

Background: Non-professional phagocytes, like epithelial cells, can recognize dead cells.
Results: Dead cells modulate the survival and proliferation of epithelial cells with which they interact via Akt-dependent and -independent signaling events.
Conclusion: Dead cells profoundly affect the viability of live cells in their vicinity.
Significance: Dead cells act as sentinels of environmental change and allow nearby cells to monitor and adapt to local stresses.
Keywords: Cell Biology, Cell Death, Cell Proliferation, Cell Signaling, Kidney, Necrosis (Necrotic Death), Renal Physiology
Abstract
Apoptosis allows for the removal of damaged, aged, and/or excess cells without harm to surrounding tissue. To accomplish this, cells undergoing apoptosis acquire new activities that enable them to modulate the fate and function of nearby cells. We have shown that receptor-mediated recognition of apoptotic versus necrotic target cells by viable kidney proximal tubular epithelial cells (PTEC) modulates the activity of several signaling pathways critically involved in regulation of proliferation and survival. Generally, apoptotic and necrotic targets have opposite effects with apoptotic targets inhibiting and necrotic targets stimulating the activity of these pathways. Here we explore the consequences of these signaling differences. We show that recognition of apoptotic targets induces a profound decrease in PTEC viability through increased responder cell death and decreased proliferation. In contrast, necrotic targets promote viability through decreased death and increased proliferation. Both target types mediate their effects through a network of Akt-dependent and -independent events. Apoptotic targets modulate Akt-dependent viability in part through a reduction in cellular β-catenin and decreased inactivation of Bad. In contrast, Akt-independent modulation of viability occurs through activation of caspase-8, suggesting that death receptor-dependent pathways are involved. Apoptotic targets also activate p38, which partially protects responders from target-induced death. The response of epithelial cells varies depending on their tissue origin. Some cell lines, like PTEC, demonstrate decreased viability, whereas others (e.g. breast-derived) show increased viability. By acting as sentinels of environmental change, apoptotic targets allow neighboring cells, especially non-migratory epithelial cells, to monitor and potentially adapt to local stresses.
Introduction
Apoptosis plays an essential role in the development and maintenance of tissues. The apoptotic program allows for the removal via phagocytosis of damaged, aged, or excess cells in a regulated manner that avoids harm to surrounding tissue (1, 2). This is accomplished through a variety of interconnected mechanisms. Notably, cells dying by apoptosis acquire new activities, both secreted and cell-associated, that enable them to modulate the function of interacting phagocytes (3–6). Although the best known of these activities is anti-inflammatory (7–12), apoptotic targets also affect other phagocyte functions related to innate and adaptive immunity, including antigen processing and presentation (13, 14), expression of co-stimulatory activity for lymphocytes (15), and the recruitment and trafficking of immune cells (16).
Importantly, the effects of apoptotic targets are limited neither to professional phagocytes nor to regulation of the immune system. Indeed, inhibition of proinflammatory responses by apoptotic targets has been described in many cell types, including non-professional phagocytes such as epithelial, neuronal, and lymphoid cells (6, 17). Moreover, exposure to apoptotic targets affects multiple aspects of cellular function, including such vital cell activities as survival and proliferation (3). To date, study of these effects has been largely focused on professional phagocytes, like macrophages (mφ).2 For example, we have shown that apoptotic targets inhibit apoptosis and maintain the viability of primary mφ cultures even in the absence of serum or other survival factors (5).
The diversity and ubiquity of the response to apoptotic targets indicate that the role of dead cells in non-immune homeostasis likely extends beyond development and maintenance of tissues. We have hypothesized that recognition and interaction with apoptotic and necrotic targets permit viable cells to monitor their immediate environment for stresses or change (18, 19). In interpreting the response of a cell to dead targets in its vicinity, several dichotomies need to be considered. Most important are the nature and function of the responding cell. For example, migratory cells can flee a hostile environment, whereas tissue-fixed cells must adapt to environmental changes. In addition, the form of cell death is important. We have shown that the signaling events induced in responding cells by apoptotic versus necrotic targets are often diametrically opposed (3, 5, 17). The different responses to these two forms of cell death can serve as a “gauge” by which surrounding cells assess the overall severity of an environmental stress: sudden and catastrophic in the case of necrotic targets versus more gradual and potentially adaptable in the case of apoptotic targets. Finally, there is the distinction between signaling events induced by “receptor-dependent recognition” versus those initiated by “engulfment.” Signaling events for which apoptotic and necrotic targets elicit opposite responses are likely triggered by distinct receptors mediating recognition, whereas signaling events for which apoptotic and necrotic targets elicit similar responses are likely triggered by the shared machinery of engulfment (i.e. phagocytosis).
These issues are exemplified by comparing the responses of mφ and epithelial cells (3, 5, 17). Like mφ, mouse kidney proximal tubular epithelial cells (PTECs) recognize apoptotic and necrotic targets via distinct and non-competing receptors, albeit with a lower binding capacity and markedly reduced phagocytosis. PTECs evince the same set of recognition-dependent responses as mφ with respect to inhibition of inflammation and modulation of mitogen-activated protein kinase (MAPK) modules. PTECs, however, differ from mφ in several important ways with respect to modulation of the prosurvival kinase Akt. First, although apoptotic targets activate Akt in mφ, they inhibit Akt in PTECs. Second, unlike mφ for which activation of Akt is linked to phagocytosis, inhibition of Akt in PTECs is independent of phagocytosis and triggered exclusively by receptor-mediated recognition. Finally, as opposed to mφ, the responses of PTECs to necrotic and apoptotic targets are oppositely directed with necrotic targets leading to activation of Akt.
Here we investigate the consequences of these signaling differences on PTEC viability as well as their dependence on Akt-dependent signaling events. Our data show that, unlike mφ for which exposure to apoptotic targets leads to an increase in viability, apoptotic targets profoundly diminish the viability of kidney PTECs. This decrease in viability is accomplished through a combination of increased responder cell death and decreased responder cell proliferation. In turn, each of these outcomes (increased death and decreased proliferation) involves a variety of interconnected target-dependent signaling events and pathways, both Akt-dependent and -independent. In sharp contrast, necrotic targets promote the survival of PTECs; they do so through effects on proliferation and survival that are opposite to those induced by apoptotic targets. Moreover, although the response of epithelial cells from different tissues is uniform with respect to necrotic targets, the response to apoptotic targets varies depending on the tissue of origin. Some epithelial cell lines, like PTECs, demonstrate decreased viability, whereas others, like mammary duct cells, show an increase in viability. Together, these data emphasize the complexity, robustness, and importance of the cellular response to dead targets. By acting as sentinels of environmental change, apoptotic and necrotic targets may allow neighboring viable cells, especially non-migratory epithelial cells, to monitor and potentially adapt to local stresses.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, all chemicals were obtained from Sigma, Invitrogen, or Fisher Scientific. Cell culture medium was obtained from Mediatech (Herndon, VA).
Antibodies
Affinity-purified polyclonal rabbit antibodies detecting the active phosphorylated or total forms of Akt (Akt1, Akt2, and Akt3), the active phosphorylated form of p90RSK, the total or phosphorylated forms of β-catenin, and the cleaved forms of caspase-8 were obtained from Cell Signaling Technology (Beverly, MA). Rabbit anti-cyclin D1 monoclonal antibody (SP4) was obtained from Abcam (Cambridge, UK). Rabbit anti-active caspase-3 monoclonal antibody (C92-605) labeled with phycoerythrin was obtained from BD Biosciences. Anti-active Akt detects Akt1 when phosphorylated at Ser473 and Akt2 and Akt3 when phosphorylated at the corresponding residues. This antibody does not recognize Akt phosphorylated at other sites nor does it recognize phosphorylated forms of related kinases such as protein kinase C or p70 S6 kinase. Anti-phospho-p90RSK detects the dually phosphorylated Thr359-X-X-X-Ser363 region (pT359XXXpS363) of p90RSK1 and cross-reacts with p90RSK3. Anti-phospho-β-catenin antibody detects β-catenin when phosphorylated at Ser33, Ser37, or Thr41. It does not recognize β-catenin when phosphorylated at other sites. Anti-total β-catenin antibody detects total endogenous levels of β-catenin irrespective of phosphorylation. Anti-cleaved caspase-8 antibody detects caspase-8 following its enzymatic cleavage at Asp387. The antibody detects the active p18 subunit as well as the p43 prodomain containing p18. Anti-cyclin D1 antibody detects total endogenous levels of cyclin D1. Anti-active caspase-3 detects the active caspase-3 heterodimer consisting of 17 and 12 kDa subunits. Horseradish peroxidase-linked donkey anti-rabbit F(ab′)2 from GE Healthcare was used as a secondary antibody for detection of Western blots by enhanced chemiluminescence.
Cell Culture
All cells were grown at 37 °C in a humidified 5% (v/v) CO2 atmosphere unless otherwise stated. The conditionally immortalized mouse kidney PTEC line (Boston University mouse proximal tubule (BU.MPT)) was maintained in high glucose Dulbecco's modified Eagle's medium (DMEM)/F-12 medium containing 10% (v/v) heat-inactivated FBS, 2 mm l-glutamine, 10 mm HEPES, 100 units/ml penicillin-streptomycin, and 10 units/ml interferon-γ (IFN-γ). BU.MPT cells were derived from a transgenic mouse bearing a temperature-sensitive mutation (tsA58) of the simian virus 40 (SV40) large tumor antigen under the control of the mouse major histocompatibility complex H-2Kb class I promoter (20–22). Under permissive conditions, defined as growth at 33–37 °C in the presence of IFN-γ, the tsA58 large tumor antigen transgene is expressed. Under non-permissive temperatures, defined as growth at 39.5 °C in the absence of IFN-γ, expression of the transgene is inhibited (by >95%), and BU.MPT cells behave like primary cultures of mouse kidney PTECs. Prior to all experiments, BU.MPT cells were serum-starved and cultured under non-permissive conditions for 24 h.
Chinese hamster ovary (CHO) epithelial cells and CHO-derived LR73 cells were grown in α-minimum Eagle's medium supplemented with 10% (v/v) heat-inactivated FBS and 2 mm l-glutamine. HeLa cells, a human cervical cancer epithelial cell line, were maintained in DMEM containing 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. MCF-7 K1 (MCF-7) cells, a human breast adenocarcinoma cell line, were maintained in DMEM containing 5% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. Opossum kidney (OK) cells, which were derived from the kidney of an adult female North American opossum and display many characteristics of kidney proximal tubular epithelial cells, were maintained in low glucose DMEM containing 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 2 mm l-glutamine. DO11.10 (DO) cells, a T-cell hybridoma line, were grown in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 units/ml penicillin/streptomycin, and 50 μm 2-mercaptoethanol.
Preparation of Apoptotic and Necrotic Cell Targets
Apoptosis of BU.MPT, CHO, LR73, HeLa, MCF-7, OK, and DO11.10 cells was induced by incubating cells in FBS-free medium containing the non-selective protein kinase inhibitor staurosporine (1 μg/ml; 3 h), incubating cells in FBS-containing medium containing the macromolecular synthesis inhibitor actinomycin D (200 ng/ml; overnight), irradiating cells with UV-B irradiation (20–50 mJ/cm2) followed by overnight incubation at 37 °C, incubating cells (DO only) in FBS-containing medium containing the glucocorticoid dexamethasone (1 μm; 24 h), or incubating cells in the presence of the false metabolite 2-deoxyglucose (500 μm) plus 0 mm dextrose or in the presence of the mitochondrial inhibitor antimycin A (2 μm) plus varying concentrations of dextrose (5, 10, and 25 mm) for 6, 24, or 48 h. Cellular ATP levels were measured with an ATP colorimetric/fluorometric assay kit (Abcam) according to the manufacturer's directions and normalized to total cellular protein. After induction of apoptosis, the remaining adherent cells were detached by addition of 5 mm EDTA and pooled with floating cells followed by three washes and resuspension in fresh FBS-free medium before use in experiments. For induction of necrosis, cells were first detached with 5 mm EDTA and suspended in the appropriate FBS-free medium. Necrosis was then induced by heating cells to 65 °C for 40 min followed by incubation at 37 °C for 2 h. Apoptotic targets were added to responder cells either directly or after fixation for 20 min with 0.4% (v/v) paraformaldehyde in PBS.
Induction of apoptosis or necrosis was confirmed by flow cytometry. Viable cells were defined as cells that were both propidium iodide (PI)- and annexin V-negative. Early apoptotic cells (intact cell membranes) were defined as PI-negative cells with annexin V staining and decreased cell size. Necrotic cells were defined as PI-positive cells of normal or increased cell size. Late apoptotic cells (non-intact cell membranes) were defined as PI-positive cells with annexin V staining and decreased cell size. Loss of membrane integrity by necrotic cells was confirmed by trypan blue staining. By these criteria, apoptotic target preparations contained ∼85% early apoptotic and ∼15% late apoptotic cells. Necrotic target preparations contained ∼95% necrotic cells. In all preparations, unless otherwise stated, viable cells, defined as PI-negative cells of normal size without annexin V staining, comprised <5% of the total cell population.
Retroviral Transfection
BU.MPT cells were infected with retroviral vectors containing either green fluorescent protein (GFP) alone (pBabe-GFP) or GFP plus a constitutively active (myristoylated) Akt construct (pBabe-GFP-myrAkt), which were kind gifts from Dr. Nissim Hay (23, 24). Myristoylation of Akt leads to its localization at the cell membrane where Akt can be activated via phosphorylation at Thr308 by the upstream kinase 3-phosphoinositide-dependent protein kinase 1 (PDK1). All downstream signaling events, both cytoplasmic and nuclear, dependent on phosphorylation of Akt at Thr308 are replicated by this constitutively active Akt construct (23, 24). Retroviruses were generated by transient transfection of the retroviral vectors into Phoenix ecotropic packaging cell lines followed by harvesting of the retrovirus-containing culture medium. Retroviral infection of BU.MPT cells was performed in the presence of 8 μg/ml Polybrene for 24 h. The pBabe-eGFP and pBabe-eGFP-myrAkt retroviral vectors as well as protocols using them have been described previously (23, 24).
Western Blot Analysis
After stimulation of BU.MPT responder cells with apoptotic or necrotic targets in the presence or absence of epidermal growth factor (EGF; 10 nm) (EMD Calbiochem), responders were washed with ice-cold PBS and then lysed in ice-cold cell lysis buffer (PBS containing 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm phenylmethylsulfonyl fluoride, and 200 μm orthovanadate). Lysates were centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatants were stored at −70 °C.
Protein samples (20 μg each as determined by the bicinchoninic acid protein assay (Pierce)) were boiled in 6× reducing sample buffer, electrophoresed on 12% SDS-polyacrylamide gels, and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked with either 2.5% (w/v) BSA or 5% (w/v) dry milk in TBS before probing with one of the primary antibodies described above. Following incubation with secondary antibody, immunoreactive bands were visualized by the luminol reaction (GE Healthcare). Equivalent loading of protein samples was monitored by staining with Ponceau S (0.25%, w/v) in 0.1% (v/v) acetic acid for 5 min.
3-(4,5-Dimethylthiazol)-2,5-diphenyltetrazolium Bromide (MTT) Assay
The number of viable BU.MPT cells following exposure to apoptotic or necrotic targets under various experimental conditions was determined using a modification of the MTT assay as described previously (25). After removing the growth medium, 200 μl of MTT dissolved in growth factor-free culture medium (1 mg/ml) was added to each well. After incubation at 37 °C for 4 h, MTT formazan crystals that formed were dissolved by adding 200 μl of 10% (w/v) SDS in 0.01 n HCl and incubating overnight at 37 °C. Aliquots from each well were read using a Bio-Rad model 680 microplate reader with a test wavelength of 570 nm and a reference wavelength of 650 nm. The relative number of viable cells for each experimental condition was expressed as a normalized value by taking the ratio of the mean absorbance (A570/650) for the experimental condition to that for untreated control BU.MPT cells.
5,6-Carboxyfluorescein Diacetate Succinimidyl Ester (CFDA-SE) Cell Proliferation Assay
BU.MPT responder cells were green labeled with CFDA-SE (4 μm; excitation wavelength, 488 nm; emission wavelength, 525 nm; Molecular Probes, Eugene, OR). CFDA-SE-labeled BU.MPT responder cells were exposed to apoptotic or necrotic target cells for 2 h. After targets were washed away, BU.MPT responders were cultured for an additional 0, 24, or 48 h; harvested with PBS supplemented with 0.4 mm EDTA; fixed with 0.4% formaldehyde; and analyzed cytofluorimetrically on a FACSCalibur instrument (BD Biosciences). The number of cell divisions BU.MPT responders had undergone was determined from the ratio of the mean CFDA-SE fluorescence at 24 or 48 h to that at 0 h by the following formula.
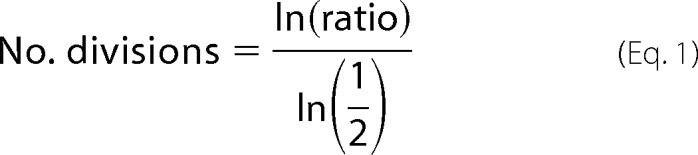 |
[3H]Thymidine Proliferation Assay
BU.MPT responder cells were grown for 24 h in the presence or absence of FBS (10%, v/v). Responder cells were exposed to apoptotic or necrotic targets either continuously throughout the 24 h or as a 2-h pulse at the start of incubation. [3H]thymidine (2 Ci/mmol; PerkinElmer Life Sciences) was added at 1 μCi/well for the final 2 h of incubation. Cells were solubilized in 0.1 n NaOH (0.5 ml/well) for 2 h after which an aliquot was added to scintillation fluid, and [3H]thymidine incorporation was measured using a β counter (Packard model 1900CA Tri-Carb liquid scintillation analyzer β counter, PerkinElmer Life Sciences).
Flow Cytometric Analysis of Apoptosis
BU.MPT responder cells undergoing apoptosis were detected using the PE Active Caspase-3 Apoptosis kit (BD Biosciences) according to kit instructions. DO targets, apoptotic or necrotic, were red prelabeled with (5,6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (8 μm; excitation wavelength, 488 nm; emission wavelength, 610 nm; Molecular Probes) to distinguish them from BU.MPT responder cells either unlabeled or GFP-labeled by retroviral infection. Samples were analyzed cytofluorimetrically on a FACSCalibur instrument (BD Biosciences).
β-Catenin/T-cell Factor (TCF) Luciferase Assay
The pTopflash (wild-type) construct was a generous gift from Carl Vogelstein (National Institutes of Health, Bethesda, MD). This construct contains a firefly luciferase gene reporter under the control of two repeats, each of which contains three copies of the wild-type TCF-binding site upstream of a thymidine kinase minimal promoter. Expression of the luciferase reporter requires the formation of an active β-catenin·TCF transcription complex. BU.MPT cells were transfected with pTopflash using LipofectamineTM LTX with PlusTM reagent (Invitrogen). To control for potential variations in well-to-well cell number and viability, cells were co-transfected with pRL-SV40, a Renilla (sea pansy; Renilla reniformis) luciferase control vector whose constitutive expression is driven by the SV40 early enhancer/promoter region (Promega, Madison, WI). After transfection, cells were treated with Li+ (0–50 mm) in the form of lithium chloride (LiCl) in the presence or absence of targets cells. Cell lysates were harvested 24 h after treatment, and the levels of firefly and Renilla luciferase control were determined by the Dual-Luciferase Reporter Assay System (Promega) in an FB12 luminometer (Zylux, Oak Ridge, TN). Each condition was repeated in triplicate wells, and the luciferase activities in cells from each well were determined independently. The firefly luciferase activity in each sample was then normalized with respect to the Renilla luciferase activity.
Statistics
Data are expressed as mean ± S.E. of the averaged values obtained from each experiment. Statistical significance was determined by a two-tailed Student's t test.
RESULTS
Apoptotic Targets Decrease Viability of Live BU.MPT Responder Cells through Induction of Responder Cell Apoptosis
We have shown previously that receptor-mediated recognition of apoptotic targets by viable BU.MPT epithelial cell responders inhibits Akt activity in responder cells (17). Because Akt is a major survival kinase, we determined the effects of apoptotic targets on the viability of BU.MPT responders (Fig. 1). BU.MPT responders were exposed to apoptotic or necrotic targets either continuously for 48 h or in the form of two 30-min pulses 24 h apart (Fig. 1A). An MTT assay was performed at 48 h to determine the relative number of viable, metabolically active cells remaining as compared with responder cells not exposed to targets. Apoptotic targets profoundly decreased the relative number of viable responders in all cases by >50%. This occurred irrespective of the presence or absence of EGF, a potent survival factor for epithelial cells. Necrotic targets, which activate Akt in BU.MPT cells, had the opposite effect and induced an increase in the relative number of viable BU.MPT responders. Notably, for both apoptotic and necrotic targets, the effects of pulsed versus continuous exposure to targets were comparable in magnitude.
FIGURE 1.

Apoptotic targets decrease viability of BU.MPT responder cells through induction of responder cell apoptosis. A, serum-starved BU.MPT responder cells in the presence or absence of EGF (50 nm) were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 either continuously or as two 30-min pulses 24 h apart, as depicted in the inset. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. After 48 h, relative cell number was determined by MTT assay. A570/650 values were normalized against those for responder cells not exposed to targets. Each data point in the graph represents the mean and S.E. from a minimum of three separate experiments. The viability of control responders was well maintained over the experimental period. As compared with freshly confluent responders at 0 h, the relative cell number for control responders at 24 and 48 h was 86.3 ± 5.0 and 75.1 ± 5.8%, respectively. Absolute A570/650 values after 48 h for control responders not exposed to targets in the absence or presence of EGF were 0.784 ± 0.026 and 0.742 ± 0.021, respectively. All experimental A570/650 values were normalized to these values as represented by the dotted line at relative cell number equal to 1.0. p < 0.00001, apoptotic targets (pulse) and apoptotic targets (continuous) versus control in the absence and presence of EGF; p < 0.05, necrotic targets (continuous) versus control in the absence and presence of EGF. B, serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 for 30 min. The source of apoptotic targets was staurosporine-treated DO11.10 cells. Induction of apoptosis in BU.MPT responders was assessed 24 h after exposure by cytofluorimetric analysis of permeabilized responders for activated caspase-3. C, the graph depicts the mean and S.E. from three separate cytofluorimetric analyses of the percentage of BU.MPT responder cells positive for activated caspase-3. p < 0.00001, apoptotic targets versus control; p = not significant, necrotic targets versus control. Tar, target(s). Error bars (A and C) denote S.E.
To evaluate whether apoptotic death contributes to the decreased viability of BU.MPT responders following their exposure to apoptotic targets, we looked for activation of caspase-3 by flow cytometry. BU.MPT responders were exposed to targets, apoptotic or necrotic, for 30 min and then examined 24 h later (Fig. 1, B and C). Under basal conditions, 17.4 ± 1.4% of BU.MPT responders were positive for activated caspase-3. Exposure to apoptotic targets increased the percentage of caspase 3-positive BU.MPT responders by almost 3-fold to 49.5 ± 4.1% (p < 0.00001). Morphologic and flow cytometric examination confirmed that responder cell death was exclusively by apoptosis (data not shown). In contrast, exposure to necrotic targets, which activate Akt, had no effect on the percentage of caspase-3-positive cells (16.2 ± 2.2%; p = not significant).
These data indicate that exposure to apoptotic targets induces the apoptotic death of viable BU.MPT responder cells. This is consistent with the observed inhibition of Akt activity in viable BU.MPT responders following receptor-mediated recognition of apoptotic targets (17). Although the primary source of apoptotic targets in these studies was murine DO T-cells induced to undergo apoptosis by treatment with actinomycin D, similar results were obtained with BU.MPT targets induced to undergo apoptosis by treatment with staurosporine (data not shown).
Apoptotic Targets Inhibit Proliferation of BU.MPT Responder Cells
Another mechanism by which apoptotic targets might contribute to a decrease in the viability of BU.MPT responders as assessed by MTT assay is through the inhibition of proliferation. Because apoptotic targets also inhibit ERK1/2, a major kinase involved in the regulation of proliferation, we hypothesized that exposure to apoptotic targets would inhibit the proliferation of BU.MPT responder cells. We also predicted that necrotic targets, which activate ERK1/2, would have an opposite effect and stimulate proliferation of BU.MPT cells.
BU.MPT responders were exposed to apoptotic or necrotic targets either continuously over 24 h or as a single 30-min pulse (Fig. 2A). [3H]Thymidine incorporation, a measure of proliferation, was determined at 24 h in both quiescent (serum-starved) and actively dividing (serum-treated) BU.MPT responders. Apoptotic targets inhibited [3H]thymidine incorporation by both quiescent and actively dividing BU.MPT responders. Inhibition was most apparent in actively dividing responders for which [3H]thymidine incorporation was decreased by >50%. Necrotic targets in contrast modestly stimulated [3H]thymidine incorporation. Stimulation was best seen in quiescent BU.MPT responders for which [3H]thymidine incorporation was increased by ∼50%. As with cell survival, these effects were comparable in magnitude for continuous versus pulsed exposure to apoptotic or necrotic targets.
FIGURE 2.

Apoptotic targets decrease viability of BU.MPT responder cells through inhibition of responder cell proliferation. A, serum-starved BU.MPT responder cells in the presence or absence of FBS (10%, v/v) were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 either continuously for 24 h or as a single 2-h pulse, as depicted in the inset. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. As a measure of proliferation, [3H]thymidine incorporation was determined during the final 2 h of a 24-h incubation. Counts per minute (cpm) were normalized against those for responder cells not exposed to targets as represented by the dotted line at relative thymidine incorporation equal to 1.0. Each data point in the graph represents the mean and S.E. from a minimum of three separate experiments. Absolute cpm for responders unexposed to targets in the absence and presence of serum were 5376 ± 830 and 8061 ± 632, respectively. p < 0.05, necrotic targets (pulse) and necrotic targets (continuous) versus control in the absence of serum; p < 0.01, apoptotic targets (pulse) and apoptotic targets (continuous) versus control in the presence of serum. B, BU.MPT responder cells (prelabeled with CFDA-SE) were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 for 2 h. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. Proliferation of BU.MPT responders was assessed at 0, 24, or 48 h after target exposure by cytofluorimetric analysis of the reduction in CFDA-SE staining. C, the graph depicts the mean and S.E. from three separate cytofluorimetric analyses of the cumulative number of cell divisions within the indicated time intervals as determined by Equation 1 given under “Experimental Procedures.” p < 0.01, apoptotic targets versus control, 0–24 h; p < 0.001, apoptotic targets versus control, 0–48 h; p = not significant, necrotic targets versus control, 0–24 and 0–48 h. Tar, target(s). Error bars (A and C) denote S.E.
To confirm inhibition of BU.MPT proliferation following exposure to apoptotic targets, we assessed cell proliferation by a second independent method. Actively dividing BU.MPT responders were labeled with CFDA-SE and then exposed to apoptotic or necrotic targets for 2 h. Proliferation was assessed 24 and 48 h later by determining the extent of halving of CFDA-SE cellular fluorescence from initial levels (Fig. 2, B and C). Under basal conditions, serum-treated BU.MPT cells underwent 1.3 ± 0.2 and 3.1 ± 0.2 cell divisions by 24 and 48 h, respectively. Exposure to apoptotic targets decreased the number of cell divisions to 0.3 ± 0.1 and 0.9 ± 0.2 at 24 and 48 h (p < 0.01), respectively. In contrast, exposure to necrotic targets did not significantly affect the number of cell divisions at either 24 or 48 h (1.4 ± 0.2 and 2.4 ± 0.7; p = not significant).
Although the primary source of apoptotic targets in these studies was actinomycin D-treated DO cells, similar results were obtained with staurosporine-treated BU.MPT targets (data not shown). These results, together with those for caspase-3 activation (Fig. 1), show that exposure to apoptotic targets reduces the number of viable BU.MPT responders through two distinct mechanisms: an increase in cell death and a decrease in cell proliferation. Necrotic cells in general had the opposite effects. Through inhibition of apoptosis and in some cases stimulation of proliferation, necrotic targets increase the number of viable BU.MPT responders.
Signaling Events Elicited in Response to Apoptotic Targets Are Long Lived
Our previous studies examining the effects of dead targets on intracellular signaling events were limited to 30 min following target exposure (17). In our studies here, we examined the viability and proliferation of BU.MPT responders up to 48 h following exposure to dead targets (Figs. 1 and 2). Similar outcomes for pulsed versus continuous exposure to targets suggested to us that the signaling events following pulsed exposure to targets may persist long after washing away of the targets. BU.MPT responders were exposed to apoptotic targets for 15 min and then extensively washed. Remarkably, even 24 h following pulsed exposure, the levels of both phosphorylated Akt and phosphorylated p90RSK (a direct downstream target of ERK1/2) were decreased as compared with those in untreated BU.MPT responder cells (Fig. 3). The persistence of Akt and ERK1/2 inhibition for as long as 24 h following a brief exposure to apoptotic targets is consistent with the prolonged and potent inhibition of responder cell survival and proliferation.
FIGURE 3.

Signaling events elicited in response to apoptotic targets are long lived. Serum-starved BU.MPT responder cells were exposed to vehicle or apoptotic targets (Apo) at a target/responder cell ratio of 1:1 for 15 min. The source of apoptotic targets was staurosporine-treated BU.MPT cells. At the indicated times following stimulation (t = 0), non-adherent cells were removed by washing, and responder cell lysates were probed with anti-active Akt and anti-active p90RSK antibodies as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt.
Mode of Induction of Apoptosis, but Not Target Cellular Origin, Influences Effect of Apoptotic Targets on Responder Cells
The anti-inflammatory effects of apoptotic targets on mφ and other phagocytes are largely independent of the type of target cell and the mode of its apoptotic death (4). We next asked whether the antisurvival effects of apoptotic targets on BU.MPT responders were similarly insensitive to target cell type or mode of apoptotic death. As shown in Fig. 4A, irrespective of the target cell species or tissue origin, apoptotic targets significantly decreased BU.MPT responder cell viability (p < 0.001), whereas necrotic targets significantly increased viability (p < 0.0001). We conclude that early apoptotic and necrotic cells without restriction to species or stimulus have characteristic and divergent effects on the viability of BU.MPT responders.
FIGURE 4.

Mode of induction of apoptosis, but not cellular origin, influences effect of apoptotic targets on responder cells. A, serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) or necrotic (Necro) targets from the indicated cell lines at a target/responder cell ratio of 10:1 for 2 h. The source of apoptotic targets was actinomycin D-treated (Act D) or staurosporine-treated (Stauro) DO11.10 cells. p < 0.001, apoptotic targets (actinomycin D) and apoptotic targets (staurosporine) versus control for all target cell types; p < 0.0001, necrotic targets versus control for all target cell types. B, serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 continuously for 24 h. Apoptosis of DO11.10 target cells was induced by treatment with actinomycin D (Act D), staurosporine (Stauro), dexamethasone (Dex), or ultraviolet irradiation (UV) and terminated by fixation or heating at the indicated times following the apoptotic stimulus, as depicted in the inset. p < 0.01, actinomycin D/fixation versus control for all times except 0 h; p < 0.001, staurosporine/fixation versus control for all times except 0 and 18 h; p < 0.001, actinomycin D/heating versus control for all times except 0 and 2 h; p = not significant, dexamethasone/fixation and UV/fixation versus control for all times. A and B, relative cell number was determined by MTT assay. A570/650 values were normalized against those for BU.MPT responder cells not exposed to targets as represented by the dotted line at relative cell number equal to 1.0. Each data point in the graphs represents the mean and S.E. from a minimum of three or more separate experiments. Absolute A570/650 values for control responders unexposed to targets were 0.515 ± 0.015 and 0.595 ± 0.026 in A and B, respectively. All experimental A570/650 values were normalized to these values. Tar, target(s). Error bars (A and B) denote S.E.
However, BU.MPT responders appeared to discriminate the mode of apoptotic target cell death (Fig. 4B). Targets induced by actinomycin D or staurosporine proved potent in decreasing the viability of viable BU.MPT responders, whereas those induced by dexamethasone or UV irradiation were without effect. Moreover, the antiviability effects of apoptotic targets on BU.MPT responders appeared to be time-dependent. We demonstrated this in the following way. At varying times following initiation of target cell apoptosis, the apoptotic process was terminated by paraformaldehyde fixation. For actinomycin D-induced targets, effectiveness increased progressively over time up to 18 h. In contrast, for staurosporine-treated targets, effectiveness was maximal within 2 h of initiation of apoptosis and then diminished over time. Similar results were obtained with actinomycin D-induced targets when the apoptotic process was terminated by heat-induced cell rupture.
Notably, at the optimal time for termination of the apoptotic process, fixed targets were equal or superior to unfixed targets (Fig. 4B). This finding suggests that target-induced effects are largely independent of released factors and dependent instead on direct physical interaction between responders and targets. We addressed this issue next.
Effects of Apoptotic Targets on BU.MPT Responder Cell Viability Are Dependent on Cell-Cell Interaction
BU.MPT responders were grown in a Transwell support system and separated from unfixed apoptotic targets by a 0.4 μm polycarbonate membrane. Prevention of physical interaction between BU.MPT responders and apoptotic targets abolished both the antisurvival (Fig. 5A) and the antiproliferative (Fig. 5B) activities of apoptotic targets. Consistent with these results, conditioned medium generated during induction of target cell apoptosis failed to inhibit the survival or proliferation of BU.MPT responders (data not shown).
FIGURE 5.

Effects of apoptotic targets on BU.MPT responder cell viability are dependent on cell-cell interaction. Serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 continuously for 24 h, as depicted in the inset, either unimpeded, enabling direct target-responder physical interaction (Control), or with separation of targets and responders by a 0.4-μm polycarbonate membrane in a Transwell support system (Transwell). The source of apoptotic targets was DO11.10 cells treated with actinomycin D (Act D), staurosporine (Stauro), or dexamethasone (Dex). After 24 h, relative cell number by MTT assay (A) or [3H]thymidine incorporation (B) was determined. All values were normalized against those for BU.MPT responder cells not exposed to targets. Each data point in the graphs represents the mean and S.E. from a minimum of three separate experiments. Absolute A570/650 values (A) for responders unexposed to targets in the absence and presence of a Transwell support system were 0.915 ± 0.023 and 0.450 ± 0.02, respectively. Absolute cpm (B) for responders unexposed to targets in the absence and presence of a Transwell support system were 2571 ± 185 and 2805 ± 47, respectively. All experimental A570/650 values (A) and cpm (B) were normalized to these values as represented by the dotted lines at relative cell number (A) and relative thymidine incorporation (B) equal to 1.0. A, p < 0.0001, apoptotic targets (actinomycin D) and apoptotic targets (staurosporine) versus no targets in the absence of Transwell; p = not significant, apoptotic targets versus no targets in the presence of Transwell. B, p < 0.05, apoptotic targets (actinomycin D), apoptotic targets (staurosporine), and apoptotic targets (dexamethasone) versus no targets in the absence of Transwell; p = not significant, apoptotic targets versus no targets in the presence of Transwell. Error bars (A and B) denote S.E.
These data also revealed a divergence between those target cell features necessary to inhibit survival versus those necessary to inhibit proliferation. Whereas dexamethasone-induced DO targets had no effect on BU.MPT responder cell survival (Fig. 5A), these targets were potent inhibitors of BU.MPT cell proliferation and equivalent in potency to actinomycin D- and staurosporine-treated targets (Fig. 5B). These data suggest that recognition of apoptotic targets may be a complex process involving more than a single receptor and/or co-receptor with different effects mediated by distinct receptors.
Effects of Apoptotic Targets on BU.MPT Responder Cell Viability Are Recognition-dependent but Independent of Phagocytosis
As described previously, apoptotic and necrotic targets have opposite effects on Akt- and ERK1/2-dependent signaling events in BU.MPT responders (17). This suggests that these signaling events depend upon unique receptor-mediated recognition rather than shared phagocytic machinery. To test this idea, we used the cytoskeletal inhibitor cytochalasin D to prevent phagocytosis. As shown, inhibition of both survival (Fig. 6A) and proliferation (Fig. 6B) in response to apoptotic targets occurred even in the absence of phagocytosis. We obtained comparable data with two concentrations of cytochalasin D, both of which inhibited mφ phagocytosis by ≥90%. Taken together, our results show that apoptotic target-mediated inhibition of BU.MPT survival and proliferation requires direct physical interaction between targets and responder cells but is independent of phagocytosis.
FIGURE 6.

Effects of apoptotic targets on BU.MPT responder cell viability are recognition-dependent but independent of phagocytosis. Serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 for 2 h, as depicted in the inset, in either the absence (Control) or the presence of cytochalasin D (Cytochalasin), an inhibitor of phagocytosis. The source of apoptotic targets was DO11.10 cells treated with actinomycin D (Act D), staurosporine (Stauro), or dexamethasone (Dex). After 24 h, relative cell number by MTT assay (A) or [3H]thymidine incorporation (B) was determined. All values were normalized against those for BU.MPT responder cells not exposed to targets. Each data point in the graphs represents the mean and S.E. from a minimum of three separate experiments. Absolute A570/650 values (A) and cpm (B) for responder cells unexposed to targets in the absence of cytochalasin D were 0.344 ± 0.012 and 2972 ± 173, respectively. All experimental A570/650 values (A) and cpm (B) were normalized to these values as represented by the dotted lines at relative cell number (A) and relative thymidine incorporation (B) equal to 1.0. A, p < 0.0001, apoptotic targets versus no targets in the presence and absence of cytochalasin D. B, p < 0.0001, apoptotic targets versus no targets in the presence and absence of cytochalasin D. Error bars (A and B) denote S.E.
Targets Induced by Chemical Anoxia Also Decrease Viability of Live BU.MPT Responder Cells
Given the apparent ability of BU.MPT responders to discriminate the mode of apoptotic death (Figs. 4 and 5), we determined the effect of targets induced by chemical anoxia, a stimulus more physiologically relevant to the kidney. As described previously (26, 27), we produced graded depletion of cellular stores of adenosine triphosphate (ATP) by two methods: inhibition of glycolysis with 2-deoxyglucose, a false metabolite, and inhibition of mitochondrial respiration with antimycin A. Both methods led to significant reductions in cellular ATP. Culture in the presence of 2-deoxyglucose (500 μm) and 0 mm dextrose for 8 h reduced target cell ATP levels to 5.5% of basal levels. Similarly, culture in the presence of antimycin A (2 μm) and dextrose (1, 5, 10, and 25 mm) for 8 h reduced target cell ATP levels to 3.3, 4.4, 5.8, and 20.8% of basal levels, respectively.
All degrees of ATP depletion resulted in substantial levels of target cell death. As assessed by flow cytometry, the percentage of targets that were non-viable 24 h after induction of ATP depletion was 89% for targets exposed to 2-deoxyglucose and 93, 94, 83, and 53% for targets exposed to antimycin A in the presence of dextrose at 1, 5, 10, and 25 mm, respectively. Dead targets appeared to be apoptotic, as assessed by decreased cell size and annexin V positivity, but other forms of cell death associated with ATP depletion, such as programmed cell necrosis, cannot be excluded. The remaining targets for all conditions of ATP depletion were viable as assessed by an unchanged cell size and a lack of staining by annexin V and PI. Targets induced by chemical anoxia were potent in inducing the death of BU.MPT responders (Fig. 7) with viability in all cases reduced by >50% (p < 0.0001). This was true regardless of the time at which the apoptotic process was terminated by paraformaldehyde fixation.
FIGURE 7.

Apoptotic targets induced by chemical anoxia decrease viability of BU.MPT responder cells. Serum-starved BU.MPT responder cells were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 continuously for 24 h. The source of apoptotic targets was DO11.10 cells treated with actinomycin D (Act D); 2-deoxyglucose (DOG) (500 μm) plus 0 mm dextrose; or antimycin A (Antimyc A) (2 μm) plus 5, 10, or 25 mm dextrose (Dex). For targets induced by chemical anoxia, the apoptotic process was terminated by paraformaldehyde fixation at 6 or 24 h following initiation of the apoptotic stimulus, as depicted in the inset. For targets induced by actinomycin D, the apoptotic process was terminated only at 24 h. Relative cell number of responders was determined by MTT assay at 48 h following exposure to targets. A570/650 values were normalized against those for BU.MPT responder cells not exposed to targets as represented by the dotted line at relative cell number equal to 1.0. Each data point in the graphs represents the mean and S.E. from a minimum of three or more separate experiments. The absolute A570/650 for control responders unexposed to targets was 0.481 ± 0.017. All experimental A570/650 values were normalized to these values. p < 0.0001, apoptotic targets versus no targets for all apoptotic stimuli and times of fixation after induction of apoptosis. Error bars denote S.E.
Viability Response of Epithelial Responders to Apoptotic Targets Correlates with Their Akt Response and Tissue of Origin
To determine whether the antisurvival response of BU.MPT responders to apoptotic targets is a general feature of epithelial cells or specific to BU.MPT cells, we evaluated a panel of epithelial cell lines derived from different tissues. As described previously (17), epithelial cells can be divided into two types: those that resemble BU.MPT cells with exposure to apoptotic targets leading to inhibition of Akt (OK and CHO cells) and those that differ from BU.MPT cells with exposure to apoptotic targets leading to activation of Akt (HeLa, LR73, and MCF-7 cells). The effect of apoptotic targets on the viability of each of these epithelial cell lines correlated with its Akt response (Fig. 8). Those epithelial cell responders, like BU.MPT cells, for which Akt was inhibited showed a significant decrease in viability (p < 0.05). In contrast, those epithelial cell responders for which Akt was activated showed significantly increased cell viability (p < 0.05). Consistent with the universal response of Akt activation in response to necrotic targets, CHO, OK, HeLa, LR73, and MCF-7 epithelial cell responder lines all showed significantly increased viability following exposure to necrotic targets (p < 0.05). These data indicate that the effect of apoptotic targets on epithelial responders is tissue-specific with activation of Akt and increased viability occurring in some epithelial cell responders, especially those derived from kidney, and inhibition of Akt and decreased survival occurring in others. Combined with the data shown in Fig. 4A, our findings suggest that the response of epithelial cells to apoptotic targets depends on the tissue origin of the responder cell but is independent of the tissue origin of the target cell.
FIGURE 8.

Viability response of epithelial responders to apoptotic targets varies with responder tissue of origin but correlates with responder Akt response. Serum-starved responders from the indicated epithelial cell lineages were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 either continuously for 24 h or as a single 2-h pulse, as depicted in the inset. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. After 48 h, relative cell number was determined by MTT assay. A570/650 values were normalized against those for the appropriate responder cell lineage not exposed to targets. Each data point in the graph represents the mean and S.E. from a minimum of three separate experiments. Responder cell lineages are grouped into those for which exposure to apoptotic targets leads to a decrease of Akt activity and those for which exposure leads to an increase of Akt activity. The tissue origin of these lineages are: kidney (OK), ovary (CHO and LR73), breast (MCF-7), and uterus (HeLa). Absolute A570/650 values for responder cells unexposed to targets were 0.177 ± 0.005 (CHO), 0.155 ± 0.006 (OK), 0.671 ± 0.023 (HeLa), 0.353 ± 0.004 (LR73), and 0.359 ± 0.051 (MCF-7), respectively. All experimental A570/650 values were normalized to these values as represented by the dotted line at relative cell number equal to 1.0. p < 0.05, apoptotic targets (pulse) versus control, CHO, OK, LR73, and MCF-7 responders; p < 0.05, apoptotic targets (continuous) versus control, CHO, OK, and MCF-7 responders; p < 0.05, necrotic targets (pulse) versus control, LR73, and MCF-7 responders; p < 0.05, necrotic targets (continuous) versus control, CHO, LR73, and MCF-7 responders. Tar, target(s). Error bars denote S.E.
Infection of BU.MPT Responders with a Constitutive Akt Construct Partially Prevents Loss of Viability following Exposure to Apoptotic Targets
To determine whether Akt inhibition is related causally to decreased BU.MPT viability following exposure to apoptotic targets, we transiently infected BU.MPT cells with a myristoylated Akt (myrAkt)-GFP retroviral construct (23, 24). Myristoylation of Akt leads to its surface membrane localization and constitutive activity (23, 24). BU.MPT cells expressing myrAkt, hereafter denoted as BU.MPT-myrAkt-GFP, were detected by co-expressed GFP. BU.MPT control cells, which were infected with a retroviral construct containing GFP alone, are denoted as BU.MPT-GFP.
Infection of BU.MPT cells with myrAkt conferred almost complete protection from UV-induced apoptosis as assessed by the percentage of responder cells positive for activated caspase-3 (Fig. 9, A and C). Whereas UV irradiation increased the percentage of caspase-3-positive BU.MPT-GFP responders from 10.9 ± 1.5 to 34.6 ± 9.1% (p < 0.05), UV irradiation of BU.MPT-myrAkt-GFP responders did not significantly increase the percentage of caspase-3-positive cells (13.5 ± 2.2 versus 9.8 ± 1.7%; p = not significant). Constitutive Akt activity also protected BU.MPT responders from loss of viability induced by exposure to apoptotic targets, but in this case the protection was only partial (Fig. 9, B and C). For BU.MPT-GFP responders, exposure to apoptotic targets increased the percentage of caspase-3-positive responder cells from 10.9 ± 1.5 to 41.8 ± 6.4% (p < 0.002). In contrast, the percentage of caspase-3-positive cells for BU.MPT-myrAkt-GFP responders was significantly reduced to 22.4 ± 3.1% compared with BU.MPT-GFP responders (p < 0.05). Protection by constitutively active Akt was incomplete because the percentage of caspase-3-positive cells remained greater for target-exposed BU.MPT-myrAkt-GFP responders compared with non-exposed responders (22.4 ± 3.1 versus 9.8 ± 1.7%; p < 0.01). We conclude that constitutive Akt activity only partially protects BU.MPT responders from apoptosis induced by exposure to apoptotic targets. These data suggest that exposure to apoptotic targets decreases the viability of BU.MPT responder cells through additional mechanisms that are independent of Akt.
FIGURE 9.

Infection of BU.MPT responders with a constitutively active Akt construct partially prevents loss of viability following exposure to apoptotic targets. BU.MPT responder cells were transiently infected with retroviral constructs containing either GFP alone (GFP) or GFP plus constitutively active myrAkt (myrAkt-GFP). A, infected BU.MPT responders were left untreated (Untreated) or induced to undergo apoptosis by exposure to UV irradiation (UV). Shown is a representative cytofluorimetric analysis for activated caspase-3 24 h after UV irradiation. B, serum-starved infected BU.MPT responders were left untreated (Untreated) or exposed to apoptotic (Apo) or necrotic (Nec) targets at a target/responder cell ratio of 10:1 for 30 min. The source of apoptotic targets was staurosporine-treated DO11.10 cells. Induction of apoptosis in BU.MPT responders was assessed 24 h after exposure to targets by cytofluorimetric analysis of permeabilized responders for activated caspase-3. C, the graph depicts the mean and S.E. from three separate cytofluorimetric analyses of the percentage of BU.MPT responder cells positive for activated caspase-3. In all cases, cytofluorimetric analysis was limited to infected BU.MPT responders whose GFP fluorescence was >1000× that of uninfected cells. The dotted line represents the percentage of caspase 3-positive responders in untreated BU.MPT responders infected with GFP alone. p < 0.05, UV-irradiated and apoptotic targets versus untreated for GFP-infected responders; p < 0.05, UV-irradiated and apoptotic targets, GFP- versus myrAkt-GFP-infected responders; p < 0.01, apoptotic targets versus untreated for myrAkt-GFP-infected responders; p = not significant, UV-irradiated versus untreated for myrAkt-GFP-infected responders. Tar, target(s); Necro, necrotic. Error bars (C) denote S.E.
Exposure of BU.MPT Responders to Apoptotic Targets Inhibits β-Catenin-dependent Transcriptional Activity
A downstream target of Akt is glycogen synthase kinase 3β (GSK3β), a kinase that plays a key role in regulating the survival and proliferation of BU.MPT (28) and other cells. Phosphorylation of GSK3β inhibits its activity and leads to stabilization of β-catenin, which then translocates to the nucleus. β-Catenin promotes the transcription of genes known to stimulate cell proliferation and inhibit apoptosis by binding to transcription factors of the TCF-lymphoid enhancer factor family.
We have shown previously that inhibition of Akt by apoptotic targets is associated with decreased phosphorylation of GSK3β (17), which should lead to increased GSK3β activity and hence decreased abundance and activity of β-catenin. We therefore explored the effect of apoptotic targets on the fate and activity of β-catenin. Exposure to apoptotic targets led to early and sustained phosphorylation of β-catenin in BU.MPT responders (Fig. 10A) as predicted. This is in accord with the decreased phosphorylation and hence greater activity of GSK3β that we have described (17). Phosphorylation of β-catenin is known to target β-catenin for ubiquitin-mediated degradation. Consistent with this, exposure to apoptotic targets led to a decrease in the cellular abundance of β-catenin (Fig. 10C). The impact of this decrease in β-catenin induced by exposure to apoptotic targets was best seen in the presence of lithium (Li+), an inhibitor of GSK3β that typically leads to increased cellular levels of β-catenin (21). The observed decrease in β-catenin was accompanied by a corresponding decrease of almost 50% (p < 0.0005) in β-catenin-dependent transcriptional activity (Fig. 10B). Transcriptional inhibition following exposure to apoptotic targets was confirmed by examining the expression of cyclin D1, a TCF-lymphoid enhancer factor-responsive gene product that plays a key role in cell cycle progression (Fig. 10C). Apoptotic targets reduced the expression of cyclin D1 under both basal and Li+-stimulated conditions. The failure of apoptotic targets to produce a decrease in β-catenin-dependent transcriptional activity in the absence of Li+ (Fig. 10B), despite decreases in cellular levels of total β-catenin and cyclin D1, may reflect either a relative insensitivity of our transcriptional assay or target-induced modulation of factors other than protein abundance that contribute to β-catenin-dependent transcriptional activity. Necrotic targets, on the other hand, consistent with their activation of Akt, had no effect on β-catenin-dependent transcriptional activity (Fig. 9B), and in the absence of Li+, they increased expression of cyclin D1 (Fig. 10C).
FIGURE 10.

Apoptotic targets inhibit β-catenin-dependent transcriptional activity. A, serum-starved BU.MPT responder cells were exposed to vehicle or apoptotic targets (Apo) at a target/responder cell ratio of 10:1 for 30 min. The source of apoptotic targets was staurosporine-treated BU.MPT cells. At the indicated times following stimulation (t = 0), non-adherent cells were removed by washing, and responder cell lysates were probed with anti-phospho-β-catenin antibody as shown. B, to assess β-catenin-dependent transcriptional activity, BU.MPT responder cells were transiently co-transfected with pTopflash and a Renilla luciferase normalization vector. 24 h after transfection, BU.MPT responders in the presence or absence of lithium (40 mm) added 1 h earlier were exposed to apoptotic (Apo) or necrotic (Necro) targets at a target/responder cell ratio of 10:1 as a single 30-min pulse as depicted in the inset. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. 24 h after exposure to targets, luciferase activities of BU.MPT responders were determined. All values were normalized against those for responder cells exposed to neither lithium nor targets. The graph depicts the mean and S.E. from three separate experiments. p < 0.005, apoptotic targets versus control in the presence of lithium. C, BU.MPT responder cells in the presence or absence of lithium (40 mm) were exposed to apoptotic (Apo) or necrotic (Nec) targets at a target/responder ratio of 10:1 continuously for 24 h. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. Targets and non-adherent cells were removed by washing, and BU.MPT responder cell lysates were probed for total cyclin D1 and β-catenin as shown. A and C, equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt. Tar, target(s). Error bars (B) denote S.E.
Apoptotic Targets Activate Caspase-8 in BU.MPT Responders
Because a constitutively active Akt construct protected BU.MPT responders only partially from the antisurvival effect of apoptotic targets (Fig. 9), we considered Akt-independent pathways by which apoptotic targets might decrease responder cell viability. We evaluated first the role of signaling pathways initiated by death receptors such as Fas for which caspase-8 is the direct mediator. Cleavage and activation of caspase-8 was observed in BU.MPT responders following exposure to apoptotic targets (Fig. 11A). Unlike inhibition of Akt, which is a more or less immediate event following exposure to apoptotic targets, cleavage of caspase-8 was delayed, becoming detectable only after 4 h and continuing to at least 24 h. To determine the importance of caspase-8-mediated cell death in our system, we used a cell-permeable chemical inhibitor of caspase-8, Z-IETD-fmk. Treatment with Z-IEDT-fmk increased the viability of BU.MPT responders exposed to apoptotic targets at both 24 and 48 h (Fig. 11B). Higher concentrations of Z-IEDT-fmk led to cellular toxicity and did not confer greater protection (data not shown). Although the magnitude of the effect was relatively small, viability at both time points was increased significantly (p < 0.05). These data are consistent with the notion that activation of caspase-8 represents but one of several parallel mechanisms by which apoptotic targets decrease responder cell viability.
FIGURE 11.

Apoptotic targets activate caspase-8 in BU.MPT responders. A, serum-starved BU.MPT responder cells were exposed to vehicle or apoptotic targets (Apo) at a target/responder cell ratio of 10:1 for 30 min. The source of apoptotic targets was staurosporine-treated BU.MPT cells. At the indicated times following stimulation (t = 0), non-adherent cells were removed by washing, and responder cell lysates were probed with anti-cleaved caspase-8 antibody as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt. B, serum-starved BU.MPT responder cells in the presence or absence of the caspase-8 inhibitor Z-IEDT-fmk (200 μm) were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 in a single 2-h pulse, as depicted in the inset. The source of apoptotic targets was actinomycin D-treated DO11.10 cells. After 24 or 48 h, relative cell number was determined by MTT assay. A570/650 values were normalized against those for responder cells not exposed to targets. Each data point in the graph represents the mean and S.E. from a minimum of three separate experiments. Absolute A570/650 values for responder cells unexposed to targets and in the absence of Z-IEDT-fmk at 24 and 48 h were 0.566 ± 0.078 and 0.661 ± 0.075, respectively. All experimental A570/650 values were normalized to these values as represented by the dotted line at relative cell number equal to 1.0. p < 0.05, apoptotic targets + Z-IEDT-fmk versus apoptotic targets at 24 and 48 h. C, BU.MPT responder cells were transiently infected with retroviral constructs containing either GFP alone (GFP) or GFP plus constitutively active myristoylated Akt (GFP-myrAkt). Serum-starved infected BU.MPT responders in the presence or absence of Z-IEDT-fmk (200 μm) were left untreated (Untreated) or exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 for 30 min. The source of apoptotic targets was staurosporine-treated DO11.10 cells. Induction of apoptosis in BU.MPT responders was assessed 24 h after exposure to targets by cytofluorimetric analysis of permeabilized responders for activated caspase-3. D, the graph depicts the mean and S.E. from three separate cytofluorimetric analyses of the percentage of BU.MPT responder cells positive for activated caspase-3. In all cases, cytofluorimetric analysis was restricted to infected BU.MPT responders whose GFP fluorescence was >1000× that of uninfected cells. The dotted line represents the percentage of caspase 3-positive responders in untreated BU.MPT responders infected with GFP alone. p < 0.01, Z-IEDT-fmk versus control, apoptotic targets, GFP-infected responders; p < 0.01, Z-IEDT-fmk versus control, apoptotic targets, myrAkt-GFP-infected responders; p < 0.01, apoptotic targets in the absence of Z-IEDT-fmk, GFP- versus myrAkt-GFP-infected responders; p < 0.005, apoptotic targets in the presence of Z-IEDT-fmk, GFP- versus myrAkt-GFP-infected responders. Tar, target(s). Error bars (B and D) denote S.E.
To determine the interaction between Akt-dependent and caspase-8-dependent pathways affecting cell viability, we assessed the degree of caspase-3 activation in BU.MPT-myrAkt-GFP versus BU.MPT-GFP responders in the presence or absence of Z-IEDT-fmk (Fig. 11, C and D). Exposure to apoptotic targets increased the percentage of caspase-3-positive BU.MPT-GFP responders from 9.5 ± 0.1 to 53.8 ± 3.8% (p < 0.002). Expression of the constitutively active myrAkt construct reduced the percentage of caspase-3-positive responders to 39.4 ± 5.2% (p < 0.01). Similarly, inhibition of caspase-8 with Z-IEDT-fmk reduced the percentage of caspase-3-positive responders to 38.6 ± 5.0% (p < 0.01). The combination of the two treatments further reduced the percentage of caspase-3-positive responders to 21.9 ± 5.7% (p < 0.01 versus myrAkt alone; p < 0.005 versus Z-IEDT-fmk alone).
The additive nature of these interventions suggests that inhibition of Akt and activation of caspase-8 contribute independently to the loss of viability induced by exposure to apoptotic targets. The failure of combined myrAkt and Z-IEDT-fmk fully to restore responder cell viability has two potential explanations. Incomplete rescue may indicate the existence of additional pathways affecting viability, or alternatively, it may reflect incomplete penetrance of the myrAkt construct and/or incomplete inhibition of caspase-8 cleavage by Z-IEDT-fmk.
Activation of p38 MAPK by Apoptotic Targets Increases Viability of BU.MPT Responders
In many cells, the same receptor can initiate both activatory and inhibitory pathways. This serves as a means for fine control. We next explored whether regulation of the response to apoptotic targets is subject to this type of dual control. Recently, it has been shown that phosphorylation of GSK3β by p38 MAPK represents an alternative Akt-independent means to inhibit GSK3β (29). We have shown previously that apoptotic targets activate p38 MAPK in BU.MPT responder cells (17). We used SB 203580, a pharmacologic inhibitor of p38 MAPK, to determine whether activation of p38 MAPK in BU.MPT responders modulates the activation of GSK3β in response to apoptotic targets. As expected, inhibition of p38 MAPK led to a decrease in levels of total β-catenin in control BU.MPT responders as well as in responders exposed to dexamethasone-induced apoptotic targets (Fig. 12A). Levels of total β-catenin in BU.MPT responders exposed to actinomycin D- or staurosporine-induced apoptotic targets were already markedly reduced, and further suppression was not detected. The functional consequences of these results were confirmed by MTT assay. Inhibition of p38 MAPK with SB 203580 significantly reduced the viability of control BU.MPT cells and BU.MPT responders exposed to apoptotic targets induced by actinomycin D, staurosporine, or dexamethasone (p < 0.001; Fig. 12B). Together, these data suggest that activation of p38 MAPK by apoptotic targets partially opposes inhibition of Akt and the resulting loss of inhibition of GSK3β. The role of Akt in BU.MPT cells appears to predominate over that of p38 MAPK because activation of p38 MAPK by apoptotic targets cannot compensate for inhibition of Akt and the resulting activation of GSK3β, with a consequent reduction in cellular abundance of β-catenin.
FIGURE 12.

Activation of p38 MAPK by apoptotic targets increases viability of BU.MPT responders. A, BU.MPT responder cells in the presence or absence of the p38 MAPK inhibitor SB 203580 (10 μm) were exposed to EGF or apoptotic (Apo) targets at a target/responder ratio of 10:1 for 30 min. The source of apoptotic targets was DO11.10 cells treated with actinomycin D (Act D), staurosporine (Stauro), or dexamethasone (Dex). 24 h after exposure, non-adherent cells were removed by washing, and BU.MPT responder cell lysates were probed for total β-catenin as shown. Equal loading was confirmed by Ponceau S staining of blotted proteins as well as by probing for total Akt. B, serum-starved BU.MPT responder cells in the absence (Control) or presence (SB 203580) of the p38 MAPK inhibitor SB 203580 (10 μm) were exposed to apoptotic (Apo) targets at a target/responder cell ratio of 10:1 continuously for 24 h, as depicted in the inset. The source of apoptotic targets was DO11.10 cells treated with actinomycin D (Act D), staurosporine (Stauro), or dexamethasone (Dex). After 48 h, relative cell number was determined by MTT assay. A570/650 values were normalized against those for responder cells not exposed to targets. Each data point in the graph represents the mean and S.E. from a minimum of three separate experiments. The absolute A570/650 for responder cells unexposed to targets and in the absence of SB 203580 was 0.532 ± 0.010. All experimental A570/650 values were normalized to these values as represented by the dotted line at a relative cell number equal to 1.0. p < 0.001, SB 203580 versus control for responders unexposed to targets or exposed to apoptotic (actinomycin D), apoptotic (dexamethasone), and apoptotic (staurosporine) targets. Error bars (B) denote S.E.
DISCUSSION
We show here that exposure of live kidney PTECs to dead target cells modulates their viability. In general, apoptotic and necrotic targets exert opposite effects when interacting with kidney PTECs. Whereas apoptotic targets profoundly decrease the viability of kidney proximal tubular epithelial responder cells, necrotic targets increase their viability. In each case, modulation of viability involves complementary effects on survival and proliferation. Thus, apoptotic targets decrease responder cell viability through a combination of increased apoptosis and decreased proliferation. Necrotic targets, on the other hand, increase responder cell viability through opposite effects, namely decreased apoptosis and increased proliferation. For both types of dead targets, the effect on viability requires direct physical interaction between targets and responders through receptor-mediated recognition and without the need for phagocytosis.
We have shown previously that kidney PTECs recognize apoptotic and necrotic targets via distinct, non-competing receptors. Receptor-mediated recognition elicits a broad array of signaling events with the capacity to affect viability. These signaling events can be divided into Akt-dependent and Akt-independent pathways (Fig. 13). This division applies to target-mediated modulation of both survival and proliferation. In general, the effects of apoptotic targets are directionally opposite (e.g. inhibition rather than activation) from those of necrotic targets. Notably, many of these signaling events are long lived, persisting for as long as 24 h following a brief 15-min exposure to targets (see Fig. 3). This is consistent with a longer term adaptive response to the surrounding environment.
FIGURE 13.

Apoptotic and necrotic targets modulate viability of epithelial responder cells by eliciting multiple recognition-dependent signaling pathways. The recognition of dead cells, either apoptotic (A) or necrotic (B), by non-professional phagocytes such as kidney PTECs is coupled with multiple signaling events that modulate cell viability. Alterations in responder cell viability occur through synergistic changes in proliferation and survival. Apoptotic and necrotic targets produce opposite effects on PTEC viability. Apoptotic targets decrease responder cell viability through a combination of decreased proliferation and increased apoptotic death. In contrast, necrotic targets increase viability by increasing proliferation and decreasing responder cell death. These recognition-dependent responses to apoptotic and necrotic targets are elicited by directionally opposite responses in the activity of multiple pathways and signaling elements, including β-catenin-dependent transcriptional activity and activity of the kinases Akt, GSK3β, ERK1/2, and p38. Pathways and signaling elements can be grouped into those that are Akt-dependent (within the keystone outline) and those that are Akt-independent (outside the keystone). For example, in response to apoptotic targets, proapoptotic effects include decreased inhibition of Bad (Akt-dependent) and increased activity of caspase-8 (Akt-independent), whereas antiproliferative effects include decreased abundance of cyclin D1 (Akt-dependent) and decreased activity of ERK1/2 (Akt-independent). Arrowheads beside the boxed signaling element indicate the overall effect of the target on the activity of the signaling element irrespective of phosphorylation status. The arrow connecting p38 and GSK3β for apoptotic targets (A) is dashed to indicate that this pathway is counter-regulatory and opposes the proapoptotic effects of Akt inhibition. The effect of Akt inhibition predominates over that of p38 activation with respect to GSK3β activity. lef, lymphoid enhancer factor.
We speculate that PTEC responders may be able to discriminate not only the mode of death (apoptotic versus necrotic) but also to a limited extent the stimulus responsible for induction of apoptotic death (see Figs. 4 and 5). Thus, certain stimuli yielded apoptotic targets capable of triggering responder cell death (staurosporine, actinomycin, and ATP depletion), whereas other stimuli were largely ineffective (UV irradiation and dexamethasone). It is important to emphasize that regardless of their effectiveness apoptotic targets never mimicked the effects of necrotic targets. The effect of apoptotic targets on PTEC responders was either lethal or neutral, whereas that of necrotic targets was to promote viability.
In the case of apoptotic targets (Fig. 13A), we have identified at least three pathways involved in the regulation of responder cell survival. Two of these pathways promote apoptosis, and one counter-regulatory pathway opposes apoptosis. Of the two pathways promoting apoptosis, one is Akt-dependent with exposure to apoptotic targets leading to decreased phosphorylation and activity of Akt. The other pathway promoting apoptosis is Akt-independent and entails activation of caspase-8. Inhibition of Akt is an immediate event occurring rapidly following exposure to apoptotic targets, whereas activation of caspase-8 is delayed for several hours (see Fig. 11A), suggesting the need for synthesis of a critical protein (discussed below). The contributions to target-induced apoptosis by these two pathways appear to be independent and roughly comparable in magnitude. Thus, at 24 h following exposure to apoptotic targets, the percentage of caspase-3-positive responders was reduced from ∼60 to ∼40% either by infection of responders with constitutively active myrAkt or by pharmacologic inhibition of caspase-8 with Z-IEDT-fmk. The combination of myrAkt and Z-IEDT-fmk was additive and further reduced the percentage of caspase-3-positive responders to ∼20%. It is unclear whether the remaining caspase-3-positive responders are attributable to a third proapoptotic pathway or whether they represent incomplete penetrance of the myrAkt construct and/or incomplete inhibition of caspase-8 cleavage by Z-IEDT-fmk.
The third pathway involved in apoptotic target-mediated regulation of responder cell survival is counter-regulatory. Activation of p38 MAPK attenuates the effects of Akt inhibition and thereby promotes the survival of responder cells. p38 MAPK acts at the level of GSK3β, a direct downstream target of Akt. Phosphorylation and inhibition of GSK3β are one of the major pathways by which Akt enhances cell survival. Although the best characterized mechanism for inhibition of GSK3β is through Akt-mediated phosphorylation at Ser9, phosphorylation at Ser389 by p38 MAPK represents an alternative Akt-independent means for inhibiting GSK3β. Both phosphorylation events promote survival through decreased degradation of β-catenin and resulting increased β-catenin-dependent transcriptional activity. We have shown previously that insulin-like growth factor II, a β-catenin-dependent gene product, serves to maintain the survival of serum-starved BU.MPT cells (21, 30). In addition, β-catenin helps to inactivate the proapoptotic protein Bax (21, 30). It should be stressed, however, that the effect of Akt inhibition predominates over that of p38 MAPK with respect to responder cell survival. Thus, the prosurvival activity of p38 MAPK was apparent only under conditions in which Akt activity was marginal or inhibited (see Fig. 12).
Like target-mediated induction of apoptosis, inhibition of proliferation involves Akt-dependent and -independent pathways. Akt-dependent inhibition is mediated at least in part by a decreased abundance of cyclin D1, a β-catenin-responsive gene product that plays a key role in cell cycle progression. Akt-independent inhibition of proliferation occurs through inhibition of ERK1/2 MAPK. The combined effects of increased apoptosis and decreased proliferation lead to a profound decrease in responder cell viability as reflected by a >50% decrease in cell number 24 h after a pulsed exposure to apoptotic targets.
Delayed activation of caspase-8 in BU.MPT responders following exposure to apoptotic targets is a provocative finding and suggests the involvement of a death receptor (DR)-dependent pathway in the reduced viability of kidney PTEC responders. DRs are members of the tumor necrosis factor receptor (TNFR) superfamily; their engagement triggers apoptosis through cleavage and activation of caspase-8. Examples of DRs include Fas (CD95), TNFR1, DR3, DR4, and DR5. Kidney PTECs have been shown to express Fas (31), although the functional relevance of Fas expression remains poorly understood. Extracellular expression of FasL, the ligand for Fas, does not occur on resting tubular cells (32). In the setting of acute renal injury, infiltrating lymphocytes expressing FasL have been suggested to play a role in tubular cell death (33). An alternative source of FasL, as shown by Linkerman et al. (34) in a cisplatin-induced model of acute kidney injury, may be the cisplatin-exposed tubular cells themselves. This process, which the authors termed “fratricide,” has clear parallels with the findings reported here. However, a number of important questions arise. Assuming a DR is involved in caspase-8 activation by apoptotic targets, the identity of the DR and the cellular source of the DR ligand (targets or bystander responder cells) need to be determined. As an example, synthesis of DR ligand may be induced in live responder cells once specific recognition of apoptotic targets exceeds a certain threshold. Alternatively, caspase-8 activation may occur independently of DRs and their ligands and represent a novel signaling event linked not to a DR but to the specific receptors responsible for recognition of apoptotic targets. In accord with the literature, resting BU.MPT cells express Fas but not FasL (31, 32). Exposure to apoptotic or necrotic targets neither altered Fas expression nor induced expression of FasL.3
The implications of our results are broad and likely extend beyond the kidney to other tissues and organs of the body. Cells of diverse lineages, including non-professional phagocytes (e.g. fibroblasts and epithelial cells) and even non-phagocytic cells such as T-lymphocytes, have the ability to recognize and signal differentially in response to apoptotic and necrotic targets. Although many of the elicited signaling events appear to be broadly conserved (for example, NFκB-dependent transcriptional activity), some vary with cell lineage. We have described the critical differences between the responses of mφ and those of kidney PTECs. Moreover, we have shown that epithelial cell responses (see Fig. 8) can differ dramatically depending on the organ of origin. These differences can best be understood in terms of the implications of neighboring cell death, both frequency and mode of death (apoptotic versus necrotic), for surviving responders. These implications are determined by multiple factors, including the lineage and function of the responding cell and the organ of residence.
With this in mind, we hypothesize that cell death plays an important role in normal tissue homeostasis. Alterations in the extent and type of cell death as triggered by such diverse events as ischemia, infection, aging, and acute injury have the potential to alert neighboring viable cells to environmental changes or stress. Within this scheme, dead cells transmit a “message” to nearby viable cells about environmental conditions within their immediate vicinity. Because the signaling responses of viable neighboring cells require physical interaction with dead targets, the effects of targets will be local and limited to their close environment. In many cases, the signaling events induced in responding cells by apoptotic versus necrotic targets are directionally opposite. Through such a dichotomy, responding cells can assess the overall severity of any environmental change. The opposite responses of kidney PTECs to apoptotic versus necrotic may be understood in the following manner. In the setting of progressive atherosclerosis, for example, chronic ischemia may lead to a diminished delivery of oxygen and nutrients. The resulting increase of apoptotic cell death will lead to a target-induced reduction in population size through a combination of increased responder cell death and decreased responder cell proliferation. When supply and demand are once again matched, apoptotic death will return to base-line levels, and a new steady state with an overall reduced population will be achieved. In contrast, an increase of necrotic cell death as may occur in the setting of vascular occlusion or infarction carries very different implications. In this the case, the environmental change is so acute and drastic that compensatory long term adaptation is not possible, and any cell survival may be of benefit.
In summary, we have shown that dead target cells, whether apoptotic or necrotic, have a broad capacity to affect the survival and proliferation of neighboring epithelial responder cells with which they interact. This broad capacity is derived from the modulation of multiple signaling pathways, both Akt-dependent and -independent, that contribute to the regulation of cell survival and proliferation. Recognition-dependent signaling events in response to apoptotic and necrotic targets tend to be directionally opposite and affect such signaling events as β-catenin-dependent transcriptional activity and kinase activity (Akt, GSK3β, ERK1/2, and p38). Together, our data emphasize the complexity and robustness of the cellular response to dead targets. By acting as sentinels of environmental change, dead target cells allow neighboring viable cells to monitor and potentially adapt to local stresses.
Acknowledgment
We thank Uzoagu A. Okonkwo for discussion and enthusiastic assistance in performing our unpublished studies relating to expression of Fas and FasL.
This work was supported, in whole or in part, by National Institutes of Health Grants DK071678 (to V. A. P.), AG024234 (to D. S. U.), and DK059529 (to J. H. S.). This work was also supported by the Genzyme Renal Innovations Program Grant (to J. S. L.), a Veterans Affairs merit award and Dialysis Clinic, Inc. Grants S-1774 and C-3176 (to W. L.), a Pennsylvania State System of Higher Education faculty professional development grant (to A. A.), Canadian Institutes of Health Research Grants MOP-67101 (to J. R.) and MOP-42391 (to J. R.), and Institute of Musculoskeletal Health and Arthritis Priority Announcement MUS-67101 (to J. R.).
V. A. Patel, L. Feng, D. J. Lee, D. Massenburg, G. Pattabiraman, A. Antoni, J. H. Schwartz, W. Lieberthal, J. Rauch, D. S. Ucker, and J. S. Levine, unpublished observations.
- mφ
- macrophage(s)
- CFDA-SE
- 5,6-carboxyfluorescein diacetate succinimidyl ester
- DR
- death receptor
- GSK3β
- glycogen synthase kinase 3β
- myrAkt
- myristoylated Akt
- MTT
- 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium bromide
- OK
- opossum kidney
- PI
- propidium iodide
- PTEC
- proximal tubular epithelial cell
- TCF
- T-cell factor
- TNFR
- tumor necrosis factor receptor
- BU.MPT
- Boston University mouse proximal tubule
- DO
- DO11.10
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- FasL
- ligand for Fas.
REFERENCES
- 1. Kerr J. F., Wyllie A. H., Currie A. R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Majno G., Joris I. (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 146, 3–15 [PMC free article] [PubMed] [Google Scholar]
- 3. Patel V. A., Longacre A., Hsiao K., Fan H., Meng F., Mitchell J. E., Rauch J., Ucker D. S., Levine J. S. (2006) Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: implications for the delayed clearance model of autoimmunity. J. Biol. Chem. 281, 4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cvetanovic M., Ucker D. S. (2004) Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 172, 880–889 [DOI] [PubMed] [Google Scholar]
- 5. Reddy S. M., Hsiao K. H., Abernethy V. E., Fan H., Longacre A., Lieberthal W., Rauch J., Koh J. S., Levine J. S. (2002) Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J. Immunol. 169, 702–713 [DOI] [PubMed] [Google Scholar]
- 6. Cvetanovic M., Mitchell J. E., Patel V., Avner B. S., Su Y., van der Saag P. T., Witte P. L., Fiore S., Levine J. S., Ucker D. S. (2006) Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J. Biol. Chem. 281, 20055–20067 [DOI] [PubMed] [Google Scholar]
- 7. Voll R. E., Herrmann M., Roth E. A., Stach C., Kalden J. R., Girkontaite I. (1997) Immunosuppressive effects of apoptotic cells. Nature 390, 350–351 [DOI] [PubMed] [Google Scholar]
- 8. Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Investig. 101, 890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDonald P. P., Fadok V. A., Bratton D., Henson P. M. (1999) Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-β in macrophages that have ingested apoptotic cells. J. Immunol. 163, 6164–6172 [PubMed] [Google Scholar]
- 10. Huynh M. L., Fadok V. A., Henson P. M. (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J. Clin. Investig. 109, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freire-de-Lima C. G., Xiao Y. Q., Gardai S. J., Bratton D. L., Schiemann W. P., Henson P. M. (2006) Apoptotic cells, through transforming growth factor-β, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 281, 38376–38384 [DOI] [PubMed] [Google Scholar]
- 12. Cocco R. E., Ucker D. S. (2001) Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albert M. L., Pearce S. F., Francisco L. M., Sauter B., Roy P., Silverstein R. L., Bhardwaj N. (1998) Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 188, 1359–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bellone M., Iezzi G., Rovere P., Galati G., Ronchetti A., Protti M. P., Davoust J., Rugarli C., Manfredi A. A. (1997) Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 159, 5391–5399 [PubMed] [Google Scholar]
- 15. Skoberne M., Beignon A. S., Larsson M., Bhardwaj N. (2005) Apoptotic cells at the crossroads of tolerance and immunity. Curr. Top. Microbiol. Immunol. 289, 259–292 [DOI] [PubMed] [Google Scholar]
- 16. Muñoz L. E., Peter C., Herrmann M., Wesselborg S., Lauber K. (2010) Scent of dying cells: the role of attraction signals in the clearance of apoptotic cells and its immunological consequences. Autoimmun. Rev. 9, 425–430 [DOI] [PubMed] [Google Scholar]
- 17. Patel V. A., Lee D. J., Feng L., Antoni A., Lieberthal W., Schwartz J. H., Rauch J., Ucker D. S., Levine J. S. (2010) Recognition of apoptotic cells by epithelial cells: conserved versus tissue-specific signaling responses. J. Biol. Chem. 285, 1829–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel V. A., Longacre-Antoni A., Cvetanovic M., Lee D. J., Feng L., Fan H., Rauch J., Ucker D. S., Levine J. S. (2007) The affirmative response of the innate immune system to apoptotic cells. Autoimmunity 40, 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patel V. A., Lee D. J., Longacre-Antoni A., Feng L., Lieberthal W., Rauch J., Ucker D. S., Levine J. S. (2009) Apoptotic and necrotic cells as sentinels of local tissue stress and inflammation: response pathways initiated in nearby viable cells. Autoimmunity 42, 317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sinha D., Wang Z., Price V. R., Schwartz J. H., Lieberthal W. (2003) Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am. J. Physiol. Renal Physiol. 284, F488–F497 [DOI] [PubMed] [Google Scholar]
- 21. Sinha D., Wang Z., Ruchalski K. L., Levine J. S., Krishnan S., Lieberthal W., Schwartz J. H., Borkan S. C. (2005) Lithium activates the Wnt and phosphatidylinositol 3-kinase Akt signaling pathways to promote cell survival in the absence of soluble survival factors. Am. J. Physiol. Renal Physiol. 288, F703–F713 [DOI] [PubMed] [Google Scholar]
- 22. Jat P. S., Noble M. D., Ataliotis P., Tanaka Y., Yannoutsos N., Larsen L., Kioussis D. (1991) Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. U.S.A. 88, 5096–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kennedy S. G., Wagner A. J., Conzen S. D., Jordán J., Bellacosa A., Tsichlis P. N., Hay N. (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11, 701–713 [DOI] [PubMed] [Google Scholar]
- 24. Eves E. M., Xiong W., Bellacosa A., Kennedy S. G., Tsichlis P. N., Rosner M. R., Hay N. (1998) Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell. Biol. 18, 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 [DOI] [PubMed] [Google Scholar]
- 26. Lieberthal W., Menza S. A., Levine J. S. (1998) Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. Renal Physiol. 274, F315–F327 [DOI] [PubMed] [Google Scholar]
- 27. Lieberthal W., Zhang L., Patel V. A., Levine J. S. (2011) AMPK protects proximal tubular cells from stress-induced apoptosis by an ATP-independent mechanism: potential role of Akt activation. Am. J. Physiol. Renal Physiol. 301, F1177–F1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doble B. W., Woodgett J. R. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thornton T. M., Pedraza-Alva G., Deng B., Wood C. D., Aronshtam A., Clements J. L., Sabio G., Davis R. J., Matthews D. E., Doble B., Rincon M. (2008) Phosphorylation by p38 MAPK as an alternative pathway for GSK3β inactivation. Science 320, 667–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Z., Havasi A., Gall J. M., Mao H., Schwartz J. H., Borkan S. C. (2009) β-Catenin promotes survival of renal epithelial cells by inhibiting Bax. J. Am. Soc. Nephrol. 20, 1919–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Safirstein R. L. (2011) Am I my brother's keeper?: fratricide in the kidney. Kidney Int. 79, 149–150 [DOI] [PubMed] [Google Scholar]
- 32. Tan K. H., Hunziker W. (2003) Compartmentalization of Fas and Fas ligand may prevent auto- or paracrine apoptosis in epithelial cells. Exp. Cell Res. 284, 283–290 [DOI] [PubMed] [Google Scholar]
- 33. Liu M., Chien C. C., Burne-Taney M., Molls R. R., Racusen L. C., Colvin R. B., Rabb H. (2006) A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 17, 765–774 [DOI] [PubMed] [Google Scholar]
- 34. Linkermann A., Himmerkus N., Rölver L., Keyser K. A., Steen P., Bräsen J. H., Bleich M., Kunzendorf U., Krautwald S. (2011) Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int. 79, 169–178 [DOI] [PubMed] [Google Scholar]