

Background: Visfatin regulates prostaglandin E2 synthesis in chondrocytes, through unknown pathways.
Results: We characterized insulin and IGF receptor signaling and Nampt activity involved in this response.
Conclusion: Proinflammatory actions of visfatin in chondrocytes implicate IR pathways, possibly through control of Nampt activity.
Significance: IR, IGF-1R, and other tyrosine kinase receptor pathways need to be considered to understand visfatin signaling.
Keywords: Insulin, Insulin-like Growth Factor (IGF), Prostaglandins, Receptor Tyrosine Kinase, Signal Transduction, Cartilage, Nampt, Osteoarthritis, Visfatin
Abstract
Visfatin (also termed pre-B-cell colony-enhancing factor (PBEF) or nicotinamide phosphoribosyltransferase (Nampt)) is a pleiotropic mediator acting on many inflammatory processes including osteoarthritis. Visfatin exhibits both an intracellular enzymatic activity (nicotinamide phosphoribosyltransferase, Nampt) leading to NAD synthesis and a cytokine function via the binding to its hypothetical receptor. We recently reported the role of visfatin in prostaglandin E2 (PGE2) synthesis in chondrocytes. Here, our aim was to characterize the signaling pathways involved in this response in exploring both the insulin receptor (IR) signaling pathway and Nampt activity. IR was expressed in human and murine chondrocytes, and visfatin triggered Akt phosphorylation in murine chondrocytes. Blocking IR expression with siRNA or activity using the hydroxy-2-naphthalenyl methyl phosphonic acid tris acetoxymethyl ester (HNMPA-(AM)3) inhibitor diminished visfatin-induced PGE2 release in chondrocytes. Moreover, visfatin-induced IGF-1R−/− chondrocytes released higher concentration of PGE2 than IGF-1R+/+ cells, a finding confirmed with an antibody that blocked IGF-1R. Using RT-PCR, we found that visfatin did not regulate IR expression and that an increased insulin release was also unlikely to be involved because insulin was unable to increase PGE2 release. Inhibition of Nampt activity using the APO866 inhibitor gradually decreased PGE2 release, whereas the addition of exogenous nicotinamide increased it. We conclude that the proinflammatory actions of visfatin in chondrocytes involve regulation of IR signaling pathways, possibly through the control of Nampt enzymatic activity.
Introduction
Visfatin, first described as an adipokine secreted by visceral fat that mimics the effects of insulin (1), was originally identified as a secreted growth factor that enhances B-cell precursor maturation, called pre-B-cell colony-enhancing factor, or PBEF (2). It was then demonstrated that this protein also has an enzymatic nicotinamide phosphoribosyltransferase (Nampt)2 activity, critical for the synthesis of NAD, an essential cofactor for cell metabolism (3). Thus, the terms visfatin, PBEF, and Nampt refer to the same protein.
Visfatin has features of classical cytokines (4) and is ubiquitously produced in several tissues (2) and in different cells such as synovial fibroblasts (5), articular chondrocytes (6), or monocytes (7), strengthening the hypothesis that visfatin plays key roles in various pathophysiological processes. Visfatin is overexpressed in several inflammatory diseases, such as atherosclerosis, rheumatoid arthritis, or osteoarthritis (OA), and could be involved in obesity-associated low grade inflammation state and metabolic syndrome (5, 7, 8). It is also up-regulated in acute lung injury (9), sepsis (10), and cancer (11). Visfatin expression is regulated by proinflammatory cytokines such as tumor necrosis factor (TNF)α or interleukin (IL)-1β, by lipopolysaccharide (LPS), and by dexamethasone (10, 12, 13). Finally, visfatin induces chemotaxis, the production of IL-1β, TNFα, IL-6, and co-stimulatory molecules by CD14+ monocytes (8). Collectively, visfatin appears to be a key cytokine involved in a large panel of chronic inflammatory diseases and immune responses. A better understanding of visfatin signaling and the pathways involved would be a major step toward the development of strategies that allow targeting this protein for anti-inflammatory purposes (11, 14).
Visfatin relevance in immune, metabolic, and stress responses depends on both extracellular (cytokine-like) and intracellular (enzymatic) functions. Visfatin was initially reported to mimic the effects of insulin by binding to the insulin receptor (IR) and by stimulating the phosphorylation of insulin-receptor substrate-1 (IRS-1) and IRS-2 and the downstream tyrosine protein kinase B (Akt) and mitogen-activated protein kinase (MAPK) (1). However, the original work describing visfatin binding to IR has been retracted (15). Therefore, IR is not considered as a visfatin receptor anymore, although there is evidence that regulation of insulin signaling pathways at the level of IRSs, Akt, and MAPK is implicated in producing the pathophysiological effects of visfatin (16–19).
A main activity of visfatin is intracellular, and its functions concentrate on NAD synthesis. NAD, an essential cofactor in a number of fundamental cellular processes, is mainly produced by salvage pathways; nicotinamide, resulting from intracellular degradation of NAD, is converted to nicotinamide mononucleotide (NMN) by Nampt and then converted back into NAD by nicotinamide mononucleotide adenylyltransferase (Nmnat) (4). Recently, Nampt activity has been linked to inflammation because its in vivo pharmacological inhibition, by using the selective inhibitor APO866, diminished the severity of arthritis in a collagen-induced arthritis model and reduced cytokine secretion in affected joints (20).
We previously reported on the important role of visfatin in matrix degradation and synthesis of the proinflammatory mediator prostaglandin E2 (PGE2) in chondrocytes (6). We hypothesized that visfatin exerts an autocrine/paracrine action on chondrocytes, supposedly through binding to its still unknown receptor, which is linked to IR signaling pathways. The original aim of this study was to confirm the role of IR pathways in the effects of visfatin on chondrocytes and to analyze the implication of other related tyrosine kinase receptors such as the insulin-like growth factor (IGF)-1 receptor. We excluded the possibility that the effects of visfatin were due to a regulation of insulin activity. We also demonstrated that Nampt activity is involved, at least in part, in visfatin-induced PGE2 release in chondrocytes.
EXPERIMENTAL PROCEDURES
Materials
All reagents were purchased from Sigma-Aldrich (Lyon, France), unless stated otherwise. Fetal calf serum (FCS) was obtained from Invitrogen (Cergy-Pontoise, France). Collagenase A, trypsin, hyaluronidase, collagenase D, and Complete protease inhibitor mixture were from Roche Diagnostics (Meylan, France). Hydroxy-2-naphthalenyl methyl phosphonic acid tris acetoxymethyl ester (HNMPA-(AM)3) was from Biomol (Tebu-Bio, Le Perray-en-Yvelines, France). Anti-IR polyclonal antibody (sc-711), anti-IR monoclonal antibody (sc-57342), and anti-type II collagen antibody were from Santa Cruz Biotechnology (from Tebu-Bio, France). The anti-human IGF-1R-blocking antibody and its isotype control were from R&D systems (Lille, France). The Western blot ECL system was from Amersham Biosciences (Orsay, France). The immunoblot polyvinylidene difluoride (PVDF) membranes for Western blot and Kaleidoscope prestained standards were from Bio-Rad (Marnes-la-Coquette, France). Recombinant mouse visfatin was from Alexis (Coger, Paris, France). APO866, a generous gift from Astellas Pharma (Munich, Germany), was provided by Alexander So.
Primary Cultures of Human Articular Chondrocytes
Human cartilages were obtained from patients undergoing joint replacement surgery for OA (21) or hip fracture at Saint-Antoine Hospital (AP-HP, Paris, France). Informed consent was obtained from each patient before surgery. The diagnosis of OA was based on clinical and radiographic evaluation according to the American College of Rheumatology criteria (22). The institutional ethics committee approved our study protocol.
Human chondrocytes were isolated by enzymatic digestion of articular cartilage from human donors according to published procedures (6). After 4 days of culture, cells were starved for 24 h in serum-free Dulbecco's modified Eagle's medium (DMEM) with 4.5 mg/liter glucose, supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 4 mm l-glutamine, and 0.3% BSA.
Protein Extraction and Western Blot
To analyze activity of IR in murine articular chondrocytes stimulated by insulin, anti-human phospho-IRβ polyclonal (1/250), anti-mouse Akt, and phospho-Akt polyclonal antibodies (1/1000, Cell Signaling, Beverly, MA) were used. Signals were detected by enhanced chemiluminescence using Fujifilm LAS-300 (Fujifilm Medical Systems, Stamford, CT). For densitometric analysis, we used the Image Gauge software (Science Lab 2004; Fujifilm) and normalized to the density of β-actin or Akt.
Primary Cultures of Immature Murine Articular Chondrocytes (iMACs)
All experiments were performed according to the protocols approved by the French and European ethics committees. Immature mouse articular chondrocytes were isolated by enzymatic digestion of articular cartilage from 6-day-old newborns from one Swiss mouse litter according to the procedure previously described (23, 24). After 6 or 7 days in culture, cells were starved for 24 h in serum-free DMEM (1 mg/liter glucose) supplemented with penicillin, streptomycin, l-glutamine, and 1% BSA. For the experiments performed on chondrocytes from IGF-1R knock-out mice because IGF-1R−/− mice are not viable after birth, chondrocytes were isolated from articular cartilage of embryonic day 19.5 embryos, in accord with the iMACs culture protocol.
RNA Extraction, Reverse Transcription, and Real-time Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from chondrocytes using the RNeasy kit (Qiagen, Hilden, Germany), and concentrations were determined spectrophotometrically. Reverse transcription was performed on 1 μg of total RNA, using the Omniscript RT kit (Qiagen). Messenger RNA (mRNA) for IR was quantified using the LightCycler LC480 (Roche Diagnostics) as described (25). Specific primers for complementary DNA (cDNA) were chosen with the LightCycler Probe Design 2 program. Primers for IR were forward, 5′-CAA-TGG-GAC-CAC-TGT-ATG-CAT-CT-3′, and reverse, 5′-GTC-CGG-CAC-GTA-CAC-AGA-AGA-3′ (58 °C) for mouse, and forward, 5′-CAG-CCC-TGT-TCC-AGG-AG-3′, and reverse, 5′-TAT-GAA-AGC-AGC-AGC-TAT-TGG-TC-3′ (60 °C) for human. Insulin receptor primers were compared with the entire genome to ensure specificity.
Protein Extraction and Western Blot
Cell lysates and immunoblots were prepared as described (26). Protein extracts from 3T3-L1 adipocytes were used as positive control of IR expression.
Immunocytofluorescence
Murine articular chondrocytes from newborns were seeded onto glass coverslips in 24-well culture plates at a density of 2 × 104 cells/coverslip. At 70–80% of confluency, cells were rinsed twice with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, and incubated with the primary antibody overnight at 4 °C. After two washes in PBS, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Invitrogen) diluted 1/1000 in PBS for 2 h at room temperature and with 1 μg/ml Hoechst 33258 for 5 min. After three washes in PBS, coverslips were mounted with Mowiol 4-88 and observed with a Nikon Diaphot 300 microscope equipped with a mercury lamp.
PGE2 Assays
PGE2 in culture medium was measured with an enzymatic immunoassay (EIA) kit from Cayman Chemical (detection limit 9 pg/ml; purchased from SPI-BIO, Montigny-le-Bretonneux, France). The PGE2 concentrations were analyzed at serial dilutions in duplicate.
Transfection of Small Interfering RNA (siRNA)
Two FlexiTube siRNAs directed against mouse IR (siRNA1: Mm_Insr_1, catalog number SI01076831 and siRNA2: Mm_Insr_2, catalog number SI01076838) were purchased from Qiagen. Murine articular chondrocytes were transfected, using the electroporation device from Amaxa set to the human chondrocyte program. Briefly, 3 × 106 cells were resuspended in 100 μl of Amaxa electroporation transfection solution, and 10 μl of 10 μm IR siRNA1 or 10 μm IR siRNA2, 5 μl of 20 μm AllStars negative control (Qiagen), or 20 μm MAPK1 positive control (Qiagen) were added. AllStars negative control siRNA has no homology to mammalian genes and has been validated using Affymetrix GeneChip arrays and a variety of cell-based assays. MAPK1 positive control siRNA is a validated siRNA directed against mouse MAPK1 mRNA (also called Erk2 and MAPK2), which drastically reduces MAPK1 expression in mouse cell lines. After electroporation, transfected cells were plated in two wells (500 μl/well) of a 12-well plate, each containing 500 μl of cell culture medium. 24 h after transfection, the cells were starved in serum-free medium overnight. The following day, cells were treated as indicated in the figure legends. Medium, RNA, or proteins were collected as described below. Cell viability was evaluated by the TACS 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (R&D Systems).
Statistical Analysis
All data are reported as means ± S.D., unless stated otherwise. Statistical analyses were performed by Welch's corrected t test to compare the mean values for two groups and with one-way analysis of variance with Bonferroni's post hoc correction to compare mean values among >2 groups, using GraphPad Prism version (GraphPad Software, San Diego, CA). p values of 0.05 or less were considered significant.
RESULTS
Expression of IR in Human Chondrocytes
We first studied IR mRNA and protein expression in human and murine chondrocytes. Primary cultures of human chondrocytes from knees of seven OA subjects and three healthy patients and quantitative real-time RT-PCR were used to assess the expression of IR in human cartilage. To detect IR protein expression, immunoblotting was performed using sc-711 antibody (27) and monoclonal sc-57342 antibody, highly specific for IR protein and devoid of cross-reactivity with IGF-1R, a structural homolog of IR abundantly expressed in cartilage. IR mRNA and protein were constitutively produced in chondrocytes from OA patients and healthy subjects. We observed strong differences in individual visfatin expression among human samples (Fig. 1, A and B).
FIGURE 1.

IR expression in articular chondrocytes. A, primary cultures of chondrocytes from healthy (H1 to H3) or osteoarthritic (OA1 to OA7) knees were starved. IR mRNA expression was assessed by real-time RT-PCR. B, IR protein expression was analyzed using immunoblotting with two specific antibodies. Sc-711 (1/200) is a widely used IR antibody; sc-57342 (1/200) is a monoclonal antibody not cross-reacting with IGF-1R. Densitometric signals of IR were normalized to β-actin and expressed as arbitrary units. MW, molecular weight. C, primary cultures of iMACs were starved, and IR protein expression was analyzed using immunocytofluorescence using sc-711. Type II collagen expression was used as positive control of cell differentiation. Specificity of staining was confirmed by omission of primary antibody. Micrographs are representative of three independent experiments performed in triplicate (size bars = 20 μm).
Expression of Functional IR in iMACs
Because only limited amounts of human cartilage were available, we used primary cultures of iMACs for more extensive studies (23). IR mRNA was constitutively expressed by iMACs (data not shown), and IR protein expression was strongly positive as detected using immunocytofluorescence (Fig. 1C). Visfatin was previously shown to stimulate IR phosphorylation and downstream signaling kinases Akt and MAPK (4). Therefore, we checked the functionality of IR in chondrocytes in response to exogenous insulin. Insulin-induced phosphorylation of IR within 5 min (Fig. 2A) and phosphorylation of Akt measured after 15 min (Fig. 2B) were maximal at 100 nm insulin. Moreover, in a kinetic study, phosphorylation of IR was maximal at 5 min (Fig. 2A), whereas Akt phosphorylation peaked at 15 min (Fig. 2B). These data indicated that insulin can activate IR pathways in chondrocytes.
FIGURE 2.

Phosphorylation of IR and Akt in iMACs treated with insulin. A, murine articular chondrocytes were starved and stimulated with different doses of insulin for 5 min or with 100 nm insulin for different time periods. Phosphorylation of IR (P-IR) was analyzed. Insulin-treated adipocytes were used as positive control (cont). B, Akt phosphorylation (P-Akt) was analyzed in murine articular chondrocytes treated with different doses of insulin for 15 min or with 100 nm insulin for different time periods. Blots are representative of three independent experiments.
IR Signaling in Visfatin-induced PGE2 Synthesis in Articular Chondrocytes
To confirm the role of IR signaling pathways in visfatin-induced PGE2 synthesis, we first checked Akt phosphorylation in response to visfatin in murine articular chondrocytes. The cells were stimulated with 5 μg/ml visfatin for 5–60 min. We found that phosphorylation of Akt peaked after 5 min (p ≤ 0.01; Fig. 3). Next, we used an siRNA strategy to interfere with IR expression. We showed that transfection with either siRNA1 or siRNA2 significantly suppressed IR expression at mRNA and protein levels. We achieved a decrease in IR mRNA of 82 and 79%, respectively, in comparison with controls (p ≤ 0.05; Fig. 4A). We checked PGE2 release in IR siRNA-transfected chondrocytes treated for 24 h with visfatin and found that IR siRNA significantly reduced visfatin-induced PGE2 release in iMACs in comparison with chondrocytes transfected with a negative control. Reduction was 48% for siRNA1 and 74% for siRNA2 (p ≤ 0.05; Fig. 4B).
FIGURE 3.
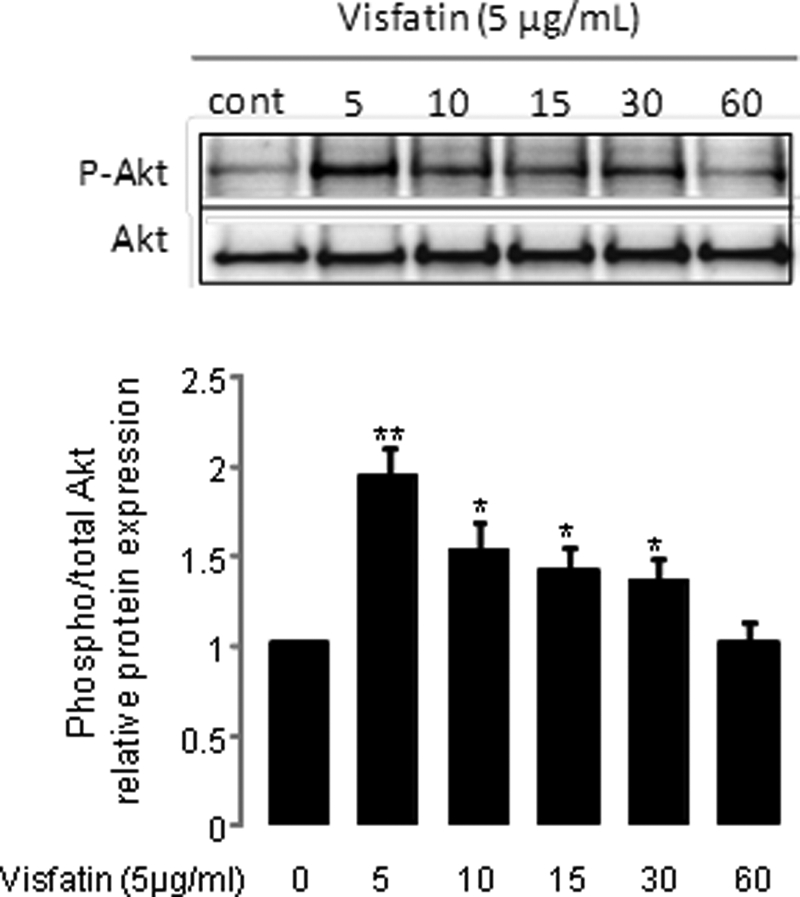
Phosphorylation of Akt in iMACs treated with visfatin. Murine articular chondrocytes were starved and stimulated with 5 μg/ml visfatin for different time periods. The blot is representative of three independent experiments. Phosphorylation of Akt was analyzed. Densitometric signals of phospho-Akt (P-Akt) were normalized to Akt and expressed as arbitrary units. **, p ≤ 0.01 and *, p ≤ 0.05 versus control (cont).
FIGURE 4.

Involvement of IR signaling pathway in visfatin-induced PGE2 release in iMACs. A, iMACs were transfected with siRNA targeting the IR gene or with negative control siRNA. IR mRNA and protein expression was analyzed using real-time RT-PCR or immunoblot, respectively, to confirm the knockdown of IR expression. Blots are representative of three independent experiments. HPRT, hypoxanthine-guanine phosphoribosyltransferase. *, p ≤ 0.05 versus control. B, cells were starved and incubated with 5 μg/ml visfatin for 24 h. PGE2 release into the media was measured by EIA. Results are expressed as the -fold change relative to the negative control-transfected murine articular chondrocytes (negative control-transfected cells released 3576 ± 265 pg/ml PGE2). Means and S.D. represent three independent experiments with two wells/condition, analyzed in duplicate. **, p ≤ 0.01 versus negative control; # p ≤ 0.05 versus 5 μg/ml treated negative control. C, iMACs were pretreated with 100 nm HNMPA-(AM)3, a specific IR inhibitor, for 24 h and were starved overnight. Cells were then incubated with 5 μg/ml visfatin in the presence of 100 nm HNMPA-(AM)3 for 24 h. Control cells released 99 ± 60 pg/ml PGE2. ***, p ≤ 0.001 versus control, ## p ≤ 0.01 versus 5 μg/ml visfatin.
We confirmed these results using the specific inhibitor HNMPA-(AM)3, widely used to block IR activity and responses of the insulin signaling pathway to visfatin (7, 28). We pretreated iMACs with 100 nm HNMPA-(AM)3 for 24 h and then maintained HNMPA-(AM)3 in the medium in the presence of either 5 μg/ml visfatin or 100 nm insulin. Interestingly, 100 nm insulin did not trigger PGE2 synthesis in iMACs. Moreover, HNMPA-(AM)3, together with 5 μg/ml visfatin for 24 h inhibited visfatin-induced PGE2 release by 60% (p ≤ 0.01; Fig. 4C), These results were concordant with the siRNA results and suggested that IR signaling in response to visfatin plays a role in articular chondrocytes.
Invalidation of Role of IGF-1R in Visfatin-induced PGE2 Synthesis
It is well described that manifold cross-talk exists among tyrosine kinase receptors, especially between IR and IGF-1R. Interestingly, IGF-1 ligand and receptor are both known for their role in cartilage physiology, in particular for stabilizing the cartilage matrix (29). Even if the affinity of visfatin to the IGF-1R is relatively low with respect to triggering an intracellular signal (1), we decided to assess the implication of IGF-1R in visfatin-induced PGE2 release in chondrocytes. First, we used primary cultures of articular chondrocytes from knock-out and wild-type IGF-1R cells. As IGF-1R−/− mice are lethal at birth, we used primary cultures of chondrocytes from embryo articular cartilage. Therefore, we previously checked whether embryo chondrocytes from IGF-1R−/− or IGF-1R+/+ exhibited the typical articular chondrocyte phenotype described in murine articular chondrocytes from newborn mice (iMACs), in terms of morphology and functionality. Knock-out and wild-type IGF-1R embryonic chondrocytes exhibited the same profile concerning type II collagen expression and visfatin-induced PGE2 synthesis as iMACs (Fig. 5A). Visfatin at 10 μg/ml for 6 h, for instance, triggered a 4-fold increase in PGE2 in embryo wild-type IGF-1R+/+ chondrocytes and a 3-fold increase in murine articular chondrocytes from newborn mice (Fig. 5A, not significant). We next assessed IR protein expression in both wild-type and knock-out IGF-1R chondrocytes using immunoblotting and found similar receptor expression in both genotypes (Fig. 5B). Then, we checked the cellular response to various doses of visfatin in IGF-1R−/− or IGF-1R+/+ chondrocytes. As expected, 10 μg/ml visfatin significantly increased PGE2 in wild-type chondrocytes (4-fold-increase; p ≤ 0.001;). Surprisingly, 10 μg/ml visfatin-induced PGE2 release was significantly increased (72%; p ≤ 0.001) in knock-out chondrocytes in comparison with wild type (Fig. 5B).
FIGURE 5.

IGF-1 receptor was not directly implicated in visfatin-induced PGE2 release in iMACs. A, articular chondrocytes from wild-type embryo IGF-1R mice and from newborn Swiss mice were starved and then stimulated with 5 or 10 μg/ml visfatin for 6 h. The control group received no visfatin. PGE2 release into the media was measured by EIA. Murine control articular chondrocytes release 46.9 ± 8.4 pg/ml. Type II collagen protein expression was analyzed using immunoblot. ***, p ≤ 0.001 versus new-born control, ## p ≤ 0.01 versus embryo control cells. NS, not significant. B, IGF-1R−/− or IGF-1R+/+ articular chondrocytes were starved and incubated with up to 10 μg/ml visfatin for 6 h. PGE2 release in wild-type cells was 21.3 ± 3.6 pg/ml. Immunoblots were performed from knock-out or wild-type IGF-1R cells to confirm IR expression using sc-711 antibody. ***, p ≤ 0.001 versus IGF-1R+/+ control, ### p ≤ 0.001 versus 10 μg/ml visfatin induced IGF-1R+/+ cells. C, murine articular chondrocytes incubated with different doses of specific blocking IGF-1R antibody or isotype antibody were treated with 5 μg/ml visfatin for 24 h. Results are expressed as the -fold change relative to the control (untreated chondrocytes released 110 ± 41 pg/ml PGE2). Means and S.D. represent three independent experiments with two wells/condition and one experiment with three wells/condition for isotype antibody, analyzed in duplicate. *, p ≤ 0.05 versus control.
Using an alternative strategy based on an IGF-1R-blocking antibody, we replicated these findings in primary cultures of iMACs exposed to visfatin. Increasing the concentration of IGF-1R-blocking antibody increased visfatin-stimulated PGE2 release. PGE2 reached a 1.9-fold maximum increase at 5 μg/ml antibody (p ≤ 0.05; Fig. 5C), whereas an isotype control antibody did not trigger significant PGE2 release (5 μg/ml nonblocking antibody triggered a 1.2-fold increased PGE2 release; not significant). Together, this demonstrated that IGF-1R is not directly involved in visfatin-induced PGE2 release in murine articular chondrocytes.
Visfatin-induced PGE2 Release in Articular Chondrocytes Is Not due to Visfatin-induced Increase in Insulin Production or Overexpression of IR
Increased insulin secretion in mouse pancreatic beta-cells treated with visfatin has been described (18). Therefore, it was possible that visfatin induces insulin secretion or IR expression in murine articular chondrocytes, which in turn may sensitize these cells to visfatin and lead to PGE2 release. To test this hypothesis, we checked whether the addition of exogenous insulin could trigger PGE2 release. We found that iMACs stimulated with insulin for 24 h did not show enhanced PGE2 release (Fig. 6). We then analyzed whether 5 μg/ml visfatin was able to induce IR mRNA expression in articular chondrocytes, but found no significant effect (Fig. 6). Hence, as visfatin did not regulate IR expression on chondrocytes and insulin was unable to increase PGE2 release, we could largely exclude the possibility that visfatin induction of PGE2 release was due to a regulation of insulin activity on its cognate receptor.
FIGURE 6.
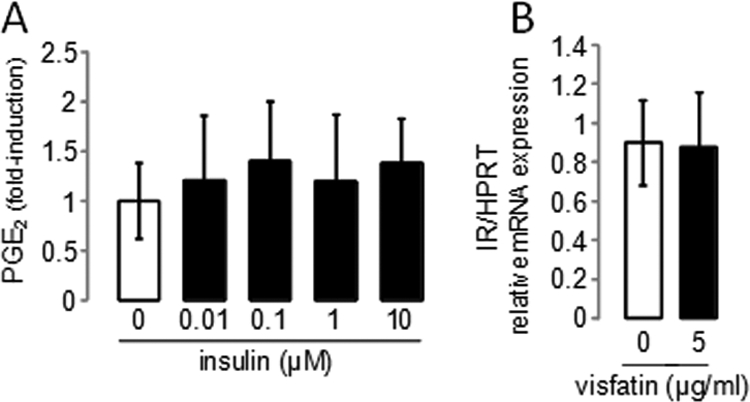
Visfatin-induced PGE2 release in articular chondrocytes is not due to visfatin-induced release of insulin or to overexpression of IR. A, iMACs were starved and then stimulated with different doses of exogenous insulin (up to 10 μm) for 24 h. The amount of the PGE2 release into the media was measured by EIA, and control cells released 20.7 ± 8.0 pg/ml PGE2. Values are the means and S.D. of three independent experiments with two wells/condition, analyzed in duplicate. B, iMACs were starved and stimulated with 5 μg/ml visfatin for 6 h. The control group received no visfatin. IR mRNA expression was analyzed using real-time RT-PCR. Results are expressed as the -fold change relative to the control. Means and S.D. represent three independent experiments analyzed in duplicate. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
Role of Nampt Activity of Visfatin in Visfatin-induced PGE2 Synthesis
As Nampt activity of visfatin has been directly involved in inflammatory processes, we tested whether synthesis of NAD could play a role, at least in part, in this response. For this purpose, we used APO866 (also known as FK866 and WK175), a specific competitive low molecular weight inhibitor of NAD synthesis (30). We pretreated iMACs for 4 h with up to 1 μm APO866 and then stimulated the cells with 5 μg/ml visfatin for 24 h in the presence of APO866. This induced a strong decrease in visfatin-induced PGE2 release of up to 43% (p ≤ 0.05; Fig. 7A). This effect was not due to cell death because we checked the viability of chondrocytes using a TACS 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Visfatin together with 1 μm APO866 induced apoptosis in 4.5% of chondrocytes, whereas visfatin induced apoptosis in 0.5% of chondrocytes (not significant; data not shown).
FIGURE 7.

Role of Nampt activity in visfatin-induced PGE2 release in iMACs. A, iMACS were pretreated for 4 h with different doses of APO866, a specific inhibitor of the Nampt activity, and stimulated with 5 μg/ml visfatin or not for 24 h. The amount of PGE2 release into the media was measured by EIA. Control cells released 96 ± 31 pg/ml PGE2. *, p ≤ 0.001 versus control, **, p ≤ 0.05 versus chondrocytes treated with 5 μg/ml visfatin. B, iMACs were stimulated with different doses of exogenous NMN for 24 h. Control cells released 19.4 ± 5.7 pg/ml PGE2. Results are expressed as the -fold change as compared with controls (untreated chondrocytes). Means and S.D. represent three independent experiments with two wells/condition, analyzed in duplicate. *, p ≤ 0.05 versus control
Finally, we tested whether the addition of exogenous NMN, mimicking the Nampt activity of visfatin, could trigger PGE2 release in iMACs. We found that iMACs stimulated with NMN for 24 h exhibited a dose-dependent release of PGE2. Induction of PGE2 was 1.6-fold at 10 μm NMN (p ≤ 0.05) and peaked with 1.8-fold at 100 μm (Fig. 7B). Collectively, this is a strong indication that NAD synthesis is implicated in visfatin-induced PGE2 release because inhibition of Nampt activity gradually decreased PGE2 release, whereas the addition of exogenous NMN increased it.
DISCUSSION
Visfatin exerts significant inflammatory effects via its Nampt activity and binding to a still hypothetical receptor. Data from the literature indicated that components in the IR signaling pathway, such as IRSs and downstream AKT and MAPK, are implicated in pathophysiological effects of visfatin. Today, understanding the visfatin signaling pathways is of high interest for the development of new pharmacological therapies. This is particularly true in OA, which is the leading cause of chronic joint disease with no effective drugs available due to a lack of knowledge in the pathophysiology of the disease. However, it has now been demonstrated that visfatin has proinflammatory and prodegradative properties suggesting that visfatin could be a target for treating OA in the future. In the present study, we tried to elucidate visfatin pathways in articular chondrocytes. We first confirmed the expression and activity of IR in both human and murine articular chondrocytes. Then, we assessed the role of IR signaling pathway in inflammatory responses using an siRNA strategy and the specific inhibitor HNMPA-(AM)3. We then excluded the implication of its close homolog IGF-1R in PGE2 production. Moreover, we invalidated a potential regulation of insulin or IR expression in visfatin-stimulated chondrocytes. Finally, we were able to demonstrate the functionality of Nampt enzymatic activity for visfatin-induced PGE2 release.
Today, the exact mechanism of extracellular visfatin on the activation of immune cells or in insulin regulation remains unclear. Various authors implicated intracellular insulin receptor signaling pathways in visfatin action. For example, Xie et al. (19) found that visfatin induced tyrosine phosphorylation of IRS-1 and IRS-2. Moreover, Adya et al. (16) showed in human umbilical vein endothelial cells that visfatin dose-dependently activated PI3K/Akt and ERK1/2 pathways that are in turn implicated in matrix metalloproteinase release. Finally, Brown et al. (18) found that visfatin triggered phosphorylation of Akt in mouse pancreatic beta-cells. Here, we confirmed the role of the IR signaling pathways in visfatin-induced PGE2 release in chondrocytes, and we hypothesized that other tyrosine kinase receptor pathways may be involved as well.
Insulin and IGF-1 are related signaling molecules that evolved from a common ancestral pathway. During evolution, the primitive receptor pathway diverged into two distinct hormonal systems that in mammals serve different but overlapping developmental and metabolic functions (31, 32). These receptors belong to the family of ligand-activated receptor kinases, and IGF-1 receptor is abundantly expressed in chondrocytes (33). Here, we described IR expression in human articular chondrocytes, confirming recent studies (34, 35). Therefore, we tested the role of IGF-1R in visfatin-induced PGE2 release in articular chondrocytes. We found that IGF-1R-deficient chondrocytes or articular chondrocytes treated with IGF-1R-blocking antibody exhibit higher visfatin-induced PGE2 release. Thus, IGF-1R does not seem to be directly involved in the response of chondrocytes to visfatin, but blocking its expression or its activity can specifically increase the level of IRSs available to other tyrosine kinases (36). Therefore, interaction of visfatin with kinase receptors other than IR or IGF-1R should be tested. In this context, the existing relationships between growth hormone receptor (GHR), IR, and IGF-1R are of particular interest. For instance, an increased IRSs phosphorylation and subsequent AKT and MAPK phosphorylation in response to growth hormone (GH) have been described in muscle (36). Therefore, binding of visfatin to GHR should be tested in articular chondrocytes. Interestingly, GH/IGF-1 deficiency causes an increased severity of articular cartilage lesions of OA (37). Thus, if visfatin binds to GHR, it may decrease GH binding to GHR, possibly leading to an increased OA severity in accord with the capacity of visfatin to increase PGE2 release in articular chondrocytes. To address this question, it is important to distinguish between articular chondrocytes of embryonic or postnatal origin that do express significant amounts of GHR (38) and articular chondrocytes from adults, where evidence for GHR expression is still limited (39).
Another way for visfatin to regulate PGE2 release in murine articular chondrocytes may involve an increased synthesis of one of its ligands, insulin or IGF-1. For example, visfatin regulates insulin secretion in mouse pancreatic beta-cells, and this effect is blocked using the specific inhibitor FK866 (18), a homolog of the APO866 inhibitor. Thus, enzymatic activity of visfatin may be implicated in insulin secretion, leading to insulin receptor activation. In our experiments, however, we failed to establish a role for insulin in increasing PGE2 release in chondrocytes. Moreover, we tested whether visfatin may induce IR expression in articular chondrocytes, and we did not find clear evidence for regulation. Another possibility is that visfatin induced IGF-1 expression, which, in very high concentrations, can bind to IR, potentially leading to increased visfatin-induced PGE2 release in chondrocytes. However, we previously published that IGF-1 is an anabolic factor that counteracts IL-1β-induced PGE2 release in rabbit articular chondrocytes (40). Moreover, we invalidated a direct role of IGF-1 by using chondrocytes from IGF-1R knock-out mice or cells treated with an IGF-1R-blocking antibody. Finally, synthesis of a mediator implicated in visfatin-induced PGE2 release in articular chondrocytes is unlikely to be implicated because phosphorylation of IRS-1 and IRS-2 in cells treated with visfatin takes more than 10 min to occur (19).
IGF-1R has been described in and outside of membrane lipid rafts and is thought to be linked to PI3K/Akt or to Erk1/2 and p38 MAPK signaling pathways in colon carcinoma cells (41). Importantly, in chondrocytes, the balance of PI3K/Akt and Erk1/2 activation determines the anabolic effect of IGF-1 as recruitment of PI3K/AKT leads to matrix production, whereas Erk signaling inhibits it (42). Recently, visfatin was found to increase lipid raft clustering in glomerular epithelial cells (43) and may therefore lead to increased IGF-1R within lipid rafts and subsequent anti-inflammatory effects of IGF-1. This may explain the increased release of PGE2 in visfatin-induced IGF-1R knock-out chondrocytes. However, visfatin also induces lipid raft redox signaling via lipid raft clustering and thereby triggers local oxidative stress (43), which may increase IGF-1 pathways via Erk1/2 (42). Interestingly, visfatin was recently described to down-regulate IGF-1-induced Akt phosphorylation independently of binding to IGF-1R by activation of Erk1/2 signaling in articular chondrocytes (44).
In the literature, production of IL-1β in response to visfatin was observed in monocytes (8). IL-1β is described as a potent inducer of PGE2 release in chondrocytes (26, 45). Therefore, it is not excluded that visfatin-induced PGE2 release depends at least in part on IL-1 production in chondrocytes. This is possible because a positive feedback loop exists through which IL-1 increases visfatin that in turn enhances PGE2 production (6, 46).
Visfatin exhibits intra- and extracellular NAD biosynthetic enzymatic activity (47). Proinflammatory effects of visfatin may be due to this Nampt activity because iNOS induction in vascular smooth muscle cells can be prevented by APO866 (48). Moreover, Brown et al. (18) showed that both visfatin and NAD activated IR, which can be prevented by FK866 inhibitor. In our study, we validated the role of Nampt activity in visfatin-induced PGE2 release. Nampt activity leads to NAD release, a cofactor for NAD-consuming enzymes such as sirtuins. Sirtuins are NAD-dependent histone deacetylases that regulate gene expression, differentiation, and development. Interestingly, interaction of Sirt1 with IRS-1 and IRS-2 has been described, and a regulatory effect of Sirt1 on insulin-induced tyrosine phosphorylation of IRS-2 through deacetylation of IRS-2 was shown recently (49). This interaction may alter the anabolic effect of insulin or IGF-1 and increase PGE2 release. Clearly, the role of Sirt1 in visfatin-induced PGE2 release needs to be clarified. Chabane et al. (50) reported that histone deacetylase inhibitors suppressed IL-1-induced COX-2 expression and subsequent PGE2 release in chondrocytes. In contrast, Sirt1 can regulate the activity of transcription factors including peroxisome proliferator-activated receptor γ (PPARγ), which displays anti-inflammatory and chondroprotective properties, notably by inhibiting PGE2 synthesis (45). Moreover, Sirt1 contributes to the inhibition of AP-1 transcriptional activity, COX-2 expression, and subsequent PGE2 release in macrophages (51).
In conclusion, because the regulation of visfatin is complex and incompletely understood, development of therapeutic strategies focused on this potent inflammatory mediator is limited. In this work, we confirmed that visfatin exerts proinflammatory action by regulating IR pathway activity and by inducing Nampt enzymatic activity. The receptor for visfatin, if there is one, remains unidentified.
Acknowledgments
We thank Alexander So for APO866 inhibitor and Alain Sautet for human cartilage samples.
This work was supported by the Fondation Arthritis and the French Society of Rheumatology.
- Nampt
- nicotinamide phosphoribosyltransferase
- NMN
- nicotinamide mononucleotide
- GH
- growth hormone
- GHR
- growth hormone receptor
- HNMPA-(AM)3
- hydroxyl-2-naphtalenyl methyl phosphonic acid tris acetoxymethyl ester
- iMAC
- immature murine articular chondrocyte
- IR
- insulin receptor
- IRS
- insulin-receptor substrate
- OA
- osteoarthritis
- Sirt
- sirtuin
- PGE2
- prostaglandin E2
- IGF-1R
- insulin-like growth factor-1 receptor
- EIA
- enzymatic immunoassay.
REFERENCES
- 1. Fukuhara A., Matsuda M., Nishizawa M., Segawa K., Tanaka M., Kishimoto K., Matsuki Y., Murakami M., Ichisaka T., Murakami H., Watanabe E., Takagi T., Akiyoshi M., Ohtsubo T., Kihara S., Yamashita S., Makishima M., Funahashi T., Yamanaka S., Hiramatsu R., Matsuzawa Y., Shimomura I. (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307, 426–430 [DOI] [PubMed] [Google Scholar]
- 2. Samal B., Sun Y., Stearns G., Xie C., Suggs S., McNiece I. (1994) Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 14, 1431–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rongvaux A., Shea R. J., Mulks M. H., Gigot D., Urbain J., Leo O., Andris F. (2002) Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 32, 3225–3234 [DOI] [PubMed] [Google Scholar]
- 4. Luk T., Malam Z., Marshall J. C. (2008) Pre-B-cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J. Leukocyte Biol. 83, 804–816 [DOI] [PubMed] [Google Scholar]
- 5. Brentano F., Schorr O., Ospelt C., Stanczyk J., Gay R. E., Gay S., Kyburz D. (2007) Pre-B-cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum. 56, 2829–2839 [DOI] [PubMed] [Google Scholar]
- 6. Gosset M., Berenbaum F., Salvat C., Sautet A., Pigenet A., Tahiri K., Jacques C. (2008) Crucial role of visfatin/pre-B-cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. 58, 1399–1409 [DOI] [PubMed] [Google Scholar]
- 7. Dahl T. B., Yndestad A., Skjelland M., Øie E., Dahl A., Michelsen A., Damås J. K., Tunheim S. H., Ueland T., Smith C., Bendz B., Tonstad S., Gullestad L., Frøland S. S., Krohg-Sørensen K., Russell D., Aukrust P., Halvorsen B. (2007) Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation 115, 972–980 [DOI] [PubMed] [Google Scholar]
- 8. Moschen A. R., Kaser A., Enrich B., Mosheimer B., Theurl M., Niederegger H., Tilg H. (2007) Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 178, 1748–1758 [DOI] [PubMed] [Google Scholar]
- 9. Ye S. Q., Simon B. A., Maloney J. P., Zambelli-Weiner A., Gao L., Grant A., Easley R. B., McVerry B. J., Tuder R. M., Standiford T., Brower R. G., Barnes K. C., Garcia J. G. (2005) Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am. J. Respir. Crit. Care Med. 171, 361–370 [DOI] [PubMed] [Google Scholar]
- 10. Jia S. H., Li Y., Parodo J., Kapus A., Fan L., Rotstein O. D., Marshall J. C. (2004) Pre-B-cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Invest. 113, 1318–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bi T. Q., Che X. M. (2010) Nampt/PBEF/visfatin and cancer. Cancer Biol. Ther. 10, 119–125 [DOI] [PubMed] [Google Scholar]
- 12. Kralisch S., Klein J., Lossner U., Bluher M., Paschke R., Stumvoll M., Fasshauer M. (2005) Hormonal regulation of the novel adipocytokine visfatin in 3T3-L1 adipocytes. J. Endocrinol. 185, R1–8 [DOI] [PubMed] [Google Scholar]
- 13. Kralisch S., Klein J., Lossner U., Bluher M., Paschke R., Stumvoll M., Fasshauer M. (2005) Interleukin-6 is a negative regulator of visfatin gene expression in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 289, E586–E590 [DOI] [PubMed] [Google Scholar]
- 14. Bao J. P., Chen W. P., Wu L. D. (2009) Visfatin: a potential therapeutic target for rheumatoid arthritis. J. Int. Med. Res. 37, 1655–1661 [DOI] [PubMed] [Google Scholar]
- 15. Fukuhara A., Matsuda M., Nishizawa M., Segawa K., Tanaka M., Kishimoto K., Matsuki Y., Murakami M., Ichisaka T., Murakami H., Watanabe E., Takagi T., Akiyoshi M., Ohtsubo T., Kihara S., Yamashita S., Makishima M., Funahashi T., Yamanaka S., Hiramatsu R., Matsuzawa Y., Shimomura I. (2007) Retraction of Fukuhara et al., Science 307 (5708) 426–430. Science 318, 565. [DOI] [PubMed] [Google Scholar]
- 16. Adya R., Tan B. K., Punn A., Chen J., Randeva H. S. (2008) Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signaling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 78, 356–365 [DOI] [PubMed] [Google Scholar]
- 17. Sun Q., Li L., Li R., Yang M., Liu H., Nowicki M. J., Zong H., Xu J., Yang G. (2009) Overexpression of visfatin/PBEF/Nampt alters whole-body insulin sensitivity and lipid profile in rats. Ann. Med. 41, 311–320 [DOI] [PubMed] [Google Scholar]
- 18. Brown J. E., Onyango D. J., Ramanjaneya M., Conner A. C., Patel S. T., Dunmore S. J., Randeva H. S. (2010) Visfatin regulates insulin secretion, insulin receptor signaling, and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J. Mol. Endocrinol. 44, 171–178 [DOI] [PubMed] [Google Scholar]
- 19. Xie H., Tang S. Y., Luo X. H., Huang J., Cui R. R., Yuan L. Q., Zhou H. D., Wu X. P., Liao E. Y. (2007) Insulin-like effects of visfatin on human osteoblasts. Calcif. Tissue Int. 80, 201–210 [DOI] [PubMed] [Google Scholar]
- 20. Busso N., Karababa M., Nobile M., Rolaz A., Van Gool F., Galli M., Leo O., So A., De Smedt T. (2008) Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PloS One 3, e2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aigner T., Sachse A., Gebhard P. M., Roach H. I. (2006) Osteoarthritis: pathobiology-targets and ways for therapeutic intervention. Adv. Drug Deliv. Rev. 58, 128–149 [DOI] [PubMed] [Google Scholar]
- 22. Altman R., Asch E., Bloch D., Bole G., Borenstein D., Brandt K., Christy W., Cooke T. D., Greenwald R., Hochberg M. (1986) Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 29, 1039–1049 [DOI] [PubMed] [Google Scholar]
- 23. Salvat C., Pigenet A., Humbert L., Berenbaum F., Thirion S. (2005) Immature murine articular chondrocytes in primary culture: a new tool for investigating cartilage. Osteoarthritis Cartilage 13, 243–249 [DOI] [PubMed] [Google Scholar]
- 24. Gosset M., Berenbaum F., Thirion S., Jacques C. (2008) Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 3, 1253–1260 [DOI] [PubMed] [Google Scholar]
- 25. Blaise R., Mahjoub M., Salvat C., Barbe U., Brou C., Corvol M. T., Savouret J. F., Rannou F., Berenbaum F., Bausero P. (2009) Involvement of the Notch pathway in the regulation of matrix metalloproteinase 13 and the dedifferentiation of articular chondrocytes in murine cartilage. Arthritis Rheum. 60, 428–439 [DOI] [PubMed] [Google Scholar]
- 26. Masuko-Hongo K., Berenbaum F., Humbert L., Salvat C., Goldring M. B., Thirion S. (2004) Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 50, 2829–2838 [DOI] [PubMed] [Google Scholar]
- 27. González-Rodríguez A., Mas Gutierrez J. A., Sanz-González S., Ros M., Burks D. J., Valverde A. M. (2010) Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes 59, 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baltensperger K., Lewis R. E., Woon C. W., Vissavajjhala P., Ross A. H., Czech M. P. (1992) Catalysis of serine and tyrosine autophosphorylation by the human insulin receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 7885–7889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schmidt M. B., Chen E. H., Lynch S. E. (2006) A review of the effects of insulin-like growth factor and platelet derived growth factor on in vivo cartilage healing and repair. Osteoarthritis Cartilage 14, 403–412 [DOI] [PubMed] [Google Scholar]
- 30. Kim M. K., Lee J. H., Kim H., Park S. J., Kim S. H., Kang G. B., Lee Y. S., Kim J. B., Kim K. K., Suh S. W., Eom S. H. (2006) Crystal structure of visfatin/pre-B-cell colony-enhancing factor 1/nicotinamide phosphoribosyltransferase, free and in complex with the anti-cancer agent FK-866. J. Mol. Biol. 362, 66–77 [DOI] [PubMed] [Google Scholar]
- 31. Kim J. J., Accili D. (2002) Signaling through IGF-I and insulin receptors: where is the specificity? Growth Horm. IGF Res. 12, 84–90 [DOI] [PubMed] [Google Scholar]
- 32. Nakae J., Kido Y., Accili D. (2001) Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 22, 818–835 [DOI] [PubMed] [Google Scholar]
- 33. Chan S. J., Plisetskaya E. M., Urbinati E., Jin Y., Steiner D. F. (1997) Expression of multiple insulin and insulin-like growth factor receptor genes in salmon gill cartilage. Proc. Natl. Acad. Sci. U.S.A. 94, 12446–12451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Claassen H., Schicht M., Brandt J., Reuse K., Schädlich R., Goldring M. B., Guddat S. S., Thate A., Paulsen F. (2011) C-28/I2 and T/C-28a2 chondrocytes as well as human primary articular chondrocytes express sex hormone and insulin receptors–Useful cells in study of cartilage metabolism. Ann. Anat. 193, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosa S. C., Rufino A. T., Judas F., Tenreiro C., Lopes M. C., Mendes A. F. (2011) Expression and function of the insulin receptor in normal and osteoarthritic human chondrocytes: modulation of anabolic gene expression, glucose transport, and GLUT-1 content by insulin. Osteoarthritis Cartilage 19, 719–727 [DOI] [PubMed] [Google Scholar]
- 36. Dominici F. P., Argentino D. P., Munoz M. C., Miquet J. G., Sotelo A. I., Turyn D. (2005) Influence of the cross-talk between growth hormone and insulin signaling on the modulation of insulin sensitivity. Growth Horm. IGF Res. 15, 324–336 [DOI] [PubMed] [Google Scholar]
- 37. Ekenstedt K. J., Sonntag W. E., Loeser R. F., Lindgren B. R., Carlson C. S. (2006) Effects of chronic growth hormone and insulin-like growth factor 1 deficiency on osteoarthritis severity in rat knee joints. Arthritis Rheum. 54, 3850–3858 [DOI] [PubMed] [Google Scholar]
- 38. Albrecht C., Helmreich M., Tichy B., Marlovits S., Plasenzotti R., Egerbacher M., Haeusler G. (2009) Impact of 3D-culture on the expression of differentiation markers and hormone receptors in growth plate chondrocytes as compared to articular chondrocytes. Int. J. Mol. Med. 23, 347–355 [DOI] [PubMed] [Google Scholar]
- 39. Doré S., Abribat T., Rousseau N., Brazeau P., Tardif G., DiBattista J. A., Cloutier J. M., Pelletier J. P., Martel-Pelletier J. (1995) Increased insulin-like growth factor 1 production by human osteoarthritic chondrocytes is not dependent on growth hormone action. Arthritis Rheum. 38, 413–419 [DOI] [PubMed] [Google Scholar]
- 40. Jacques C., Béréziat G., Humbert L., Olivier J. L., Corvol M. T., Masliah J., Berenbaum F. (1997) Posttranscriptional effect of insulin-like growth factor-I on interleukin-1β-induced type II-secreted phospholipase A2 gene expression in rabbit articular chondrocytes. J. Clin. Invest. 99, 1864–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Remacle-Bonnet M., Garrouste F., Baillat G., Andre F., Marvaldi J., Pommier G. (2005) Membrane rafts segregate pro- from anti-apoptotic insulin-like growth factor-I receptor signaling in colon carcinoma cells stimulated by members of the tumor necrosis factor superfamily. Am. J. Pathol. 167, 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin W., Park J. I., Loeser R. F. (2009) Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 284, 31972–31981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boini K. M., Zhang C., Xia M., Han W. Q., Brimson C., Poklis J. L., Li P. L. (2010) Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim Biophys. Acta 1801, 1294–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yammani R. R., Loeser R. F. (2012) Extracellular nicotinamide phosphoribosyltransferase (NAMPT/visfatin) inhibits insulin-like growth factor-1 signaling and proteoglycan synthesis in human articular chondrocytes. Arthritis Res. Ther. 14:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li X., Afif H., Cheng S., Martel-Pelletier J., Pelletier J. P., Ranger P., Fahmi H. (2005) Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J. Rheumatol. 32, 887–895 [PubMed] [Google Scholar]
- 46. Liu P., Li H., Cepeda J., Zhang L. Q., Cui X., Garcia J. G., Ye S. Q. (2009) Critical role of PBEF expression in pulmonary cell inflammation and permeability. Cell Biol. Int. 33, 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Revollo J. R., Körner A., Mills K. F., Satoh A., Wang T., Garten A., Dasgupta B., Sasaki Y., Wolberger C., Townsend R. R., Milbrandt J., Kiess W., Imai S. (2007) Nampt/PBEF/Visfatin regulates insulin secretion in beta-cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romacho T., Azcutia V., Vázquez-Bella M., Matesanz N., Cercas E., Nevado J., Carraro R., Rodríguez-Mañas L., Sánchez-Ferrer C. F., Peiró C. (2009) Extracellular PBEF/NAMPT/visfatin activates proinflammatory signaling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia 52, 2455–2463 [DOI] [PubMed] [Google Scholar]
- 49. Zhang J. (2007) The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J. Biol. Chem. 282, 34356–34364 [DOI] [PubMed] [Google Scholar]
- 50. Chabane N., Zayed N., Afif H., Mfuna-Endam L., Benderdour M., Boileau C., Martel-Pelletier J., Pelletier J. P., Duval N., Fahmi H. (2008) Histone deacetylase inhibitors suppress interleukin-1β-induced nitric oxide and prostaglandin E2 production in human chondrocytes. Osteoarthritis Cartilage 16, 1267–1274 [DOI] [PubMed] [Google Scholar]
- 51. Zhang R., Chen H. Z., Liu J. J., Jia Y. Y., Zhang Z. Q., Yang R. F., Zhang Y., Xu J., Wei Y. S., Liu D. P., Liang C. C. (2010) SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J. Biol. Chem. 285, 7097–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]