

Abstract
Underlying mechanisms for how bacterial infections contribute to active resolution of acute inflammation are unknown1-4. Here, we performed exudate leukocyte trafficking and mediator-metabololipidomics of murine peritoneal Escherichia coli (E. coli) infections with temporal identification of pro-inflammatory (prostaglandins and leukotrienes) and specialized pro-resolving mediators (SPM). In self-resolving E. coli exudates (105 CFU), the dominant SPM identified were resolvin (Rv) D5 and protectin D1 (PD1), which at 12 h were significantly greater than levels in exudates from higher titer E. coli (107 CFU) challenged mice. Germ-free mice displayed endogenous RvD1 and PD1 levels higher than in conventional mice. RvD1 and RvD5 (ng/mouse) each reduced bacterial titers in blood and exudates, E. coli-induced hypothermia and increased survival, demonstrating the first actions of RvD5. With human polymorphonuclear neutrophils (PMN) and macrophages, RvD1, RvD5, and PD1 each directly enhanced phagocytosis of E. coli, and RvD5 counter-regulated a panel of pro-inflammatory genes, including NF-κB and TNF-α. RvD5 activated the RvD1 receptor, GPR32, to enhance phagocytosis. With self-limited E. coli infections, RvD1 and the antibiotic ciprofloxacin accelerated resolution, each shortening resolution intervals (Ri). Host-directed RvD1 actions enhanced ciprofloxacin’s therapeutic actions. In 107 CFU E. coli infections, SPM (RvD1, RvD5, PD1) together with ciprofloxacin also heightened host antimicrobial responses. In skin infections, SPM enhanced vancomycin clearance of Staphylococcus aureus. These results demonstrate that specific SPM are temporally and differentially regulated during infections and that they are anti-phlogistic, enhance containment and lower antibiotic requirements for bacterial clearance.
The acute inflammatory response is a protective mechanism that is evolved to eliminate invading organisms. It should ideally be self-limited and lead to complete resolution, returning to homeostasis1-4. Evidence has emerged indicating that resolution of acute inflammation is an active process with biosynthesis of specialized pro-resolving mediators (SPM), e.g. resolvins (recently reviewed4,5). During natural resolution, PMN required for anti-microbial defense2 stop further infiltration, apoptose and are removed from the inflammatory site by specific macrophages (MΦ)6-10. In sterile inflammation, resolving exudates biosynthesize SPM from essential fatty acids including lipoxins (LX) from arachidonic acid, E-series resolvins (Rv) from omega-3 eicosapentaenoic acid (EPA), D-series Rv, protectins (PD) and maresins from docosahexaenoic acid (DHA) (reviewed in4,11). In humans, SPM production is also temporally regulated12,13, and SPM selectively control inflammation by stimulating resolution without immunosuppression, and are organ protective in eye, kidney, lung and periodontal diseases4,14.
Self-resolving E. coli infections versus higher titer E. coli challenges: E. coli infections are an urgent worldwide health concern as in a recent outbreak in Germany. In the United States, E. coli infections account for ~270,000 cases/year with underreporting estimated at ~20-fold15. Here we used a well-established model of murine peritonitis, relevant to human infections16, to identify specific SPM that may be directly involved in resolving infections. E. coli inoculation at 105 CFU/mouse i.p. evoked a self-limited host response (Fig. 1a). PMN infiltration reached maximum at ~12 h followed by decline. Monocytes/MΦ gradually increased from 2 h to 72 h; most of the exudate mononuclear cells at later time points were MΦ (~90% CD14+F4/80+ cells; Fig. S1a), a finding consistent with anti-phlogistic actions of MΦ (e.g. clearing apoptotic PMN)6,7. To provide quantitative analysis of resolution components with E. coli infection, we used resolution indices4, since they give unbiased assessment of progress during resolution and are now in wide use (cf. 9,17). Resolution indices were Tmax ~12h and T50 ~28h, giving a resolution interval (Ri) of 16h (see Methods). In sharp contrast, inoculation at 107 CFU/mouse delayed resolution with sustained PMN infiltration (Fig. 1b). PMN continued to increase until 72 hours and the major mononuclear cell type at later intervals was monocytes (~90 % CD14+F4/80−; Fig. S1a), with only ~6% MΦ. Also with the higher inoculum of 107 CFU, bacteria counts in both blood and lavages remained elevated at 24-48h, whereas with lower E. coli inoculum bacteria were cleared by 24h (Fig. S1b). Thus, high inoculum of 107 CFU evoked excessive PMN accumulation and limited MΦ in exudates that reflect delayed resolution of infection. Importantly, the lower E. coli inoculum gave self-limited profiles (Ri=16h), permitting differential analysis.
Figure 1. Profiling of SPM in E. coli infections.

Mice were inoculated with E. coli at (a) 105 or (b) 107 CFU by intraperitoneal injection, and peritoneal leukocytes enumerated. Results are expressed as mean±s.e.m., n=4-6. See Methods for calculation of resolution indices. *p<0.05, **p<0.01, vs. 24h; #p<0.05, ##p<0.01, ###p<0.001, vs. 48h. (c) Time course of SPM and related pathway markers. Results are expressed as mean±s.e.m. of n=5 separate time courses. *p<0.05, **p<0.01, self-resolving (105 CFU) vs. higher titer (107 CFU) E. coli exudates. (d) Representative MRM chromatograph of eicosanoid, resolvin and protectin pathway products from naive germ-free mice. Each LM was identified based on published LC-MS-MS30 (see Table S1). (e) SPM and pathway markers in colons of germ-free and conventional mice; representative of 3 mice. (f) Representative MS-MS of RvD1 (from germ-free mice) and RvD5 (from E. coli-infected mice); a=[M-H] (parent ion).
Infection-resolution metabololipidomics: Profiling metabololipidomics targeted on local acting lipid mediators (LM) was carried out with E. coli exudates using mass spectrometry-based LM-lipidomics targeting 5 LM metabolomes, e.g. leukotrienes, resolvins and protectins (Figs. 1c and S1). In self-resolving peritonitis (105 CFU), biosynthetic pathway markers for protectin D1 (PD1) and maresins (MaR1), namely 17-HDHA and 14-HDHA, were identified and elevated at the peak of PMN infiltration ~12h in resolving exudates (see Table S1 for LM identification). By comparison, mice that received higher titer E. coli (107 CFU) gave increased levels of proinflammatory LTB4 and reduced 17-HDHA and 14-HDHA levels at 12-48h (Figs. 1c & S1d). Within the initial phase (4h), RvD5 (7S,17S-dihydroxy-docosa-4Z,8E,10Z,13Z,15E,19Z-hexaenoic acid), a pivotal biosynthetic marker18 accumulated in self-resolving exudates (120±39 pg/exudate), ~3 times more than in exudates from higher titer E. coli infections (40±11 pg RvD5/exudates, p<0.05) (Figs. 1c & S1). At later interval 12h, RvD5 remained elevated (326±39 pg/exudate) in self-resolving exudates. It is noteworthy that PD1 levels were also significantly greater in self-resolving exudates (1066±247 pg/exudate) than in higher titer E. coli-challenged (468±116 pg/exudate). In these exudates, RvD5 and PD1 were most abundant of the DHA metabolome. Human PMN transform 17-H(p)DHA via two distinct biosynthetic pathways to RvD5 and/or PD1 (Fig. S2)4,18. These pathways were activated on infection and utilized endogenous essential fatty acids without supplement to biosynthesize potent novel mediators (vide infra). Their presence suggested specific role(s) in resolving infection.
RvD5 and PD1 each accumulated while RvD1 rapidly disappeared in exudates from E. coli infections. To monitor metabolic flux of RvD1 during E. coli infections, RvD1 was administered with E. coli (105 CFU) into peritoneum (Fig. S3a). At 12-24h post-inoculation, only 5-10% RvD1 was recovered from peritoneal exudates. Along these lines, with human macrophages ~40-50% of RvD1 was lost within 0.5-2.0 h accompanied by an increase in its further metabolite dihydro-RvD1. (Fig. S3b). Hence, these are dynamic pathways in infectious exudates.
We calculated ratios for pro-resolving vs. inflammatory mediators, i.e. RvD5/LTB4 and PD1/LTB4. In self-resolving exudates these ratios at 12 and 24h were greater than those in exudates from higher (107 CFU) E. coli infections (Fig. S1e), indicating that differential LM exudate profiles were present with these E. coli infections. To access their potential endogenous roles, we profiled these pathways in germ-free mice19 (Fig. 1d). In colons of naive germ-free mice, lower amounts of LTB4 were identified and increased levels of endogenous DHA products 14-HDHA, 17-HDHA, RvD1, as well as PD1 (Figs. 1e&f, S4). Hence, both endogenous and infected tissues produced D-series resolvins and PD1.
Since D-series resolvins, in particular RvD5, were one of the more abundant SPM, we sought to determine its impact in E. coli infections. RvD5 given in physiologic range, i.e. nanograms/mouse with E. coli (107 CFU) significantly enhanced phagocyte containment of E. coli in vivo (160% increase) compared to mice challenged with E. coli alone (Fig. 2a). RvD1 shared this action, registering 42% increase. Of note, RvD1 or RvD5 markedly reduced blood and exudate bacterial counts (Fig. 2b). Infected mice developed hypothermia, giving 2.4±1.2°C decrements in body (surface) temperature. Both RvD1 and RvD5 prevented this (Fig. 2c&S5a). The higher lethal dose E. coli (2.5×107 CFU) gave only 25% survival) and RvD1 significantly increased survival to ~66% (Figs. 2d and S5b, p<0.05). Here, RvD5 significantly reduced pro-inflammatory cytokines KC and TNFα in exudates and RvD1 decreased IL-1β (Fig. S5c). Thus, both RvD1 and RvD5 stimulated phagocyte E. coli ingestion, lowered bacterial titers, protected from hypothermia and increased survival.
Figure 2. RvD1 and RvD5 protect mice during infection: enhancing bacterial killing and preventing hypothermia.

Mice were inoculated with E. coli (107 CFU) together with RvD1 or RvD5 methyl ester (100 ng), and peritoneal exudates collected 24h later. (a) Intracellular E. coli levels. MFI denotes mean fluorescence intensity. Results are expressed as mean of n=3-5. (bottom panel) Representative histograms. (b) Bacterial titers. Results are expressed as mean of n=4-5. (c) Changes in body temperatures expressed as mean of Δ°C (24h-0h) n=4-10. *p<0.05, **p<0.01, vs. E. coli alone; ##p<0.01, vs. naïve mice. (d) % survival of E. coli inoculated mice (2.5×107 CFU) alone or with RvD1 (100 ng/mouse). *p<0.05 log-rank (Mantel-Cox) test, n=12 each group.
Given these in vivo findings, we next questioned whether SPM have a direct impact on bacterial containment with isolated human cells. RvD1, RvD5 and PD1 each potently enhanced human MΦ phagocytosis of fluorescent E. coli ~40-70% increases in pM to nM ranges (Fig. 3a-c). We also tested whether RvD5 activates human RvD1 receptor GPR32 expressed in a beta-arrestin reporter system20, and found that RvD5 directly activates this receptor (Fig. S6a). RvD5-enhanced phagocytic activity also proved dependent on GPR32. Both RvD1 and RvD5 increased MΦ phagocytosis of E. coli, an action further enhanced with GPR32 overexpression compared to mock transfected cells (Fig. S6b).
Figure 3. SPM enhance human macrophage phagocytosis of E. coli.
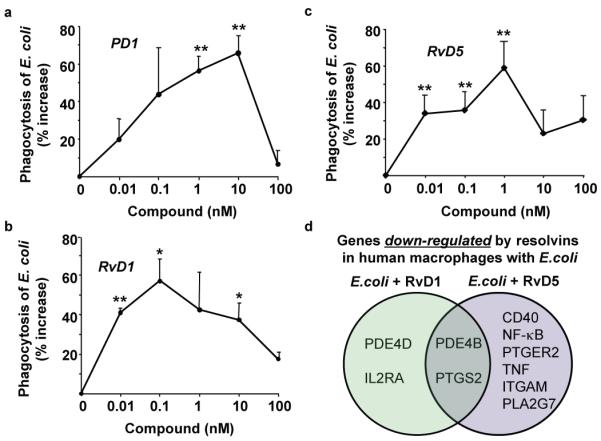
(a-c) MΦ phagocytosis of fluorescent E. coli in the presence of PD1, RvD1 or RvD5. Results are percent increase above vehicle and expressed as mean±s.e.m., d=3-4, n=3 (PD1) or 4 (RvD1 and RvD5). *p<0.05, **p<0.01, E. coli plus SPM vs. E coli alone. (b) Venn diagrams of genes regulated by resolvins in human MΦ incubated with E. coli. CD40 (TNFRSF5, TNF receptor superfamily member 5), ITGAM (CD11b), PDE (phosphodiesterase), PLA2 (phospholipase A2), PTGS2 (cyclooxygenase-2), PTGER2 (prostaglandin E2 receptor, EP2).
Gene array analysis with human MΦ (> 90 genes) showed that E. coli up-regulated a panel of inflammation-related genes, including NF-κB, phosphodiesterase 4B (PDE4B), COX-2 (PTGS2) and TNFα. These results are consistent with Gram-negative lipopolysaccharide (LPS) stimulation of MΦ that up-regulates COX-221, phosphodiesterase 4B (PDE4B) and TNFα22. When RvD5 (1 nM) was incubated with MΦ and E. coli, many of these genes were down-regulated. RvD1 down-regulated, e.g., PDE4B and COX-2 (Fig. 3d and Table S2). Ablation of PDE4B protects mice from LPS-induced septic shock23 and inhibition of COX enhances bacterial clearance of penicillin-resistant Streptococcus pneumoniae24. Therefore, RvD1 and RvD5 each counter-regulate COX and proinflammatory signaling components initiated by E. coli that might contribute to their actions in enhancing non-phlogistic phagocytosis (Fig. 3, Table S2) and bacterial clearance in vivo (Fig. 2).
Similar findings were obtained with human PMN, where RvD1, RvD5 and PD1 each stimulated PMN anti-microbial mechanisms, increasing E. coli ingestion and intracellular reactive oxygen species (ROS) production for killing during E. coli challenge (Fig. S7). These SPM did not exhibit direct anti-bacterial activities (Fig. S8). Together, these results characterize anti-phlogistic and antibacterial actions of RvD5, RvD1 and PD1 in that they enhance phagocytic activity of human phagocytes without evoking pro-inflammatory responses of these cells.
SPM and antibiotics: Antibiotic-resistant bacteria are a global concern, increasing health-care costs. Ciprofloxacin, for example, is the only antibiotic recommended by the World Health Organization for management of bloody diarrhea25. Unfortunately, rapid increases in bacterial resistance to ciprofloxacin reduces treatment options25. Hence, new strategies are needed. Given that specific SPM enhanced bacterial containment (Figs. 2, 3), we questioned whether SPM-directed host responses would embellish antibiotic treatment. To this end, we determined resolution indices for ciprofloxacin and RvD1. Given along with E. coli (105 CFU), RvD1 (50 ng) reduced maximal PMN (Ψmax), and shortened resolution interval (Ri) from ~20h to ~12h (Fig. 4a). Ciprofloxacin alone at equi-doses also reduced Ri to ~11h and initiated resolution at an earlier point Tmax ~6h. RvD1 plus ciprofloxacin further accelerated the onset of resolution with Tmax ~4h, and reduced Ri to ~5h (Fig. 4a and Table S3).
Figure 4. SPM and antibiotics accelerate resolution and enhance bacterial killing.

(a-c) Mice were inoculated with E. coli (105 CFU), RvD1 methyl ester (50 ng), and ciprofloxacin (Cipro, 50 ng). (a) Exudate PMN numbers and resolution indices. (b) Bacterial titers. (c) 14-HDHA and PD1 levels determined using LC-MS-MS-based LM-lipidomics. Results are expressed as mean±s.e.m. *p<0.05, **p<0.01, vs. E. coli alone; #p<0.05, ##p<0.01, vs. E. coli+RvD1; §p<0.05, vs. E. coli+Cipro. Gray, E. coli alone; blue, E.coli+RvD1; green, E.coli+Cipro; dark blue, E.coli+RvD1+Cipro. (d) Mice were inoculated with E. coli (107 CFU), SPM panel (RvD1, RvD5 and PD1; 50 ng each) and Cipro (25 μg). Body temperatures and bacterial titers were determined at 24h. Results are expressed as mean±s.e.m. *p<0.05, **p<0.01, ***p<0.001 vs. E. coli alone; #p<0.05, ##p<0.01 vs. E. coli+SPM; §p<0.05, vs. E. coli+Cipro. (e,f) Murine dorsal pouches were given live S. aureus (105 CFU) alone, plus SPM (RvD1, RvD5, PD1 at 100 ng each per mouse), vancomycin (Vanco, 2.5 μg), or both SPM and vancomycin by intra-pouch injection, pouch exudates were collected at 24h. (e) PMN and bacterial counts (CFU). Results are expressed as mean±s.e.m. *p<0.05, **p<0.01 vs. S. aureus alone; #p<0.05, vs. S. aureus+SPM; §§p<0.01, vs. S. aureus+Vanco. (f) Skin-pouch biopsies (Magnifications 40X). Arrows denote PMN infiltration into pouch linings. PC, pouch cavity. L, linings. Animal numbers are denoted within brackets (a) or bars (b-e).
RvD1 plus ciprofloxacin further reduced bacterial titers in blood and exudates at 4h, compared to ciprofloxacin alone (Fig. 4b). Similar results were obtained at 12h with exudates, whereas there were no significant differences between Cipro and Cipro plus RvD1 in blood. RvD1 actions were more pronounced at 4h in blood. A panel of cytokines/chemokines was monitored (Fig. S9A), and RvD1 and ciprofloxacin each gave differential actions; RvD1 significantly reduced IL-1β, IL-6 and increased IL-10 at 4h, and IFNγ at 24h. Ciprofloxacin alone reduced IL-12 levels at 4h (Fig. S9B). We also assessed LM metabololipidomic profiles: RvD1 plus ciprofloxacin significantly increased both 14-HDHA and PD1 at 4h (Figs. 4c, S9C&D), whereas neither alone was effective. At 12h, RvD1 or ciprofloxacin alone increased endogenous PD1. In addition, RvD1 or ciprofloxacin alone at 4 h did not enhance macrophage containment of E. coli, but significantly increased phagocytic activity when both were administered (Fig. S9E). Thus, host-directed RvD1 responses plus bacteria-directed antibiotic accelerated resolution via enhanced E. coli killing and clearance, as well as selectively regulated both endogenous LM and cytokines.
Since the specific SPM identified in infectious exudates were from DHA metabolome (Fig. 1), we tested RvD1, RvD5 and PD1 together (50 ng each) plus ciprofloxacin (25 μg/mouse) in higher titer E. coli (107 CFU) infections. Sub-optimal doses of ciprofloxacin (25 μg, Fig. S10A) still protected mice from hypothermia and reduced bacterial titers, actions that were enhanced by the SPM panel (Fig. 4d). In addition, treatment with both ciprofloxacin and SPMs significantly increased phagocyte ingestion of E coli, whereas neither treatment alone proved effective (Fig. S10B).
SPM enhance actions of vancomycin in S. aureus-initiated infections: Staphylococcus aureus is an emerging cause of various skin infections and a high percentage of hospital-acquired infections are caused by antibiotic-resistant S. aureus25. To assess a second system, we determined the impact of SPM and vancomycin in S. aureus-initiated infections in murine dorsal skin pouches. SPM panel (RvD1, RvD5 and PD1, 100 ng each) and/or sub-optimal doses of vancomycin (2.5 μg) were administrated with S. aureus (105 CFU) via intra-pouch injections. SPM or vancomycin alone at 4h each decreased exudate bacterial counts by ~10-fold. Treatment with both further reduced bacterial counts by ~100-fold (Fig. S11 A,B). Similar results were obtained at 24 h; SPM and vancomycin together gave significantly lower bacterial counts compared to those from each treatment alone (Fig. 4e). In S. aureus-infected mice, histologic analysis demonstrated PMN infiltration and Gram stain positives within linings surrounding the pouch cavities (Figs. 4f & S11c) that were markedly reduced by SPM, vancomycin and their use together (Fig. 4f).
A panel of cytokines/chemokines was also monitored. SPM and vancomycin at 4h significantly reduced S. aureus-induced IL-6 and GM-CSF in pouch exudates (Fig. S11), whereas neither alone was effective. It is noteworthy that vancomycin reduced LTB4 and increased the Rv precursor 17-HDHA (Fig. S11). Other SPM, including resolvin E1, protect mice from E. coli pneumonia and acute lung injury26 and 15-epi-LXA4, an arachidonic acid-derived SPM, enhances resolution of E. coli pulmonary inflammation27 and macrophage phagocytosis28. Hence, it appears that specific SPM pathways are used by the host during Gram-positive and Gram-negative bacterial infections.
Here, we carried out LM-metabolomics with infectious-inflammatory exudates and identified specific pro-resolving mediators that were also present in germ-free mice. RvD1 and RvD5 each accelerated resolution of E. coli infections and increased survival. SPM given together with ciprofloxacin shortened resolution intervals, stimulated phagocyte containment of E. coli, enhanced bacterial killing, and protected mice from hypothermia. These are the first actions identified for RvD5. With human macrophages, both RvD1 and RvD5 stimulated phagocytosis of E. coli in a GPR32-dependent manner. SPM also enhanced antibiotic effectiveness in clearing Gram-positive S. aureus skin infections. Together, these results indicate that stimulation of targeted host resolution responses by pro-resolving mediators in conjunction with bacterial-directed antibiotics lowers the required antibiotic doses for bacterial clearance. They illustrate new opportunities to address antibiotic resistance via lowering antibiotic use by also targeting host resolution programs.
Method Summary
Microbial-initiated inflammation in vivo
Peritonitis
Mice were given SPM or vehicle together with live E. coli (serotype O6:K2:H1; 105 or 107 CFU) or saline injections (mock infection) i.p. Ciprofloxacin was given 1 hr after E. coli. At designated points, mice were euthanized, peritoneal exudate and blood were collected. Cellular composition. intracellular E coli, bacterial titers and body temperatures were determined.
Resolution indices were calculated as in4: Ψmax, the maximal PMN numbers; Tmax, the time point when PMN reach maximum; R50; 50% of maximal PMN; T50, the time point when PMN reduce to 50% of maximum; Ri (resolution interval), T50 -Tmax, the time period when 50% PMN are lost from exudates.
Murine dorsal skin pouches
Mice were given SPM and/or vancomycin with live S. aureus (serotype (b)c1; 105 CFU) by intra-pouch injection. Pouch exudates were collected, and bacterial counts determined. Skin punch biopsies were collected for Gram and hematoxylin-eosin staining.
Mediator-Metabololipidomics
LC/MS/MS-based LM-lipidomics were carried out using either an ABI QTrap 3200 or Qtrap 5500 (AB Sciex). LM were profiled via multiple reaction monitoring (MRM) and identified using retention time and at least 6 diagnostic ions29.
RvD5 synthesis
RvD5 was prepared by incubating 7(R/S)-HDHA with soybean 15-LO followed by chiral HPLC separation29. For methyl ester preparation, RvD5 was treated with excess ethereal diazomethane and purified using reverse phase HPLC.
Macrophage and PMN phagocytosis
Human PMN or GM-CSF-differentiated MΦ were incubated with SPM or vehicle for 15 min at 37°C, followed by fluorescent-labeled E. coli at a 50:1 ratio (E. coli:MΦ) for 60 min (phagocytosis) or 120 min (gene arrays).
Statistical analysis
Statistical analyses were performed using one-way ANOVA or the Student’s t test; p≤0.05 was taken as significant. Percent survival was analyzed by log-rank test (GraphPad).
Methods
Bacteria growth
E. coli (serotype O6:K2:H1) were cultured in LB broth and harvested at mid-log phase (OD600 ~0.5, 5×108 CFU/ml), washed in sterile saline before inoculation into mouse peritoneum.
Microbial-initiated peritonitis & leukocyte differentials
Mice were anesthetized with isoflurane and experiments were carried out with male FVB mice (6 to 8 weeks; Charles River; lab diet containing essential fatty acids from supplier). Microbial-initiated peritonitis was performed in accordance with the Harvard Medical Area Standing Committee on Animals (protocol no. 02570). Briefly, mice were anesthetized, test compounds or vehicle controls were injected into the peritoneal cavity together with live E. coli (105 or 107 CFU). The antibiotic ciprofloxacin (Sigma) was given 1 hour after E. coli injection. At designated points, mice were euthanized (overdose of isoflurane), blood was collected by heart puncture, and peritoneal exudate was collected by lavaging with 5 ml PBS. Cellular composition. Exudates were taken for differential leukocyte counts determined by nuclear morphology with light microscopy using Cytofuge (Statspin, Norwood, MA) stained with Wright Giemsa stain. For flow cytometry, aliquots of exudate cells were incubated with anti-mouse CD16/32 blocking Ab (0.5 μg/0.5×106 cells, 5 min) and then incubated (20 min, 4°C) with individual antibody or combinations of FITC-conjugated anti-mouse CD14 (clone rmC5-3) for mononuclear cells and PE-conjugated anti-mouse F4/80 Ab (clone BM8) for MΦ or PE-conjugated anti-mouse Ly-6G (clone RB6-8C5) for PMN, to determine leukocyte sub-types (FACS Canto II). Antibodies were from BD Biosciences (San Jose, CA) and eBioscience (San Diego, CA). Intracellular E. coli levels were determined using a FITC-conjugated anti-E. coli antibody (GTX40856; GeneTex). Bacterial counts. Aliquots of lavage or blood were used for serial dilution, plated onto LB agar plates, and cultured overnight at 37°C. Body temperature was monitored using an infrared thermometer (Fluke).
Resolution indices were calculated4: Ψmax, the maximal PMN numbers in the exudates; Tmax, the time point when PMN numbers reach maximum; R50; 50% of maximal PN numbers; T50, the time point when PMN numbers reduce to 50% of maximum; Ri (resolution interval), T50 -Tmax, the time period when 50% PMN are lost from the exudates.
Murine dorsal skin pouches
Pouches were raised for 6 days30. Mice were given test SPM and/or vancomycin with live S. aureus (serotype (b)c1; 105 CFU) by intra-pouch injection. At designated points, mice were euthanized, intra-pouch exudate and blood were collected, and bacterial counts determined. Skin punch biopsies were collected for both Gram and hematoxylin-eosin staining.
Germ-free mice
Germ-free 12-week-old C57BL/6J male mice were maintained in flexible film isolators under a 12-h light cycle and fed an autoclaved chow diet (Labdiet, St Louis, MO) ad libitum. Mice were sacrificed by cervical dislocation and colon were removed. All tissues were snap frozen in liquid nitrogen and stored at -80C. Animal protocols were approved by the Research Animal Ethics Committee in Gothenburg, Sweden.
Mediator-metabololipidomics
All incubations and in vivo samples (i.e. exudates) were stopped with two volumes of cold methanol, containing deuterated internal standards (d8-5S-HETE, d4-LTB4 and d4-PGE2; Cayman Chemical, Ann Arbor, MI) were added to each sample to obtain the extraction recoveries for specific lipid mediators. Samples were taken for solid-phase extraction and LC/MS/MS-based mediator lipidomics using an HPLC-UV (Agilent 1100) coupled to an ion-trap mass spectrometer in some experiments (QTrap 3200; Applied Biosystems/Sciex); for others an ABI Qtrap 5500 equipped with a C18 column (Agilent Eclipse Plus, 4.6 mm × 50 mm × 1.8 μm) was used. The mobile phase consisted of methanol/water/acetic acid (60/40/0.01;v/v/v) and was ramped to 80/20/0.01 (v/v/v) over 5 min, to 95/5/0.01 (v/v/v) over the next 3 min and to 100/0/0.01 (v/v/v) over 6 min before returning to 60/40/0.01 (v/v/v) at a flow rate of 0.4 ml/min. Lipid mediators were profiled using multiple reaction monitoring (MRM) and identified by direct comparison with synthetic and biogenic standards using retention time and 6 diagnostic ions for matching criteria29. Linear calibration curves were determined using mixtures of lipid mediator standards: d8-5S-HETE (r2= 0.9939, n=7), d4-LTB4 (r2 = 0.9995, n=7), d4-PGE2 (r2 = 0.9969, n=7), RvD1 (r2= 0.9954, n=7), RvD2 (r2= 0.9931, n=7), RvE1 (r2= 0.9941, n=7), LXA4 (r2= 0.9941, n=7), LXB4 (r2= 0.9959, n=7), PGE2 (r2= 0.8370, n=7), PGD2 (r2= 0.9893, n=7), 20-OH-LTB4 (r2= 0.9960, n=7), LTB4 (r2= 0.9949, n=7), PD1 (r2= 0.9948, n=7), 17-HDHA (r2= 0.9931, n=7), 14-HDHA (r2= 0.9940, n=7), 7-HDHA (r2= 0.9964, n=7), 4-HDHA (r2= 0.9950, n=7), 15-HETE (r2= 0.9978, n=7), 12-HETE (r2= 0.9975, n=7), 5-HETE (r2= 0.9905, n=7) at 50, 100, 200, 400 and 800pg. Quantification was carried out based on the peak area of the Multiple Reaction Monitoring (MRM) transition and the linear calibration curve for each. Synthetic deuterium labeled RvD1 was prepared by Dr. N. Petasis (USC) for tracking and quantitation of RvD1 in the P01(GM095467).
RvD5 synthesis and isolation
Biogenic RvD5 was prepared by incubating 7(R/S)-HDHA (Cayman Chemical) with soybean 15-LO (Sigma-Aldrich). Briefly, 7(R/S)-HDHA (50 μM) was suspended in 50mM sodium borate buffer (pH 9.3) and 1000 units of 15-LOX added. After 5 minutes, the reaction was quenched by adding the same volume of cold methanol and reduced with excess amount of sodium borohydride. Reaction mixtures were acidified and directly injected to HPLC to separate 7S,17S-dihydroxy-containing product termed RvD5. Characteristic RvD5 chromophore of lambda(max)=242nm was used for UV monitoring29. For methyl ester preparation, RvD5 was treated with excess ethereal diazomethane, taken to dryness with nitrogen gas, suspended in methanol and isolated using HPLC. RvD5 reaction mixture was separated by chiral column (Chiralcel AD-RH, 150mm × 2.1mm) equipped HPLC (Agilent HP 1100 Chemstation with DAD) with gradient eluent, methanol:water:acetic acid=95:5:0.01 to 100:0:0.01 for 10 minutes at the flow rate of 200 μL/min.
Macrophage phagocytosis and gene arrays
Human MΦ were obtained by differentiating PBMC, collected from healthy volunteers, with GM-CSF (10 ng/ml) in culture medium for 7 days. Before each experiment (24 hours), cells were adhered on a 24-well plate (0.1×106 cells/well) in culture medium deprived of GM-CSF. In parallel, E. coli (serotype O6:K2:H1) harvested at mid-log phase (OD600 ~0.5; ~5×108 CFU/ml) were labeled with fluorescence dye (BacLight, Molecular Probes, Carlsbad, CA). MΦ were exposed to vehicle (DPBS+/+) alone or compounds (PD1, RvD1, RvD5) for 15 min at 37°C, followed by incubation with fluorescent-labeled E. coli at 50:1 ratio (E. coli: MΦ) for 60 min at 37 °C. Plates were gently washed, extracellular E. coli quenched by trypan blue, and phagocytosis determined by measuring total fluorescence (Ex 495/Ex535 nm) using a fluorescent plate reader (Molecular Probes), and confirmed by flow cytometry (FACS Canto II).
For inflammatory gene arrays, human MΦ (2×106 cells) were adhered to 10-cm2 petri dishes. Next, PD1, RvD1, RvD5 (1 nM) or vehicle was added for 15 min at 37°C, followed by addition of E. coli (E. coli:MΦ 50:1) for 120 min at 37°C. Total RNA was isolated and inflammatory gene array carried out following manufacturer’s instruction (SA Biosciences). Results were analyzed using RT2 profiler PCR array data analysis (SA Biosciences).
GPR32 receptor β-arrestin reporter system
CHO cells engineered to stably coexpress human GPR32 linked to the Pro-Link peptide and β-arrestin tagged with the β-galactosidase EA (custom constructs, DiscoveRx, Fremont, CA) denoted CHO-GPR32 were routinely grown in medium with selection antibiotics, and plated in 96-well plates (at 20,000 or 10,000 cells/well) at twenty-four hours prior to experiments. Receptor activation was determined by increased chemiluminescence upon conversion of hydrolysable substrate (PathHunter EFC Detection kit™) catalyzed by β-gal using a luminometer (Molecular Probes).
PMN phagocytosis and generation of intracellular ROS
Human PMN were obtained from healthy volunteers according to Partners Human Research Committee Protocol #1999-P-001297. Adherent human PMN (5×106 cells/well in a 24-well plate) were incubated with RvD5, RvD1 or PD1 (0.1-100nM) or vehicle for 15min prior to incubation with E. coli (JM109; 50:1 ratio) for 60 min at 37°C. Plates were gently washed, extracellular E. coli quenched by trypan blue, and phagocytosis determined by measuring total fluorescence (Ex 495/Ex535 nm) using a fluorescent plate reader (Molecular Probes), and confirmed by flow cytometry (FACS Canto II). To assess intracellular ROS generation, PMN (5×106 cells/well in a 24-well plate) were pre-incubated with 5 μM carboxy-H2DCFDA (C400; Invitrogen) for 30min, followed by incubation with SPM for 15 min, and then E. coli (50:1 ratio) for 60 min. Probe oxidation was determined using a fluorescent plate reader.
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis were performed using one-way ANOVA or the Student’s t test and p≤0.05 was considered to be significant.
Supplementary Material
Acknowledgements
The authors thank Mary Small for expert assistance with manuscript preparation, Dr. Jim Lederer (Dept. of Surgery, BWH) for cytokine measurements, Dr. Jesmond Dalli (CET&RI, BWH) for helpful discussions, and Dr. Nicos Petasis for preparation of deuterium-labeled RvD1.
This work was supported in part by NIH grants P01GM095467 and R01GM38765 (CNS).
Abbreviations
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- GF
Germ-free
- LC-MS-MS
liquid chromatography tandem mass spectrometry
- LM
lipid-derived mediators
- LOX
lipoxygenase
- LT
leukotriene
- LX
lipoxin
- PG
prostaglandins
- Mϕ
macrophage
- Maresins
macrophage mediators in resolving inflammation
- MFI
mean fluorescence intensity
- PMN
polymorphonuclear leukocyte
- PD/NPD1
protectin D1/ neuroprotectin D1, 10R,17S-dihydroxydocosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid
- SPM
specialized pro-resolving mediators
- Rv, Resolvins
bioactive omega -3 derived resolution phase interaction products
- RvD1, resolvin D1:
7S,8R,17S-trihydroxydocosa-4Z,9E,11E,13Z,15E,19Z-hexaenoic acid
- RvD5, resolvin D5:
7S,17S-dihydroxy-docosa-4Z,8E,10Z,13Z,15E,19Z-hexaenoic acid
- ROS
reactive oxygen species
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions NC, GBF & SFO contributed to experimental design, carried out experiments and data analyses. TV and SFO performed metabololipidomics and lipid mediator analyses. FB carried out experiments with germ-free mice and contributed to manuscript composition. BAS carried out dermatopathology. All authors contributed to manuscript presentation and figure preparation. NC and CNS carried out overall experimental design and CNS conceived of the overall research plan.
Author Information Reprints and permissions information is available at www.nature.com/reprints.
Competing Financial Interests C.N.S. is an inventor on patents [resolvins] assigned to BWH and licensed to Resolvyx Pharmaceuticals. C.N.S. is a scientific founder of Resolvyx Pharmaceuticals and owns equity in the company. C.N.S.’ interests were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
References
- 1.Houck JC, editor. Chemical Messengers of the Inflammatory Process. Elsevier/North-Holland Biomedical Press; 1979. [Google Scholar]
- 2.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–776. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN. Resolution phases of inflammation: novel endogenous anti-inflammatory and pro-resolving lipid mediators and pathways. Annu. Rev. Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 5.Stables MJ, Gilroy DW. Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 2011;50:35–51. doi: 10.1016/j.plipres.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Henneke P, Golenbock DT. Phagocytosis, innate immunity, and host-pathogen specificity. J. Exp. Med. 2004;199:1–4. doi: 10.1084/jem.20031256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi AG, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro-Xavier RA, et al. A new strategy for the identification of novel molecules with targeted proresolution of inflammation properties. J. Immunol. 2010;184:1516–1525. doi: 10.4049/jimmunol.0902866. [DOI] [PubMed] [Google Scholar]
- 10.Schif-Zuck S, et al. Satiated-efferocytosis generates pro-resolving CD11blow macrophages: Modulation by resolvins and glucocorticoids. Eur. J. Immunol. 2011;41:366–379. doi: 10.1002/eji.201040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Caterina R. n-3 fatty acids in cardiovascular disease. N. Engl. J. Med. 2011;364:2439–2450. doi: 10.1056/NEJMra1008153. [DOI] [PubMed] [Google Scholar]
- 12.Morris T, et al. Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol. 2009;183:2089–2096. doi: 10.4049/jimmunol.0900477. [DOI] [PubMed] [Google Scholar]
- 13.Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 2011;121:569–581. doi: 10.1172/JCI42545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spite M, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mead PS, et al. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klingensmith ME, Soybel DI. In: The Physiological Basis of Modern Surgical Care. Miller TA, Rowlands BJ, editors. Mosby Year Book; 1998. pp. 478–490. [Google Scholar]
- 17.Xu YN, Zhang Z, Ma P, Zhang SH. Adenovirus-delivered angiopoietin 1 accelerates the resolution of inflammation of acute endotoxic lung injury in mice. Anesth. Analg. 2011;112:1403–1410. doi: 10.1213/ANE.0b013e318213fbd3. [DOI] [PubMed] [Google Scholar]
- 18.Serhan CN, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. J. Exp. Med. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnamoorthy S, et al. Resolvin D1 binds human phagocytes with evidence for pro-resolving receptors. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1660–1665. doi: 10.1073/pnas.0907342107. doi:doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grkovich A, Johnson CA, Buczynski MW, Dennis EA. Lipopolysaccharide-induced cyclooxygenase-2 expression in human U937 macrophages is phosphatidic acid phosphohydrolase-1-dependent. J. Biol. Chem. 2006;281:32978–32987. doi: 10.1074/jbc.M605935200. [DOI] [PubMed] [Google Scholar]
- 22.Jin SL, Lan L, Zoudilova M, Conti M. Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J. Immunol. 2005;175:1523–1531. doi: 10.4049/jimmunol.175.3.1523. [DOI] [PubMed] [Google Scholar]
- 23.Link A, Selejan S, Maack C, Lenz M, Böhm M. Phosphodiesterase 4 inhibition but not beta-adrenergic stimulation suppresses tumor necrosis factor-alpha release in peripheral blood mononuclear cells in septic shock. Crit. Care. 2008;12:R159. doi: 10.1186/cc7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stables MJ, et al. Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood. 2010;116:2950–2959. doi: 10.1182/blood-2010-05-284844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.World Health Organization Antibiotics Resistance. 2011 Feb; Factsheet No. 194. 2011, doi: http://www.who.int/mediacentre/factsheets/fs194/en/
- 26.Seki H, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010;184:836–843. doi: 10.4049/jimmunol.0901809. doi:doi: 10.4049/jimmunol.0901809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El Kebir D, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009;180:311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prescott D, McKay DM. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;301:G487–G497. doi: 10.1152/ajpgi.00042.2011. [DOI] [PubMed] [Google Scholar]
- 29.Yang R, Chiang N, Oh SF, Serhan CN. Metabolomics-lipidomics of eicosanoids and docosanoids generated by phagocytes. Curr. Protoc. Immunol. 2011;95:14.26.11–14.26.26. doi: 10.1002/0471142735.im1426s95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winyard PG, Willoughby DA, editors. Inflammation Protocols. Humana; 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.