechanism of inhibition, we next determined if the aptamer could inhibit cleavage of small synthetic substrates. Factor IXa was incubated with varied concentrations of aptamer 9.3t or 9.3tM before being added to synthetic substrate. As expected, increasing concentrations of aptamer 9.3t decreased the rate of substrate cleavage. Unexpectedly, substrate cleavage did not go to zero; even when the aptamer was in a molar excess to factor IXa, ∼10% residual substrate cleavage remained (Fig. 6A).
FIGURE 6.

Aptamer partially blocks factor IXa cleavage of synthetic substrate. A, factor IXa was incubated with aptamer 9.3t (●) or 9.3tM (○) before being added to synthetic substrate. The rate of substrate cleavage is plotted against the concentration of the aptamer. The curve shape suggests that the residual activity is not due to factor IXa that does not have aptamer bound. B, factor IXa (200 nm) was incubated with 400 nm aptamer 9.3t (●) or 9.3tM (○) before being added to DEGR-CK. Fluorescence at 490 nm (excitation at 330 nm) was monitored, and the change in fluorescence relative to the maximal change is plotted versus time.
Aptamer Slows Factor IXa Incorporation of Active Site Probe
We next examined the ability of the aptamer to inhibit an active site probe from binding to factor IXa. DEGR-CK can incorporate into the active site of factor IXa (32). Incorporation can be measured as a change in the fluorescent environment of the dansyl group. The chloromethyl ketone group is smaller than the p-nitroanilide group of the chromogenic substrate. Factor IXa (200 nm) was incubated with aptamer 9.3t or 9.3tM (400 nm) in the absence of calcium for 15 min before addition of 20 μm DEGR-CK. Incorporation was monitored by following the fluorescence at 490 nm (excitation at 330 nm). Data were fit to Equation 10.
Aptamer decreased Fmax, and although it did not completely block incorporation of DEGR-CK into the active site (Fig. 6B), aptamer 9.3t did decrease the rate to ∼30% of that observed with the mutant aptamer (from 3.2 to 1.1 s−1).
DISCUSSION
Functionally, factor IXa binds to factor VIIIa on a surface and converts factor X to factor Xa. Interfering with any of those functions, including lipid binding, factor VIIIa binding, or factor X cleavage, can block factor IXa activity. When the aptamer was incubated with factor IXa, factor VIIIa-dependent activation of factor X on a lipid surface was blocked in a dose-dependent fashion. The data could be fitted to a model in which the aptamer bound to factor IXa with a Kd of ∼0.2 nm. This compares tolerably well to values of 0.6 nm measured in a double-filter binding assay and 0.9 nm determined from factor IXa cleavage of factor X in the absence of factor VIIIa. Taken together, these values are in reasonable agreement with a previous publication that used surface plasmon resonance to measure a dissociation constant of ∼0.4 nm (31).
Monitoring the complete time course of factor IX activation showed that the aptamer did not alter factor IX activation by factor XIa. The aptamer did reduce factor IX activation by factor VIIa-tissue factor to ∼50% of the rate seen in its absence. This result was somewhat surprising given that, in a previous study, Gopinath et al. (31) reported a greater inhibition of factor IX activation by factor VIIa-tissue factor. The relative difference in our conclusions may arise from differences in the conditions used in the assays. We used 1 nm tissue factor with 2 nm factor VIIa and a ratio of factor IX to factor VIIa-tissue factor of 1000:1, and we monitored the reaction for up to 120 min by Western blotting so that the presence of other proteins did not interfere. Gopinath et al. used less tissue factor (0.5 nm) with more factor VIIa (800 nm) and a ratio of factor IX to factor VIIa-tissue factor of 17,400:1, and they assessed at a 30-min time point by Coomassie Blue staining. The loss of factor IX could not be easily assessed because the tissue factor reagent ran at the same place on the gel as factor IX. If staining of the cleavage product were not linear at low concentrations of protein, then the reduced rate of activation that we observed would not be inconsistent with the results of Gopinath et al. given the differences in methodology.
To determine whether the aptamer was interfering with factor IXa binding to factor VIIIa, the complex of factors IXa and VIIIa was allowed to form, and the aptamer was then added immediately before the start of the assay. If the aptamer and factor VIIIa are competing for a binding site on factor IXa, then the aptamer should not bind as well to the preformed complex as it does to factor IXa alone. However, the aptamer was not any less effective at inhibiting the preformed factor IXa-factor VIIIa complex, which suggests that the aptamer interacts with factor IXa at a site different from the sites of factor VIIIa interaction.
When factor VIIIa was removed from the reaction, the aptamer still blocked factor IXa activation of factor X, so the aptamer could be blocking either factor IXa binding to lipid or factor IXa binding to substrate. To help sort the two possibilities, a solution-phase reaction was studied. The aptamer blocked formation of a factor IXa-antithrombin complex for as long as 12 h. This result suggests that the aptamer acts by inhibiting substrate binding and cleavage rather than by inhibiting factor IXa binding to a surface.
One possible explanation for these observations would be that the aptamer binds to factor IXa so as to block access to the active site. Thus, we were surprised to observe that the aptamer did not completely block factor IXa cleavage of synthetic substrate, although the rate was reduced to 10% of the initial value. Titrating the aptamer into factor IXa suggested that all of the factor IXa in the reaction was bound to the aptamer (as expected because the Kd for complex formation was <1 nm and the concentration of factor IXa was 200 nm). Furthermore, saturating concentrations of the aptamer were not able to completely block incorporation of DEGR-CK into the active site of factor IXa, although the rate was reduced. This result is consistent with the observation that no aptamers directed toward a proteolytic active site have yet been identified and suggests that an exosite necessary for macromolecular substrate binding is blocked.
Exosites on coagulation proteins are important for providing the substrate specificity of those factors (33). There are a number of exosite inhibitors that target and can potently inhibit the function of many proteins in the coagulation cascade (33). An aptamer targeted to be an exosite inhibitor of thrombin completely blocks conversion of fibrinogen to fibrin (34) but does not inhibit cleavage of a small molecular weight substrate (35). Ixolaris, a protein from the salivary gland of ticks, binds to an exosite on factor Xa, and although it completely blocks factor Xa/Va activation of prothrombin, it enhances factor Xa cleavage of small synthetic substrates (36). A fusion of two peptides targeted to exosites of factor VIIa results in a molecule that completely inhibits factor X activation but only partially inhibits small substrate cleavage (37, 38). In this study, the action of aptamer 9.3t against factor IX behaved as an exosite inhibitor (completely blocked macromolecular substrate cleavage but only partially blocked small substrate interactions). To the best of our knowledge, this aptamer represents the first inhibitor that targets the extended substrate-binding site (exosite) of factor IXa. The mechanism of this aptamer can be distinguished from nitrophorin-2 because that molecule appears to block lipid binding, possibly through interaction with the light chain of factor IX (39).
Aptamer binding to a heavy chain site is consistent with the observed difference of factor XIa activation and factor VIIa-tissue factor activation. Factor XIa activation of factor IX is mediated in large part through factor XIa exosite interactions with the Gla domain of factor IX (40, 41). These interactions would not be changed by aptamer binding to the heavy chain. By contrast, the factor VIIa-tissue factor complex binds to the catalytic domain of its substrate to activate factor IX (42, 43), so it is not unreasonable that aptamer binding to the catalytic domain (heavy chain) of factor IX might alter activation by factor VIIa-tissue factor.
Among the macromolecular interactions blocked by the aptamer is factor IXa interaction with the natural inhibitor antithrombin. Although this might seem to argue against use of the aptamer in a clinical setting, data from preclinical studies on this molecule as well as from clinical studies of other active site inhibitors suggest otherwise. Active site inhibitors that, as a side effect, block antithrombin inactivation are currently available for thrombin (dabigatran) and factor Xa (rivaroxaban). With these inhibitors, the major adverse events are not thrombosis, which would be expected if factor Xa and thrombin are available because they are not inhibited, but rather the inhibitors are too effective, with bleeding, frequently major bleeding, being the dominant adverse effect (27). Early phase clinical studies with this aptamer have shown that it is effective, suggesting that the ability of the aptamer to block factor IXa activity outweighs any negative consequences of blocking antithrombin inhibition (21).
In conclusion, these data show that the factor IXa aptamer 9.3t blocks factor IXa interaction with macromolecular substrate but does not completely block interaction with small molecular weight active site probes. These results suggest 1) that aptamers may be particularly useful for probing extended surfaces on target proteins and for blocking substrate-binding exosites of enzymes and 2) that the factor IXa aptamer in clinical development acts as a potent anticoagulant in patients by inhibiting factor X binding to the factor IXa-factor VIIIa complex.
This work was supported, in whole or in part, by National Institutes of Health Grant HL065222. This work was also supported by the Coagulation Research Trust Fund from the University of North Carolina.
- aPTT
- activated partial thromboplastin time
- DEGR-CK
- dansyl-Glu-Gly-Arg chloromethyl ketone.
REFERENCES
- 1. Geerts W. H., Bergqvist D., Pineo G. F., Heit J. A., Samama C. M., Lassen M. R., Colwell C. W. (2008) Prevention of venous thromboembolism: American College of Chest Physicians evidence-Based clinical practice guidelines (8th Edition). Chest 133, 381S–453S [DOI] [PubMed] [Google Scholar]
- 2. Bauer K. A. (2002) Selective inhibition of coagulation factors: advances in antithrombotic therapy. Semin. Thromb. Hemost. 28, Suppl. 2, 015–024 [DOI] [PubMed] [Google Scholar]
- 3. Yavari M., Becker R. C. (2008) Anticoagulant therapy during cardiopulmonary bypass. J. Thromb. Thrombolysis 26, 218–228 [DOI] [PubMed] [Google Scholar]
- 4. Millar J. A. (2008) Rational thromboprophylaxis in medical inpatients: not quite there yet. Med. J. Aust. 189, 504–506 [DOI] [PubMed] [Google Scholar]
- 5. McNeely T. B., Griffith M. J. (1985) The anticoagulant mechanism of action of heparin in contact-activated plasma: inhibition of factor X activation. Blood 65, 1226–1231 [PubMed] [Google Scholar]
- 6. Béguin S., Dol F., Hemker H. C. (1991) Factor IXa inhibition contributes to the heparin effect. Thromb. Haemost. 66, 306–309 [PubMed] [Google Scholar]
- 7. Spanier T. B., Chen J. M., Oz M. C., Edwards N. M., Kisiel W., Stern D. M., Rose E. A., Schmidt A. M. (1998) Selective anticoagulation with active site-blocked factor IXa suggests separate roles for intrinsic and extrinsic coagulation pathways in cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 116, 860–869 [DOI] [PubMed] [Google Scholar]
- 8. Spanier T. B., Oz M. C., Minanov O. P., Simantov R., Kisiel W., Stern D. M., Rose E. A., Schmidt A. M. (1998) Heparinless cardiopulmonary bypass with active site-blocked factor IXa: a preliminary study on the dog. J. Thorac. Cardiovasc. Surg. 115, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 9. Refino C. J., Himber J., Burcklen L., Moran P., Peek M., Suggett S., Devaux B., Kirchhofer D. (1999) A human antibody that binds to the γ-carboxyglutamic acid domain of factor IX is a potent antithrombotic in vivo. Thromb. Haemost. 82, 1188–1195 [PubMed] [Google Scholar]
- 10. Tuerk C., Gold L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505–510 [DOI] [PubMed] [Google Scholar]
- 11. Ellington A. D., Szostak J. W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346, 818–822 [DOI] [PubMed] [Google Scholar]
- 12. Sullenger B. A., Gallardo H. F., Ungers G. E., Gilboa E. (1990) Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 63, 601–608 [DOI] [PubMed] [Google Scholar]
- 13. Long S. B., Long M. B., White R. R., Sullenger B. A. (2008) Crystal structure of an RNA aptamer bound to thrombin. RNA 14, 2504–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nimjee S. M., Rusconi C. P., Sullenger B. A. (2005) Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 56, 555–583 [DOI] [PubMed] [Google Scholar]
- 15. Rusconi C. P., Scardino E., Layzer J., Pitoc G. A., Ortel T. L., Monroe D., Sullenger B. A. (2002) RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419, 90–94 [DOI] [PubMed] [Google Scholar]
- 16. Rusconi C. P., Roberts J. D., Pitoc G. A., Nimjee S. M., White R. R., Quick G., Jr., Scardino E., Fay W. P., Sullenger B. A. (2004) Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol. 22, 1423–1428 [DOI] [PubMed] [Google Scholar]
- 17. Nimjee S. M., Keys J. R., Pitoc G. A., Quick G., Rusconi C. P., Sullenger B. A. (2006) A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol. Ther. 14, 408–415 [DOI] [PubMed] [Google Scholar]
- 18. Dyke C. K., Steinhubl S. R., Kleiman N. S., Cannon R. O., Aberle L. G., Lin M., Myles S. K., Melloni C., Harrington R. A., Alexander J. H., Becker R. C., Rusconi C. P. (2006) First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation 114, 2490–2497 [DOI] [PubMed] [Google Scholar]
- 19. Oney S., Nimjee S. M., Layzer J., Que-Gewirth N., Ginsburg D., Becker R. C., Arepally G., Sullenger B. A. (2007) Antidote-controlled platelet inhibition targeting von Willebrand factor with aptamers. Oligonucleotides 17, 265–274 [DOI] [PubMed] [Google Scholar]
- 20. Chan M. Y., Rusconi C. P., Alexander J. H., Tonkens R. M., Harrington R. A., Becker R. C. (2008) A randomized, repeat-dose, pharmacodynamic, and safety study of an antidote-controlled factor IXa inhibitor. J. Thromb. Haemost. 6, 789–796 [DOI] [PubMed] [Google Scholar]
- 21. Chan M. Y., Cohen M. G., Dyke C. K., Myles S. K., Aberle L. G., Lin M., Walder J., Steinhubl S. R., Gilchrist I. C., Kleiman N. S., Vorchheimer D. A., Chronos N., Melloni C., Alexander J. H., Harrington R. A., Tonkens R. M., Becker R. C., Rusconi C. P. (2008) Phase 1b randomized study of antidote-controlled modulation of factor IXa activity in patients with stable coronary artery disease. Circulation 117, 2865–2874 [DOI] [PubMed] [Google Scholar]
- 22. Cohen M. G., Purdy D. A., Rossi J. S., Grinfeld L. R., Myles S. K., Aberle L. H., Greenbaum A. B., Fry E., Chan M. Y., Tonkens R. M., Zelenkofske S., Alexander J. H., Harrington R. A., Rusconi C. P., Becker R. C. (2010) First clinical application of an actively reversible direct factor IXa inhibitor as an anticoagulation strategy in patients undergoing percutaneous coronary intervention. Circulation 122, 614–622 [DOI] [PubMed] [Google Scholar]
- 23. McCord D. M., Monroe D. M., Smith K. J., Roberts H. R. (1990) Characterization of the functional defect in factor IX Alabama. Evidence for a conformational change due to high affinity calcium binding in the first epidermal growth factor domain. J. Biol. Chem. 265, 10250–10254 [PubMed] [Google Scholar]
- 24. Griffith M. J., Noyes C. M., Church F. C. (1985) Reactive site peptide structural similarity between heparin cofactor II and antithrombin III. J. Biol. Chem. 260, 2218–2225 [PubMed] [Google Scholar]
- 25. Hope M. J., Bally M. B., Webb G., Cullis P. R. (1985) Biochim. Biophys. Acta 812, 55–65 [DOI] [PubMed] [Google Scholar]
- 26. Mayer L. D., Hope M. J., Cullis P. R. (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 858, 161–168 [DOI] [PubMed] [Google Scholar]
- 27. Perka C. (2011) Preoperative versus postoperative initiation of thromboprophylaxis following major orthopedic surgery: safety and efficacy of postoperative administration supported by recent trials of new oral anticoagulants. Thromb. J. 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Griffith M. J., Breitkreutz L., Trapp H., Briet E., Noyes C. M., Lundblad R. L., Roberts H. R. (1985) Characterization of the clotting activities of structurally different forms of activated factor IX. Enzymatic properties of normal human factor IXaα, factor IXaβ, and activated factor IX Chapel Hill. J. Clin. Invest. 75, 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Telgt D. S., Macik B. G., McCord D. M., Monroe D. M., Roberts H. R. (1989) Mechanism by which recombinant factor VIIa shortens the aPTT: activation of factor X in the absence of tissue factor. Thromb. Res. 56, 603–609 [DOI] [PubMed] [Google Scholar]
- 30. Wong I., Lohman T. M. (1993) A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. U.S.A. 90, 5428–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gopinath S. C., Shikamoto Y., Mizuno H., Kumar P. K. (2006) A potent anticoagulant RNA aptamer inhibits blood coagulation by specifically blocking the extrinsic clotting pathway. Thromb. Haemost. 95, 767–771 [PubMed] [Google Scholar]
- 32. Lollar P., Fass D. N. (1984) Inhibition of activated porcine factor IX by dansyl-glutamyl-glycyl-arginyl chloromethyl ketone. Arch. Biochem. Biophys. 233, 438–446 [DOI] [PubMed] [Google Scholar]
- 33. Bock P. E., Panizzi P., Verhamme I. M. (2007) Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost. 5, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nimjee S. M., Oney S., Volovyk Z., Bompiani K. M., Long S. B., Hoffman M., Sullenger B. A. (2009) Synergistic effect of aptamers that inhibit exosites 1 and 2 on thrombin. RNA 15, 2105–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Müller J., Freitag D., Mayer G., Pötzsch B. (2008) Anticoagulant characteristics of HD1–22, a bivalent aptamer that specifically inhibits thrombin and prothrombinase. J. Thromb. Haemost. 6, 2105–2112 [DOI] [PubMed] [Google Scholar]
- 36. Monteiro R. Q., Rezaie A. R., Ribeiro J. M., Francischetti I. M. (2005) Ixolaris: a factor Xa heparin-binding exosite inhibitor. Biochem. J. 387, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberge M., Santell L., Dennis M. S., Eigenbrot C., Dwyer M. A., Lazarus R. A. (2001) A novel exosite on coagulation factor VIIa and its molecular interactions with a new class of peptide inhibitors. Biochemistry 40, 9522–9531 [DOI] [PubMed] [Google Scholar]
- 38. Roberge M., Peek M., Kirchhofer D., Dennis M. S., Lazarus R. A. (2002) Fusion of two distinct peptide exosite inhibitors of factor VIIa. Biochem. J. 363, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Y., Ribeiro J. M., Guimarães J. A., Walsh P. N. (1998) Nitrophorin-2: a novel mixed-type reversible specific inhibitor of the intrinsic factor-X activating complex. Biochemistry 37, 10681–10690 [DOI] [PubMed] [Google Scholar]
- 40. Aktimur A., Gabriel M. A., Gailani D., Toomey J. R. (2003) The factor IX γ-carboxyglutamic acid (Gla) domain is involved in interactions between factor IX and factor XIa. J. Biol. Chem. 278, 7981–7987 [DOI] [PubMed] [Google Scholar]
- 41. Ogawa T., Verhamme I. M., Sun M. F., Bock P. E., Gailani D. (2005) Exosite-mediated substrate recognition of factor IX by factor XIa. The factor XIa heavy chain is required for initial recognition of factor IX. J. Biol. Chem. 280, 23523–23530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen S. W., Pellequer J. L., Schved J. F., Giansily-Blaizot M. (2002) Model of a ternary complex between activated factor VII, tissue factor, and factor IX. Thromb. Haemost. 88, 74–82 [PubMed] [Google Scholar]
- 43. Hamaguchi N., Roberts H., Stafford D. W. (1993) Mutations in the catalytic domain of factor IX that are related to the subclass hemophilia Bm. Biochemistry 32, 6324–6329 [DOI] [PubMed] [Google Scholar]