

Abstract
HIV-1 protease is an important target for the treatment of HIV/AIDS. However, drug resistance is a persistent problem and new inhibitors are needed. An approach toward understanding enzyme chemistry, the basis of drug resistance, and the design of powerful inhibitors is to establish the structure of enzymatic transition states. Enzymatic transition structures can be established by matching experimental kinetic isotope effects (KIEs) with theoretical predictions. However, the HIV-1 protease transition state has not been previously resolved using these methods. We have measured primary 14C and 15N KIEs and secondary 3H and 18O KIEs for native and multidrug-resistant HIV-1 protease (I84V). We observed 14C KIEs (14V/K) of 1.029 ± 0.003 and 1.025 ± 0.005, 15N KIEs (15V/K) of 0.987 ± 0.004 and 0.989 ± 0.003, 18O KIEs (18V/K) of 0.999 ± 0.003 and 0.993 ± 0.003, and 3H KIEs (3V/K) KIEs of 0.968 ± 0.001 and 0.976 ± 0.001 for the native and I84V enzyme, respectively. The chemical reaction involves nucleophilic water attack at the carbonyl carbon, proton transfer to the amide nitrogen leaving group, and C-N bond cleavage. A transition structure consistent with the KIE values involves proton transfer from the active site Asp-125 (1.32 Å) with partial hydrogen bond formation to the accepting nitrogen (1.20 Å) and partial bond loss from the carbonyl carbon to the amide leaving group (1.52 Å). The KIEs measured for the native and I84V enzyme indicate nearly identical transition states, implying that a true transition-state analogue should be effective against both enzymes.
Keywords: aspartyl protease, protease mechanism, transition-state structure, drug design
Human immunodeficiency virus-1 (HIV-1) protease is an essential enzyme for the HIV-1 viral life cycle and is the target of nine drugs approved by the U.S. Food and Drug Administration (FDA) for the treatment of HIV/AIDS (1). HIV-1 protease inhibitors are administered as part of a drug combination in a treatment termed highly active antiretroviral therapy (HAART), which has become the most effective therapeutic strategy since the discovery of the virus. Nevertheless, mutations in viral enzymes that reduce drug affinity but not catalytic activity continue to breed resistance to HAART. Several mutations in HIV-1 protease have been identified, and resistance to all nine FDA-approved drugs has been reported (1, 2). Accordingly, attempts to improve inhibitor quality continue with a common goal of increasing the specificity and affinity of enzyme-drug interactions.
Meek and colleagues published critical insights into the HIV-1 protease reaction mechanism in the early 1990s (3), which contributed substantially to the first generation of clinical HIV-1 protease inhibitors. Subsequent generations of inhibitors relied on crystallographic information and medicinal chemistry to improve binding affinity and oral bioavailability (2). While these approaches have led to many effective HIV inhibitors, none of these drugs are free from the development of resistance. Biologically active mutants of HIV-1 protease recognize and cleave the same precursor polypeptides as their native counterparts; therefore, there remains a common structure-function relationship among them that has not been exploited in existing drugs.
HIV-1 protease is a homodimer of 99 amino acid subunits, each of which contribute a catalytic aspartate to the enzyme’s active site (Asp-25 and Asp-125) (4, 5). The catalytic mechanism and structural details of the HIV-1 protease reaction are illustrated in Fig. 1 (structures 1–7). The cycle initiates with water activation by hydrogen-bonding interactions with the active site aspartate residues (1) (6, 7). Bell-shaped pH-rate profiles (8–12) have revealed an acidic pH optimum, and structural studies (13, 14) suggest that the reactant-bound enzyme contains a single protonated Asp (with a di-Asp, net charge of −1) facilitating a general acid-base mechanism, which is common among many Asp proteases (7, 15). Nucleophilic attack by the water generates a reversible gem-diol intermediate, (3) which has been observed structurally (16–18) and identified experimentally (19, 20). A series of kinetic studies have concluded that the breakdown of 3 to generate the products (7) is the rate-liming chemical step (9–11, 19, 21), denoted by k5. Prior 15N isotope effect studies suggest that N-protonation contributes to k5 (22), but little additional experimental evidence distinguishes whether the rate-limiting chemical step is proton transfer to the leaving nitrogen (3 → 5), breakage of the C-N bond (5 → 7), or a concerted N-protonation and C-N bond breakage (3 → 7) and thus which transition state (4, 6, or a concerted 4/6) has the highest energy barrier.
Fig. 1.

Chemical mechanism of the reaction catalyzed by HIV-1 protease. Structures along the pathway are indicated as follows: (1) enzyme-substrate complex; (2) water attack TS; (3) tetrahedral gem-diol intermediate; (4) proline N-protonation TS; (5) protonated amide intermediate; (6) cleavage of scissile C-N bond TS; and (7) enzyme-product complex. For transition structure 2, the r(C-O) bond distance is defined as the distance between the oxygen of the attacking water and the carbonyl carbon of the peptide. For transition structure 4, the r(N-H) is defined as the bond distance between the nitrogen on the proline, and the proton on the catalytic aspartate and r(H-O) is defined as the bond distance between the oxygen and proton on the catalytic aspartate. Finally, in transition structure 6, r(C-N) is the bond distance of the scissile bond of the peptide.
Theoretical studies of the chemical mechanism of HIV-1 protease have included electronic structure calculations (23–26), quantum mechanics/molecular mechanics (QM/MM) studies (27–29), and molecular dynamic simulations (30–32). Early ab initio calculations by Lee et al. proposed that the irreversible step of the protease reaction comes after the formation of 3 (24). Later, Okimoto et al. used Hartree–Fock (HF) and a simple model system to calculate that the second step of the reaction consists of a stepwise rate-limiting proton transfer (4) followed by cleavage of the C-N bond (6) (23). A more recent QM/MM molecular dynamics study on the entire enzyme characterized the two reaction barriers and confirmed earlier findings that 4 is rate limiting (21 kcal/mol barrier) (29). Additional insights have stemmed from molecular dynamic simulations, which have shown that Asp-125 is responsible for the initial protonation of the carbonyl oxygen and that Asp-25 subsequently protonates the amide nitrogen (29, 30). Some general conclusions from these studies are that 3 is a stable intermediate in the reaction and that the bystander water, believed to maintain hydrogen bonding within the active site, has a high binding energy (27, 32).
A powerful technique in determining the chemical details of a reaction mechanism is experimentally measured kinetic isotope effects (KIEs) used to constrain theoretical predictions (33). This method for elucidating enzymatic transition structures has provided sufficiently robust transition-state knowledge to permit design and synthesis of powerful transition-state mimics (34). Here, we apply transition-state analysis to the study of the chemical mechanism of HIV-1 protease. Experimentally measured intrinsic KIEs are used to evaluate candidate transition-state structures originating from the distinct chemical barriers and intermediates in the reaction cycle of HIV-1 protease. The availability of a geometrically accurate transition structure provides chemical resolution of the reaction and will aid in the future development of transition-state analogs of the enzyme. Understanding transition-state features for native and resistant enzymes also provides insights into the mechanism of resistance.
Results and Discussion
Kinetic Isotope Effects.
We have measured a family of KIEs at atomic positions surrounding the cleavage site of the heptapeptide substrate Acetyl-Ser-Gln-Asn-Phe*Pro-Val-Val-NH2 (* denotes the scissile bond). The individual isotopic substitutions and measured KIEs are listed in Table 1 and shown Fig. 2A. Additionally, KIEs were measured for the multidrug-resistant variant (I84V), which is illustrated in Fig. 2B (see further discussion below). KIEs were determined by comparing 3H/14C ratios from isotopic peptides and reference peptides bearing remote radiolabels (Table 1). We observed primary carbonyl 14C KIEs (14V/K) of 1.029 ± 0.003 and 1.025 ± 0.005, primary 15N KIEs (15V/K) of 0.987 ± 0.004 and 0.989 ± 0.003, secondary 18O KIEs (18V/K) of 0.999 ± 0.003 and 0.993 ± 0.003, and secondary 3H KIEs (3V/K) KIEs of 0.968 ± 0.001 and 0.976 ± 0.001 for the native and I84V enzyme, respectively. Derived from the mechanism in Fig. 1 (assuming that any step up to bond cleavage may be isotope-sensitive), Eq. 1 can be used to represent the observed KIE (22, 35), where Keq3 represents the equilibrium constant for formation of the 3 and k5 is the rate constant for the formation of 7. The forward commitment (cf) represents a probability of substrate bound in complex 1 being catalytically hydrolyzed rather than being released from the Michaelis complex:
| [1] |
Table 1.
Experimental and theoretical kinetic isotope effects
| Peptide substrates |
Experimental KIEs*† |
Theoretical structures and KIE predictions |
|||||||
| Heavy |
Light |
Native |
I84V |
8 |
9 |
10 |
11 |
12 |
13 |
| Ac-SQN[1-14C]F‡PVV-NH2 | [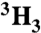 ]Ac-SQNF‡PVV-NH2 ]Ac-SQNF‡PVV-NH2
|
1.029(3) | 1.025(5) | 1.073 | 1.017 | 1.020 | 1.019 | 1.057 | 1.025 |
[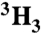 ]Ac-SQNF‡[15N]PVV-NH2 ]Ac-SQNF‡[15N]PVV-NH2
|
[14C]Ac-SQNF‡PVV-NH2 | 0.987(3) | 0.989(3) | 1.000 | 1.002 | 0.996 | 0.992 | 1.015 | 0.995 |
[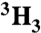 ]Ac-SQN[18O]F‡PVV-NH2 ]Ac-SQN[18O]F‡PVV-NH2
|
[14C]Ac-SQNF‡PVV-NH2 | 0.993(3) | 0.999(3) | 0.991 | 0.995 | 0.994 | 0.994 | 0.996 | 0.995 |
| Ac-SQN[α-3H]F‡PVV-NH2 | [14C]Ac-SQNF‡PVV-NH2 | 0.968(1) | 0.976(1) | 0.938 | 0.922 | 0.919 | 0.908 | 0.874 | 0.917 |
KIE, kinetic isotope effect.
*Experimental errors are shown in parentheses (x10-3) following the KIE values and reflect the standard error of the mean resulting from n ≥ 18 independent measurements.
†KIEs obtained using the [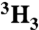 ]-Ac remote radiolabel are corrected for a 3H effect that was measured to be 0.974(2) and 0.976(2) for the native and I84V enzyme, respectively.
]-Ac remote radiolabel are corrected for a 3H effect that was measured to be 0.974(2) and 0.976(2) for the native and I84V enzyme, respectively.
‡Denotes the location of the scissile bond.
Fig. 2.

Kinetic isotope effects measured for native and I84V HIV-1 protease. (A) Peptide structure indicating the location of isotopic substitutions used for KIE measurements. KIE values are listed for each isotope (values for I84V are in bold). (B) Previously determined crystal structure (17) illustrating the location of the Ile-84 residue that is altered to Val in the drug-resistant mutant.
In our experimental approach, the KIE is measured on V/K and reports on isotope-sensitive steps up to and including the first irreversible step of each catalytic cycle (35, 36). A high probability of 1 being converted to products presents a virtually irreversible step prior to chemistry, which can mask expression of the KIE on the chemical step (the intrinsic KIE), the value that reports on the transition state. The reportedly high Km for this peptide (37) and the observation that the 15N KIE value is within the limit of all calculated transition-state models for this reaction (see discussion below) suggest that the forward commitment can be considered negligible in our analysis, simplifying Eq. 2 of the observed isotope effects to the product of the equilibrium isotope effect (EIE) on formation of 3 and the intrinsic KIE determined by the rate-limiting transition state
| [2] |
Theoretical Structures.
One difficulty inherent in using the fixed parameter method in calculating KIEs for a multistep enzymatic reaction is that the different chemical steps are electronically similar at points along the reaction coordinate, resulting in similar KIEs for 14C and 15N. For example, a late transition structure of 4 (short r(N-H)) is electronically similar to an early transition structure of 6(short r(C-N))—consequently, both of the theoretical structures are also similar to intermediate 5. Depending upon the model and method used in the calculations, redundancies exist in the KIE predictions for some of the transition structures for each of the chemical steps involved in the HIV-1 protease reaction. Therefore, the fixed parameter method of locating a single transition structure that matches all experimental KIEs is not appropriate for studying the HIV-1 protease mechanism. It is important to note, though, that a number of proton transfer transition structures obtained from using various methods and models exhibit calculated KIEs that most closely match the experimental values (Tables S1–S7 show examples of predicted IEs for several structures with fixed bond distances).
Theoretical structures used for the final calculation of KIEs include an Ala-Pro dipeptide as the substrate—an alanine residue was used in place of the experimental phenylalanine for simplicity—in addition to several important residues in the active site, including the two catalytic aspartates, the nucleophilic water, as well as the structural water (Fig. 3). Theoretical structures were derived from a published crystal structure of HIV-1 protease co-crystallized with a gem-diol intermediate (17). Calculations were performed using both ONIOM (M06-2X/6-31+G**:am1) and (B3LYP/6-31G*:am1) as implemented in Gaussian 09 (38). Theoretical structures were located as local minima for the starting peptide and structures 3 and 5. For each chemical step of the HIV-1 protease reaction, IEs were calculated for each of the structures based upon the lowest energy conformation of the starting material peptide and an intermediate or transition structure similar to the conformational geometry found in the published crystal structure.
Fig. 3.

Theoretical structures calculated for each step of the HIV-1 protease chemical mechanism using ONIOM(am1:M06-2X/6-31+G**). Calculated structures include: (8) TS for attack of water on the carbonyl; (9) gem-diol intermediate; (10) proton transfer from Asp-25 to proline; (11) protonated amide intermediate; (12) scissile bond cleavage. Distances are shown in angstroms.
To address the difficulties innately associated with the protease reaction, all theoretical structures for transition states 2, 4, and 6 were located as first order saddle points on the potential energy surface. By explicitly including a portion of the active site, we are assuming that geometry constraints are not required to mimic the active site functionality and that the saddle points will represent the on-enzyme transition structures. The theoretical structures from the ONIOM (M06-2X/6-31+G**:am1) method (Fig. 3) were used for the calculation of KIEs shown in Table 1 (theoretical structures and KIE values calculated from ONIOM (B3LYP/6-31G*:am1) are included in the supporting information). Both computational methods give similar results; however, 15N KIEs derived from B3LYP calculations are smaller than expected for most structures (near unity). This observation is consistent with previous studies in the literature that show B1LYP and B3LYP underpredict 15N KIEs (39–41). Furthermore, the M06-2X functional gives more accurate results than B3LYP in bond breaking and forming reactions (42).
Theoretical structures 8, 10, and 12 represent the unconstrained first order saddle points for transition states 2, 4, and 6, respectively (Fig. 3). Transition structure 8 corresponds to the first step of the reaction and indicates that attack of the catalytic water is late—r(C-O) is 1.79 Å. From the intermediate gem-diol (9), the second step of the reaction involves proton transfer from the aspartyl residue to the nitrogen on proline and occurs at r(N-H) of 1.26 Å as shown in 10. The third chemical step involves the breaking of the scissile C-N bond of protonated amide (11), which occurs relatively late (r(C-N) is 2.02 Å, as shown in transition structure 12). A concerted transition structure could not be located for direct formation of 7 from intermediate 3. Therefore, the protonation and cleavage of the amide are considered to be stepwise, though we cannot entirely rule out the possibility of a concerted reaction.
KIE Predictions.
In accordance with the experimental observation of H218O exchange (19), IEs for 10, 11, and 12 were calculated as the product of the EIE for formation of 9 and the KIE of the transition structure (Eq. 2) (22). Predicted 14C and 15N KIEs for the attack of water on the peptide from transition structure 8 are both too large and are poor matches to the measured values; though it is unlikely that 2 is KIE-determining because of the experimental observation supporting the reversibility of this step (19). Similarly, 14C and 15N KIE predictions for the breaking of the C-N bond of the amide peptide in 12 were also too large to match the experimental values. Clearly the proton transfer transition structure 10 most closely matches the experimentally measured KIEs, though the 14C and 15N KIEs are smaller than predicted by approximately 1%. Additionally, the EIEs for the protonated amide intermediate (11) are a relatively close match to the experimentally measured values. This is unsurprising because structures 10 and 11 are nearly identical with the exception of the r(N-H) distance of 1.26 Å and 1.12 Å, respectively.
The prediction of KIEs for proton transfer often requires a more extensive study than provided by density functional theory (DFT). However, when the experimental data and computational approach also include heavy atom isotope effects associated with proton transfer, predictive ability is improved. Regardless, the ability of any given method to reliably predict isotope effects to greater accuracy than the 1% shown here is questionable. To further explore the nature of the proton transfer, we applied a simplified variational transition-state theory approach by varying the r(N-H) and r(H-O) bond distances of transition structure 10 to locate a structure with KIE predictions that most closely match experimental values. Using this approach, we located transition structure 13 (Fig. 4) at r(N-H) of 1.20 Å and r(H-O) 1.32 Å, which gave the best match to experimental KIEs, bringing the predictions for 14C and 15N within 0.7 and 0.8%, respectively (Table 1).
Fig. 4.

Theoretical structure for proton transfer from Asp-25 to proline calculated by fixing the r(N-H) bond length at 1.20 Å using ONIOM(am1:M06-2X/6-31+G**). Predicted KIEs derived from this transition structure most closely match the experimentally measured values.
Experimental KIEs were measured for the carbonyl 18O and the phenylalanine α-3H, neither of which are directly involved in bond breaking or forming steps of the mechanism. Hydrogen bonding to the carbonyl oxygen of the peptide is the factor with the largest influence on the 18O KIE in the mechanism. Hydrogen-bonding networks change as the chemical mechanism progresses; however, each of the oxygens at the reactive center are continuously hydrogen bonded to one of the catalytic aspartates, making each step of the reaction electronically similar, resulting in a null change in the 18O KIE after substrate binding. An inverse α-3H KIE is attributed to the breaking of the interaction in the starting material peptide of the lone pair of electrons on oxygen to the σ∗ orbitals of the adjacent hydrogens upon the binding of the peptide to the enzyme. Predictions for α-3H are inverse at every step of the reaction and depend more upon the conformation of the peptide upon binding than upon the chemistry occurring at the adjacent carbonyl position (Fig. 3 and Table 1).
Proton Transfer.
An important aspect of proton transfer transition structure 13 is that the model contains three protons directly associated with the substrate (Figs. 4 and 5). The most stable conformation of the proline is a five-membered boat, placing the lone pair on the nitrogen in position to accept the proton from Asp-25. Stretching of the scissile C-N amide bond is associated with the imaginary frequency for the transition structure of proton transfer, suggesting that amide bond cleavage is associated with this step. However, little change occurs between 10, 13, and 11 (in order of decreasing r(N-H)), with r(C-N) changing only slightly from 1.51, 1.52, to 1.53 Å, respectively. Hydrogen bonding to both oxygens of the gem-diol occurs solely with Asp-125 and is maintained throughout the reaction, consistent with previous dynamic, crystallographic, and neutron diffraction studies (15).
Fig. 5.

Electrostatic potential, NBO charges, and inhibitor scaffolds. (A) Structure of the clinical inhibitor indinavir bound to the HIV-1 protease active site obtained from the PDB structure 2AVO (Left) and the proton transfer transition structure 13 (Right). (B) Electrostatic potential maps (blue = partial positive, red = partial negative) extrapolated from single-point energy calculations (M06-2X/6-31+G**) of indinavir (Left) and 13 (Right). NBO charges are indicated in parentheses. (C) Structural interactions with the catalytic aspartates and indinavir (Left) and a potential proton transfer TS scaffold (Right).
Despite significant exploration, a concerted transition structure was not located for proton transfer and C-N bond cleavage; though it is plausible that the steps occur concurrently. From an examination of simultaneously varying r(C-N) and r(N-H), a concerted transition structure would have to exist with r(C-N) equal to or less than 1.70 Å to be consistent with the measured KIEs. However, structures with r(C-N) less than 2.10 Å had no associated imaginary frequency. A match of predicted KIEs to the experimental values from a possible concerted transition structure is unlikely.
Drug Resistance.
For an inhibitor to be effective against biologically relevant variants of HIV-1 protease, binding interactions should be maximized with essential regions of the active site. The observation that the transition structures are nearly identical in the native and I84V enzyme (as indicated by experimental KIE data, Fig. 2A and Table 1) suggests that transition-state interactions should be a focus of inhibitor design. Although many powerful HIV-1 protease inhibitors have been developed, drug resistance continues to arise and attempts to understand mechanisms of drug resistance persist. The variant used in our experiments contains a mutation at an active site Ile residue (I84V), as illustrated in Fig. 2B. This variant has displayed resistance to all nine FDA-approved inhibitors (2, 43) and has been reported to cause up to a 32-fold reduction in inhibitor binding affinity (44). Our observation that the I84V transition structure is identical to that for the native enzyme indicates that the drug-resistant behavior that arises is independent from transition-state interactions. A stable mimic of transition structure 13 shown in Fig. 4 should be an effective inhibitor both the native and I84V HIV-1 protease, and likely an effective inhibitor against all biologically relevant HIV-1 protease variants.
An electrostatic potential and natural bond orbital (NBO) charge comparison of 13 and a well-characterized clinical inhibitor, indinavir, is shown in Fig. 5. Electrostatic potential maps provide useful blueprints for inhibitor design since they provide shape and charge details of the reactant at the moment of the transition state. The map of indinavir reveals a discrepancy common among the transition structure and the existing FDA-approved HIV-1 protease inhibitors. All clinically approved inhibitors posses the gem-diol mimic but lack the charge character that results from the additional proton present in 13 (Fig. 5 B and C). The electrostatic maps in Fig. 5B reveal that transition structure 13 has three protons with NBO charges of +0.549, +0.564 (diol OH’s), and +0.504 (proline N), where the indinavir has one proton within the diol mimic with an NBO charge of +0.514. A transition-state analogue scaffold proposed from the electrostatic potential map in Fig. 5B is shown in Fig. 5C, Right, with a focus on transition structure features. Although the catalytic aspartic acids provide a proton to both the transition-state mimic and indinavir upon inhibitor binding, the crucial feature of the proposed transition-state inhibitor scaffold is the increased protonation state of the nitrogen and should be a focus of future inhibitor design.
Conclusions.
Using a combination of experimentally measured KIEs and theoretical calculations, we have resolved the structure of the rate-limiting transition state in the HIV-1 protease reaction. Our findings confirm previous proposals that proton transfer is rate limiting (22, 23, 29). We have established an electrostatic map structure to serve as a blueprint for improved inhibitor design. Inhibitors of HIV-1 protease are usually referred to as transition-state analogues; however, no transition structures have been previously reported from any combination of comprehensive KIE analysis and computational approaches as established here. Additionally, we have confirmed that a highly drug-resistant mutant form of the HIV-1 protease enzyme shares nearly identical transition-state features with the native enzyme, indicating that drug resistance in this mutant arises from alterations in the enzyme distant from transition-state interactions. Similarity in the transition-state features suggests that a robust transition-state mimic would serve as an effective inhibitor of both the native and I84V enzyme and likely other mutants that share common transition-state features.
Materials and Methods
Isotopic Labeling and Synthesis of Peptides.
Peptide substrates (Fig. 2A and Table 1) were synthesized by sequentially coupling 9-Fluorenylmethyloxycarbonyl (Fmoc)-protected amino acids onto a cross-linked ethoxylate acrylate resin (CLEAR)-amide resin (100–200 mesh 0.43 mmol/g) followed by N-acetylation, resin cleavage, precipitation, and purification. Isotopic labels were incorporated at the remote positions by acetylation with either [ ]-acetic anhydride (Ac2O) (purchased) or [1-14C] Ac2O (purchased), and isotopic labels at the scissile positions were incorporated by coupling the appropriate labeled amino acid from the following: Fmoc-[1-14C]Phe-OH (purchased), Fmoc-[15N]Pro-OH (purchased), Fmoc-[α-3H]Phe-OH (synthesized), or Fmoc-[
]-acetic anhydride (Ac2O) (purchased) or [1-14C] Ac2O (purchased), and isotopic labels at the scissile positions were incorporated by coupling the appropriate labeled amino acid from the following: Fmoc-[1-14C]Phe-OH (purchased), Fmoc-[15N]Pro-OH (purchased), Fmoc-[α-3H]Phe-OH (synthesized), or Fmoc-[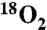 ]Phe-OH (synthesized). Peptides were purified by semipreparative reverse-phase high-performance liquid chromatography (HPLC) using a linear gradient (3–40% acetonitrile [AcN] in water with 0.1% trifluoroacetic acid [TFA] over 40 min). The purified nonradioactive peptides were characterized by electrospray ionization mass spectrometry (MS-ESI), and radioactive peptides were confirmed by co-elution with the nonradioactive peptide using analytical reverse-phase HPLC over the same linear gradient.
]Phe-OH (synthesized). Peptides were purified by semipreparative reverse-phase high-performance liquid chromatography (HPLC) using a linear gradient (3–40% acetonitrile [AcN] in water with 0.1% trifluoroacetic acid [TFA] over 40 min). The purified nonradioactive peptides were characterized by electrospray ionization mass spectrometry (MS-ESI), and radioactive peptides were confirmed by co-elution with the nonradioactive peptide using analytical reverse-phase HPLC over the same linear gradient.
Fmoc-[1-14C]Phe-OH, [3H]Ac2O, and [1-14C]Ac2O were purchased from American Radiolabeled Chemicals, Inc., and Fmoc-[15N]-proline was purchased from Cambridge Isotope Laboratories, Inc.
Synthesis of Fmoc-[18O]2-Phe-OH.
18O incorporation into phenylalanine was accomplished by exchange in acidic H218O (97%, Cambridge Isotope Laboratories, Inc.) as previously described (45) (also see SI Materials and Methods). [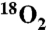 ]Phe-OH was then reacted with 9-Fluorenylmethyl N-succinimidyl carbonate (Fmoc-Osu) (dissolved in ice-cold acetone) under basic conditions (NaHCO3 to H218O [97%] to pH 9). The reaction was monitored by loss of ninhydrin reactivity and thin-layer chromatography (TLC) and complete conversion to Fmoc-[
]Phe-OH was then reacted with 9-Fluorenylmethyl N-succinimidyl carbonate (Fmoc-Osu) (dissolved in ice-cold acetone) under basic conditions (NaHCO3 to H218O [97%] to pH 9). The reaction was monitored by loss of ninhydrin reactivity and thin-layer chromatography (TLC) and complete conversion to Fmoc-[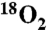 ]-Phe-OH was confirmed by MS.
]-Phe-OH was confirmed by MS.
Synthesis of Fmoc-[α-3H]-Phe-OH.
[α-3H]-Phe-OH was synthesized by reductive amination of phenylpyruvate through a series of enzymatic 3H transfer reactions (Fig. S1). A reaction mixture of 4 mM [1-3H]glucose, 50 mM adenosine triphosphate (ATP), 160 μM nicotinamide adenine dinucleotide (NAD+), 250 mM NH4Cl, 10 mM MgCl2, and 10 mM phenylpyruvate in 50 mM Tris-HCl pH 8.5 was prepared and the following enzymes were added sequentially (0.5 units each): (i) phenylalanine dehydrogenase (Sigma), (ii) glucose-6-phosphate dehydrogenase (Sigma), and (iii) hexokinase (Sigma). The reaction was completed in approximately 45 min at room temperature. Purification of the phenylalanine product was accomplished by reverse-phase HPLC over a linear gradient (5–50% AcN in water and 0.1% TFA over 20 min) on a C18 column (Waters Delta Pak, 300 × 3.9 mm, 15 μm, 300 Å), with the peak corresponding to phenylalanine eluting at approximately 25% AcN. [α-3H]Phe-OH was then reacted with Fmoc-Osu (dissolved in ice-cold acetone) under basic conditions (NaHCO3 to H2O to pH 9), and conversion to Fmoc-[α-3H]-Phe-OH was confirmed by loss of ninhydrin reactivity and TLC analysis.
Protease Expression and Purification.
Both HIV-1 protease constructs (native and I84V) bearing the five background mutations Q7K, L33I, L63I (for restricted auto-proteolysis), and C67A and C95A (for restricted Cys thiol oxidation) were provided in pET-11a vectors. The constructs were transformed into BL21(DE3) Escherichia coli cells and expressed and purified from inclusion bodies according to an established protocol (46).
Kinetic Isotope Effect Measurements.
KIEs were measured using the competitive isotopes method (47). The KIE on V/K was determined by the relative change in the ratio of light and heavy peptides (each bearing either 14C or 3H radiolabels) in the unreacted substrate versus remaining substrate after multiple reaction cycles. Peptides were radiolabeled with either 3H or 14C as shown in Table 1. KIEs were measured by mixing the heavy and light peptides such that the counts-per-minute (cpm) ratio of 3H∶14C was 3∶1 (150,000 cpm∶50,000 cpm), with a total peptide concentration kept at 0.5 mM in a 200 μL reaction volume (GAMT-NEDT pH 6.0; see SI Materials and Methods). A measure of 50 μL was taken immediately to determine the ratio of the radiolabels in unreacted peptides, R0 (R = scissile bond heavy isotope/scissile bond light isotope). Approximately 150 nM protease was added (0.5 μL of 1 mg/mL stock) to the remaining 150 μL and the reaction was stopped at approximately 70% completion (f = 0.7). The unreacted substrates were purified from products over 0.5 mL AG1-X8 anion exchange columns (equilibrated with two column volumes 0.1 N NaOH and two column volumes of water), which bind the carboxylate generated in the product peptides (22). The ratios of isotopes of the unreacted substrates at f = 0(R0) and f = 0.7(Rf) were determined by liquid scintillation counting (the counting protocol is outlined in SI Text), and the KIEs were determined from Eq. 3 (48):
| [3] |
Each reported KIE results from a minimum of three repeats of six reactions (n≥18), which were determined from the average of 10 cycles of scintillation counting at 10 min per sample.
Computational Methods.
The mechanism of HIV-1 protease was studied using the QM/MM two-layer ONIOM (M06-2X/6-31+G**:AM1) method as implemented in Gaussian 03 and 09 (G03 and G09) (38, 49). A model system of the active site containing 102 atoms was used in the calculations and was derived from the crystal structure of HIV-1 protease co-crystallized with the gem-diol intermediate (17). All starting materials and stable intermediates were located as local minima and frequency calculations performed on the optimized structures contained no imaginary frequencies. Transition structures were located as first order saddle points and contained only one imaginary frequency. Additionally, structures for each chemical step of the protease reaction were explored with geometric constraints (as nonstationary points) to determine the range of possible predicted KIEs for each step of the reaction. Forward and reverse intrinsic reaction coordinate calculations were performed as implemented in G09 from each transition structure to verify that each structure lies on the relevant reaction path.
The isotope effects for each of the theoretical structures were calculated from conventional transition-state theory by the method of Bigeleisen and Mayer (50–52) from the scaled theoretical vibrational frequencies. Tunneling corrections were applied on all atoms using a one-dimensional infinite parabolic barrier model, and a truncated infinite parabola model was used for proton transfers (53).
Natural bond orbital (NBO) analysis and single-point energy calculations were carried out on the optimized proton transfer transition structure 13 and the clinical inhibitor indinavir from Protein Data Bank structure 2AVO (54) in G09 (M06-2X/6-31+G**). Electrostatic potential maps were visualized in Gaussview 5.0 (isovalue = 0.04) from density and potential cubes acquired from the single-point energy checkpoint files in G09.
Supplementary Material
Acknowledgments.
We thank Dr. John M. Louis, Laboratory of Chemical Physics, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health for generously providing the protease expression constructs and taking time to discuss expression strategies. We thank Dr. Emilie J. Wang for assistance in protease expression experiments. This work was supported by National Institutes of Health research Grant GM41916 (V.L.S.) and the Einstein Center for AIDS Research NIH AI-51519.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202808109/-/DCSupplemental.
References
- 1.Wensing AM, van Maarseveen NM, Nijhuis M. Fifteen years of HIV protease inhibitors: Raising the barrier to resistance. Antiviral Res. 2010;85:59–74. doi: 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Mitsuya H, Maeda K, Das D, Ghosh AK. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv Pharmacol. 2008;56:169–197. doi: 10.1016/S1054-3589(07)56006-0. [DOI] [PubMed] [Google Scholar]
- 3.Meek TD, Rodriguez EJ, Angeles TS. Use of steady-state kinetic methods to elucidate the kinetic and chemical mechanisms of retroviral proteases. Methods Enzymol. 1994;241:127–156. doi: 10.1016/0076-6879(94)41063-1. [DOI] [PubMed] [Google Scholar]
- 4.Navia MA, et al. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989;337:615–620. doi: 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- 5.Wlodawer A, et al. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science. 1989;245:616–621. doi: 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- 6.Brik A, Wong CH. HIV-1 protease: Mechanism and drug discovery. Org Biomol Chem. 2003;1:5–14. doi: 10.1039/b208248a. [DOI] [PubMed] [Google Scholar]
- 7.Northrop DB. Follow the protons: A low-barrier hydrogen bond unifies the mechanisms of the aspartic proteases. Acc Chem Res. 2001;34:790–797. doi: 10.1021/ar000184m. [DOI] [PubMed] [Google Scholar]
- 8.Short GF, III, et al. Probing the S1/S1′ substrate binding pocket geometry of HIV-1 protease with modified aspartic acid analogues. Biochemistry. 2000;39:8768–8781. doi: 10.1021/bi000214t. [DOI] [PubMed] [Google Scholar]
- 9.Szeltner Z, Polgar L. Rate-determining steps in HIV-1 protease catalysis: The hydrolysis of the most specific substrate. J Biol Chem. 1996;271:32180–32184. doi: 10.1074/jbc.271.50.32180. [DOI] [PubMed] [Google Scholar]
- 10.Polgar L, Szeltner Z, Boros I. Substrate-dependent mechanisms in the catalysis of human-immunodeficiency-virus protease. Biochemistry. 1994;33:9351–9357. doi: 10.1021/bi00197a040. [DOI] [PubMed] [Google Scholar]
- 11.Hyland LJ, Tomaszek TA, Jr, Meek TD. Human immunodeficiency virus-1 protease 2. Use of pH rate studies and solvent kinetic isotope effects to elucidate details of chemical mechanism. Biochemistry. 1991;30:8454–8463. doi: 10.1021/bi00098a024. [DOI] [PubMed] [Google Scholar]
- 12.Ido E, Han HP, Kezdy FJ, Tang J. Kinetic studies of human immunodeficiency virus type-1 protease and its active-site hydrogen-bond mutant A28S. J Biol Chem. 1991;266:24359–24366. [PubMed] [Google Scholar]
- 13.Wang YX, et al. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry. 1996;35:9945–9950. doi: 10.1021/bi961268z. [DOI] [PubMed] [Google Scholar]
- 14.Smith R, Brereton IM, Chai RY, Kent SBH. Ionization states of the catalytic residues in HIV-1 protease. Nat Struct Biol. 1996;3:946–950. doi: 10.1038/nsb1196-946. [DOI] [PubMed] [Google Scholar]
- 15.Coates L, et al. The catalytic mechanism of an aspartic proteinase explored with neutron and X-ray diffraction. J Am Chem Soc. 2008;130:7235–7237. doi: 10.1021/ja801269x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das A, et al. X-ray snapshot of HIV-1 protease in action: Observation of tetrahedral intermediate and short ionic hydrogen bond SIHB with catalytic aspartate. J Am Chem Soc. 2010;132:6366–6373. doi: 10.1021/ja100002b. [DOI] [PubMed] [Google Scholar]
- 17.Kovalevsky AY, Chumanevich AA, Liu F, Louis JM, Weber IT. Caught in the act: The 1.5 A resolution crystal structures of the HIV-1 protease and the I54V mutant reveal a tetrahedral reaction intermediate. Biochemistry. 2007;46:14854–14864. doi: 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar M, Prashar V, Mahale S, Hosur MV. Observation of a tetrahedral reaction intermediate in the HIV-1 protease-substrate complex. Biochem J. 2005;389:365–371. doi: 10.1042/BJ20041804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyland LJ, et al. Human immunodeficiency virus-1 protease. 1. Initial velocity studies and kinetic characterization of reaction intermediates by 18O isotope exchange. Biochemistry. 1991;30:8441–8453. doi: 10.1021/bi00098a023. [DOI] [PubMed] [Google Scholar]
- 20.Kipp DR, Silva RG, Schramm VL. Mass-dependent bond vibrational dynamics influence catalysis by HIV-1 protease. J Am Chem Soc. 2011;133:19358–19361. doi: 10.1021/ja209391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter DJ, Hanlon MH, Furfine ES. HIV-1 protease: Characterization of a catalytically competent enzyme-substrate intermediate. Biochemistry. 2002;41:1302–1307. doi: 10.1021/bi0116543. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez EJ, Angeles TS, Meek TD. Use of N-15 kinetic isotope effects to elucidate details of the chemical mechanism of human immunodeficiency virus-1 protease. Biochemistry. 1993;32:12380–12385. doi: 10.1021/bi00097a015. [DOI] [PubMed] [Google Scholar]
- 23.Okimoto N, Tsukui T, Hata M, Hoshino T, Tsuda M. Hydrolysis mechanism of the phenylalanine-proline peptide bond specific to HIV-1 protease: Investigation by the ab initio molecular orbital method. J Am Chem Soc. 1999;121:7349–7354. [Google Scholar]
- 24.Lee H, Darden TA, Pedersen LG. An ab initio quantum mechanical model for the catalytic mechanism of HIV-1 protease. J Am Chem Soc. 1996;118:3946–3950. [Google Scholar]
- 25.Venturini A, Lopez-Ortiz F, Alvarez JM, Gonzalez J. Theoretical proposal of a catalytic mechanism for the HIV-1 protease involving an enzyme-bound tetrahedral intermediate. J Am Chem Soc. 1998;120:1110–1111. [Google Scholar]
- 26.Park H, Suh J, Lee S. Ab initio studies on the catalytic mechanism of aspartic proteinases: Nucleophilic versus general acid/general base mechanism. J Am Chem Soc. 2000;122:3901–3908. [Google Scholar]
- 27.Suresh CH, Vargheese AM, Vijayalakshmi KP, Mohan N, Koga N. Role of structural water molecule in HIV protease-inhibitor complexes: A QM/MM study. J Comput Chem. 2008;29:1840–1849. doi: 10.1002/jcc.20961. [DOI] [PubMed] [Google Scholar]
- 28.Hensen C, et al. A combined QM/MM approach to protein—ligand interactions: Polarization effects of the HIV-1 protease on selected high affinity inhibitors. J Med Chem. 2004;47:6673–6680. doi: 10.1021/jm0497343. [DOI] [PubMed] [Google Scholar]
- 29.Piana S, Bucher D, Carloni P, Rothlisberger U. Reaction mechanism of HIV-1 protease by hybrid carparrinello/classical MD simulations. J Phys Chem B. 2004;108:11139–11149. [Google Scholar]
- 30.Chatfield DC, Brooks BR. HIV-1 protease cleavage mechanism elucidated with molecular-dynamics simulation. J Am Chem Soc. 1995;117:5561–5572. [Google Scholar]
- 31.Trylska J, Grochowski P, McCammon JA. The role of hydrogen bonding in the enzymatic reaction catalyzed by HIV-1 protease. Protein Sci. 2004;13:513–528. doi: 10.1110/ps.03372304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okimoto N, et al. Molecular dynamics study of HIV-1 protease-substrate complex: Roles of the water molecules at the loop structures of the active site. J Am Chem Soc. 2000;122:5613–5622. [Google Scholar]
- 33.Schramm VL. Enzymatic transition-state analysis and transition-state analogs. Methods Enzymol. 1999;308:301–355. doi: 10.1016/s0076-6879(99)08015-5. [DOI] [PubMed] [Google Scholar]
- 34.Schramm VL. Enzymatic transition states, transition—state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. New York: Garland Science; 2007. pp. 253–323. [Google Scholar]
- 36.Northrop DB. On the meaning of Km and V/K in enzyme kinetics. J Chem Educ. 1998;75:1153–1157. [Google Scholar]
- 37.Moore ML, et al. Peptide—substrates and inhibitors of the HIV-1 protease. Biochem Biophys Res Commun. 1989;159:420–425. doi: 10.1016/0006-291x(89)90008-9. [DOI] [PubMed] [Google Scholar]
- 38.Frisch MJ, et al. Gaussian09. Wallingford, CT: Gaussian, Inc; 2009. [Google Scholar]
- 39.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 40.Silva RG, Hirschi JS, Ghanem M, Murkin AS, Schramm VL. Arsenate and phosphate as nucleophiles at the transition states of human purine nucleoside phosphorylase. Biochemistry. 2011;50:2701–2709. doi: 10.1021/bi200279s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh V, Schramm VL. Transition-state structure of human 5′-methylthioadenosine phosphorylase. J Am Chem Soc. 2006;128:14691–14696. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 43.Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 protease: Structure, dynamics, and inhibition. Adv Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- 44.Gulnick SV, et al. Kinetic characterization and cross—resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 45.Murphy RC, Clay KL. Synthesis and back exchange of 18O labeled amino acids for use as internal standards with mass spectrometry. Biol Mass Spectrom. 1979;6:309–314. doi: 10.1002/bms.1200060709. [DOI] [PubMed] [Google Scholar]
- 46.Sayer JM, Agniswamy J, Weber IT, Louis JM. Autocatalytic maturation, physical/chemical properties, and crystal structure of group N HIV-1 protease: Relevance to drug resistance. Protein Sci. 2010;19:2055–2072. doi: 10.1002/pro.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkin DW, Leung HB, Schramm VL. Synthesis of nucleotides with specific radiolabels in ribose. Primary 14C and secondary 3H kinetic isotope effects on acid-catalyzed glycosidic bond hydrolysis of AMP, dAMP, and inosine. J Biol Chem. 1984;259:9411–9417. [PubMed] [Google Scholar]
- 48.Melander L, Saunders WH. Reaction Rates Of Isotopic Molecules. Malabar, FL: Kreiger; 1980. pp. 95–123. [Google Scholar]
- 49.Frisch MJ, et al. Gaussian03. Wallington, CT: Gaussian, Inc; 2004. [Google Scholar]
- 50.Bigeleisen J, Mayer MG. Calculation of equilibrium constants for isotope exchange reactions. J Chem Phys. 1947;15:261–267. [Google Scholar]
- 51.Wolfsbert M. Theoretical evaluation of experimentally observed isotope effects. Acc Chem Res. 1972;5:225–233. [Google Scholar]
- 52.Anisimov V, Paneth P. ISOEFF98: A program for studies of isotope effects using Hessian modifications. J Math Chem. 1999;26:75–86. [Google Scholar]
- 53.Bell RP. The Tunnel Effect in Chemistry. London: Chapman and Hall; 1980. pp. 60–63. [Google Scholar]
- 54.Liu F, et al. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J Mol Biol. 2005;354:789–800. doi: 10.1016/j.jmb.2005.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.