

Background: Gβ5−/− mice have defective retinal outer plexiform layer and lack b-wave in electroretinogram (ERG).
Results: Mice lacking RGS7 and RGS11 recapitulate ERG and morphological defects of Gβ5−/− mice.
Conclusion: RGS7 and RGS11 are required for normal DBC morphology and function.
Significance: Despite its ability to complex with conventional Gγ subunits, the role of Gβ5 in retina is mediated exclusively by R7 RGS proteins.
Keywords: G Proteins, Gene Knock-out, Retina, RGS Proteins, Signaling, ERG, Gβ5, RGS11, RGS7, Depolarizing Bipolar Cell
Abstract
Two members of the R7 subfamily of regulators of G protein signaling, RGS7 and RGS11, are present at dendritic tips of retinal depolarizing bipolar cells (DBCs). Their involvement in the mGluR6/Gαo/TRPM1 pathway that mediates DBC light responses has been implicated. However, previous genetic studies employed an RGS7 mutant mouse that is hypomorphic, and hence the exact role of RGS7 in DBCs remains unclear. We have made a true RGS7-null mouse line with exons 6–8 deleted. The RGS7−/− mouse is viable and fertile but smaller in body size. Electroretinogram (ERG) b-wave implicit time in young RGS7−/− mice is prolonged at eye opening, but the phenotype disappears at 2 months of age. Expression levels of RGS6 and RGS11 are unchanged in RGS7−/− retina, but the Gβ5S level is significantly reduced. By characterizing a complete RGS7 and RGS11 double knock-out (711dKO) mouse line, we found that Gβ5S expression in the retinal outer plexiform layer is eliminated, as is the ERG b-wave. Ultrastructural defects akin to those of Gβ5−/− mice are evident in 711dKO mice. In retinas of mice lacking RGS6, RGS7, and RGS11, Gβ5S is undetectable, whereas levels of the photoreceptor-specific Gβ5L remain unchanged. Whereas RGS6 alone sustains a significant amount of Gβ5S expression in retina, the DBC-related defects in Gβ5−/− mice are caused solely by a combined loss of RGS7 and RGS11. Our data support the notion that the role of Gβ5 in the retina, and likely in the entire nervous system, is mediated exclusively by R7 RGS proteins.
Introduction
RGS6,2 RGS7, RGS9, and RGS11 are members of the R7 subfamily of regulators of G protein signaling (1), whose stable expression in vivo requires an obligate partner Gβ5 (2). In addition to the signature RGS domain located near the C terminus, these R7 RGS proteins contain DEP, DHEX, and GGL domains (1, 3), wherein the GGL domain mediates the interaction with Gβ5 (4). We previously reported two depolarizing bipolar cell (DBC)-related defects in Gβ5−/− mice, namely the absence of an electroretinogram (ERG) b-wave and the failure of the number of triadic ribbon synapse at retinal outer plexiform layer (OPL) to increase during postnatal development (5). Because Gβ5 exists in a photoreceptor-specific long form (Gβ5L) and a more broadly expressed short form (Gβ5S) (6), it was also determined that DBC-related defects resulted not from a combined loss of Gβ5L and Gβ5S, but specifically from the loss of postphotoreceptor Gβ5S expression (5). Given the obligate relationship between Gβ5 and R7 RGS proteins, we and others have employed RGS7- and RGS11-targeted mutant mice to show that these two RGS proteins play a role in DBC light responses in what appears to be a functionally redundant manner (7–9). However, the exact role of RGS7 was not unequivocally determined because of the discovery that the RGS7 mutant mice were hypomorphic and not null, due to an in-frameshift caused by exon 10 deletion that leads to the expression of a truncated RGS7 protein lacking 27 amino acids in the interdomain and the first six residues of the GGL domain (7).
Determining the function of RGS7 in DBCs is important in several ways. First, RGS7 is expressed widely in the nervous system, and its physiological role is poorly understood. Second, whereas the G protein pathway utilized within rod and cone photoreceptors is well understood, DBC relies on a G protein pathway containing mGluR6, Gαo, and TRPM1, but many of the details and regulation of this pathway are unclear (10). Third, the OPL ultrastructural defects seen in the Gβ5−/− mice may be due to loss of both RGS7 and RGS11 within DBCs or the loss of RGS7 alone, which cannot be determined without the development of a true RGS7 knock-out mouse line. Fourth, and importantly, although Gβ5 is most likely acting by stabilizing R7 RGS proteins, the possibility that it acts via an RGS-independent mechanism, e.g. through demonstrated interactions with conventional Gγ subunits (6, 11–13), has neither been confirmed nor ruled out. To address these issues, we have generated the first true RGS7-null mouse line, which resulted in a smaller body size and delayed electroretinogram (ERG) b-waves. By generating a RGS7 and RGS11 double knock-out (711dKO) line, we have recapitulated DBC-related structural and functional defects of Gβ5−/− mice in retinal OPL. These data demonstrate that Gβ5S works through stabilizing RGS7 and RGS11 in DBC dendrites and that RGS7 alone is as important as RGS11 in the development and function of the first vision synapse. To extend the applicability of the finding in DBC to other retinal neurons and to address whether Gβ5S can be stabilized in the absence of R7 RGS proteins by other molecules such as canonical Gγ subunits, we generated a triple knock-out mice (tKO) lacking RGS6, RGS7, and RGS11 in which we found that Gβ5S in the retina is undetectable. Together, our data firmly establish that Gβ5S works not as a conventional Gβ subunit, but exclusively through its interaction with R7 RGS proteins.
EXPERIMENTAL PROCEDURES
RGS7 Inactivation
A backbone targeting vector, pDNDF-7, which contains a neomycin cassette flanked by tandem FRT sites (NEO), two diphtheria toxin cassettes (DTA), and two multiple cloning sites (MCS) in the following order: DTA-MCS1-NEO-MCS2-DTA, was generated by modifying pBluescript II SK (Agilent Technologies, Santa Clara, CA). The MCS1 contains XhoI, BglII, and NheI sites, and the MCS2 contains NotI, KpnI, and AvrII sites. The 3.3-kb right homology arm was amplified by PCR from a BAC clone containing 214.3 kb of mouse genomic DNA (RP23-387K4; BACPAC Resources, Oakland, CA) with primers RGS7RAKpnF (5′-GG ggt acc AGC CAG CTC TCA CTG GAC AT-3′) and RGS7RAAvr2R (5′-TTA cct agg GCA AAA ACA GCC ATC TCC AT-3′) using Phusion DNA polymerase (New England Biolabs, Ipswich, MA), sequence-verified, and subcloned into MCS2 with KpnI and AvrII sites. The 2.8-kb left homology arm was similarly amplified by PCR using primers RGS7LAXhoF (5′-A CCG ctc gag AGG AGT CTT GCG TCT TGC AT-3′) and RGS7LANheR (5′-CTA gct agc GCA CAA GGT TCA ACA TCC AA-3′) from the same BAC clone, sequence-verified, and subcloned into XhoI and NheI sites of MCS1. The resulting RGS7 targeting vector was linearized with BsiWI digestion. A commercialized service electroporated the targeting vector into a hybrid 129SvEv/C57BL/6N mouse embryonic stem (ES) cell line, and ES cell clones with homologous recombination were injected into C57BL/6 blastocysts to generate chimeras (inGenious Targeting Laboratory, Stony Brook, NY).
Animal Genotyping
The animal use protocol was reviewed and approved by Virginia Commonwealth University Institutional Animal Care and Use Committee (VCU IACUC). The RGS7 knock-out (RGS7−/−) mice were genotyped by PCR using tail gDNA with the following three primers: RGS7-X7-F (5′-TTT ATG CAA GCA GAA GCA CAA GC-3′), RGS7-Gen-R (5′-TGC AAA TGT CCA GTG AGA GC-3′), and NEO-Seq-R3 (5′-GGG GGA ACT TCC TGA CTA GG-3′) at 60 °C annealing temperature. The wild-type (WT) RGS7 allele gave a 418-bp product, and the knock-out allele generated a 282-bp product. The genotyping of RGS11 knock-out (RGS11−/−) mice was as described previously (7). The genotyping of RGS6 knock-out (RGS6−/−) mice was as described previously (14). The nob1 mice were genotyped as described previously (15).
Antibodies and Western Blotting
GAPDH antibody was from Cell Signaling Technologies (2118L; Danvers, MA); RGS6/7 antibody was from Santa Cruz Biotechnology (H-190; Santa Cruz, CA). RGS11 peptide antibody (VCU008) was as described previously (7). RGS6 antibody was as described (14). Gβ5 antibody CT215 was from Dr. Mel Simon of Caltech. mGluR6 antibody was made in guinea pig by Neuromic (GP13105; Edina, MN). PKCα antibody was from Sigma (P4334). CtBP2 antibody is from BD Biosciences (612044). All secondary antibodies were from Invitrogen. Western blotting and quantification of protein expression level were conducted as described previously (7).
Immunohistochemistry
Animals were killed by CO2 inhalation, and the eyeballs were enucleated immediately. After removal of cornea and lens the resulting eyecups were immersion-fixed in 4% paraformaldehyde in 1× PBS at room temperature for 15 min. This short fixation time ensured good mGluR6 and Gβ5 signals at the OPL. After cryoprotection in 30% sucrose in 1× PBS, the eyecups were embedded in TBS (Richard Allan Scientific, Kalamazoo, MI), sectioned at 20-μm thickness, and stained using appropriate primary and secondary antibodies. The dilution of each antibody is given in figure legends or as described previously (7).
ERG
Animals were dark-adapted for a minimum of 8 h prior to recordings. Under infrared illumination, mice were anesthetized using a mixture of ketamine/xylazine (150 mg/10 mg per kg, intraperitoneal). Pupils were dilated using 1% tropicamide and 2.5% phenylephrine topical eye drops (Bausch & Lomb, Tampa, FL). Body temperature was maintained at 35–37 °C using a heating plastic coil surrounding the mouse body. Under scotopic conditions, both eyes were recorded simultaneously on a UTAS E-3000 system (LKC Technologies, Gaithersburg, MD) as described (16). Analysis of ERG recordings was conducted as described (7). The b-wave implicit time is defined as the time difference between flash onset and the time b-wave is at its maximum. It was determined and plotted as a function of flash strength.
Electron Microscopy
Enucleated eyeballs were immersion-fixed at room temperature overnight in 2% paraformaldehyde/2% glutaraldehyde in 0.1 m cacodylic acid, pH 7.4, and processed as described (16). Ultrathin sections (100 nm) were prepared using a Sorvall MT-6000 ultramicrotome. Sections were stained with uranyl acetate and lead citrate and imaged using a Joel JEM-1230 transmission electron microscope. Quantification of OPL synaptic triads and ribbons from four 3-month-old 711dKO and three age-matched WT control mice were performed from 20–25 random and nonoverlapped 38 × 38 μm2 transmission electron microscopic images near the meridian as described previously (5). All images were obtained with retinal sections containing the optic nerve head. The percentage of rod spherules with triads and ribbons from WT and 711dKO retinas was subjected to two-tailed Student's t test assuming unequal variance, and p value of <0.01 is used to signify differences between the two groups.
RESULTS
Deletion of Exons 6–8 Produces Runty RGS7-null Mouse
The RGS7 mutant mouse with exon 10 deletion expresses a partial loss-of-function truncated RGS7 protein (7, 8). To avoid targeting the DEP domain that is alternatively spliced (data not shown, but see Ref. 17) and not to repeat the mishap of targeting a single RGS7 exon, we chose to delete multiple exons from exons 6–8 using the targeting construct shown in Fig. 1A. Upon confirmation of homologous recombination, targeted ES cells were used to generate three chimeras. The mating of chimeras to C57BL/6 animals produced heterozygous RGS7 knock-out mice, which were interbred to generate homozygous knock-out (RGS7−/−) mice. Protein extracts derived from retina and cerebellum of RGS7−/− mice lack detectable levels of RGS7 (Fig. 1, B and C). Further verifying that a true RGS7-null is at hand, the RGS7−/− mouse appears runty from postnatal day 3 (P3) compared with its WT and heterozygous knock-out littermates (Fig. 1D), and the weight deficit is more apparent during P15–19, when there is no weight gain in RGS7−/− mice (Fig. 1D), a phenotype similar to that of the Gβ5−/− mouse. A notable feature of RGS7−/− mice that distinguishes them from Gβ5−/− mice is their apparent normal viability and fertility, despite their smaller sizes.
FIGURE 1.

RGS7−/− mice have a lower body weight. A, partial map of the mouse RGS7 gene, targeted exons, and the targeting construct containing negative and positive selection markers in DTA and NEO, respectively. Upon homologous recombination, exons 6–8 are replaced with the NEO marker, whereas the DTA markers are concurrently lost. Restriction enzyme sites in parentheses are present in the backbone pDND7-F vector and were introduced by PCR during the amplification of homology arms. B and C, Western blotting analysis of protein extracts derived from retina (20 μg, B) and cerebellum (10 μg, C) in littermate wild-type (+/+), heterozygous (+/−), and homozygous (−/−) RGS7 knock-out mice using anti-RGS6/7 (H-190, 1:2,000) antibody indicates the complete loss of RGS7 in RGS7−/− mice. Anti-GAPDH (1:50,000) serves as loading control. Asterisk denotes RGS6 signal picked up by the H-190 antibody. D, weights of RGS7+/+ (WT, n = 4), RGS7+/− (HET, n = 6), and RGS7−/− (KO, n = 4) mice recorded from P1 to P23 at 2-day intervals, showing that KO mice have an early noticeable smaller body weight (p < 0.01, P3–P23) compared with those of WT and HET littermates.
RGS7−/− Mice Exhibit Delayed ERG b-Wave That Ameliorates with Age
The RGS7−/− mouse eyes open at around P15, ∼24–48 h later than WT littermates. When ERG recordings were conducted at P16, a prolonged b-wave implicit time was readily notable in RGS7−/− mice at all light intensities tested (Fig. 2A, P16). Such differences progressively disappear as RGS7−/− mice age, shown by less pronounced delay in the b-wave in these mice at P30 (Fig. 2A, P30) and no differences from WT mice at 2 months of age (Fig. 2A, 2 months). With the loss of RGS7, we surmised that the RGS11 or RGS6 level might be up-regulated to compensate in RGS7−/− retina, but quantitative Western blot analysis revealed no such projected increase in RGS11 or RGS6 level (data not shown). Quantification of the Gβ5S level did reveal a significant ∼27 ± 7% decrease in retinas of RGS7−/− mice (Fig. 2, C and D), suggesting that expression of RGS6, RGS7, and RGS11 may not have significant overlaps in the retina and/or that in neurons where they are co-expressed, they may be independently regulated and targeted.
FIGURE 2.

RGS7−/− mice exhibit an age-dependent delayed ERG b-wave implicit time as well as loss of Gβ5S protein in the retina. A, ERG b-wave implicit time, measured as the time difference between flash onset and the time b-wave is at its peak, was significantly delayed (*, p < 0.01) after eye opening at P16 in RGS7−/− mice (KO, n = 8) compared with WT littermate controls (n = 8). At P30, the KO (n = 6) implicit time delay was improved compared with WT (n = 6), showing a slight trend, but this delay was no longer statistically significant. At 2 months of age (2mth), the b-wave implicit time of KO (n = 8) mice is indistinguishable from that of WT animals (n = 6). Error bars are S.E. B, representative ERG traces from WT (black) and KO (gray) animals at P16 are shown with flash intensity indicated at left in unit of log cd s m−2. C and D, Gβ5S protein level in the retina was decreased in adult RGS7−/− mouse at P30. C, representative immunoblot shows the level of Gβ5S from two WT and two RGS7−/− mice retinal protein extracts (20 μg) using anti-Gβ5 (CT215, 1:2,000) and anti-GAPDH (1:50,000) antibodies. D, quantification of Gβ5S protein level, normalized to GAPDH signals, shows a significant 27 ± 7% reduction in the RGS7−/− (n = 6) versus WT (n = 4) mice (*, p < 0.01). Error bars are S.E.
711dKO Mice Lack ERG b-Waves, and Gβ5S Is Undetectable in DBC Dendrites
To examine the role of R7 RGS proteins in DBC function, RGS7−/− mice were mated with RGS11−/− mice to produce the first true RGS7/RGS11 double KO (711dKO) line, which also appears viable and fertile. Because the expression of Gβ5S at the OPL appears as puncta and co-localizes with mGluR6, as shown previously (8) and in Fig. 3, we found that such Gβ5S puncta were undetectable in the OPL of 711dKO mice (Fig. 3). This confirms that within DBC dendrites Gβ5S interacts only with RGS7 and RGS11, and not with RGS6. Consistent with this finding, we found that the ERG b-wave is absent in 711dKO mice at all light intensities tested under scotopic (Fig. 4A) and photopic conditions (data not shown). To ascertain that these mice indeed lack ERG b-waves, we compared their responses with those from a naturally occurring mutant strain called nob1 (18), within which the TRPM1 channels responsible for DBC light responses (10, 19) were recently reported to be missing at DBC dendritic tips (10, 20). We found no differences when we superimposed the ERG response of nob1 (18) with that of 711dKO mice (data not shown). Together, we conclude that removing RGS7 and RGS11 completely abolishes light responses of retinal DBCs, akin to the defects found in Gβ5−/− (5) and other mice lacking ERG b-waves.
FIGURE 3.

Absence of Gβ5 immunoreactivity in mGluR6-containing puncta in retinas of 711dKO mice. Retinal sections from WT control and 711dKO (DKO) mice stained for metabotropic glutamate receptor 6 (mGluR6, 1:200) and Gβ5 (CT215, 1:250) demonstrate superimposed signals in the WT but absence of Gβ5 signal in the DKO. Scale bar, 10 μm.
FIGURE 4.

RGS7 and RGS11 dKO mice have no ERG b-waves and contain ultrastructural defects in the retinal OPL. A, representative scotopic ERG responses from 2-month-old WT control and 711dKO (DKO) mice with flash intensity indicated at left in units of log cd s m−2. B, immunohistochemical staining of control and DKO retinal sections for CtBP2 (1:2,000, white) and PKCα (1:100,000, red) demonstrating reduction in rod bipolar cell dendritic arborization. BV, blood vessel. Scale bar, 5 μm. C, representative transmission electron microscopic images of the OPL from control and DKO mice. Scale bar, 2 μm. RS, rod spherule. Asterisk denotes abnormal structure. D and E, percentage of rod spherules with normal synaptic triads (D) and ribbons (E) in the DKO (n = 4, 80 images) versus control (n = 3, 65 images) retinas (*, p < 0.01). Error bars are S.D.
711dKO Mice Have OPL Morphological Defects
Because Gβ5−/− mice display DBC dendritic arborization defects that could be detected by immunohistochemisty, 711dKO retinal sections were further examined by double labeling for PKCα and CtBP2, which mark rod bipolar cells and synaptic ribbons, respectively. As shown in Fig. 4B, the dendrites of 711dKO DBCs appear stunted compared with those of WT controls. To quantify the extent of OPL structural defect, transmission electron microscopy was performed, and the percentage of rod spherules with ribbons and triads at the OPL of adult 711dKO and WT control mice were compared. Adult 711dKO mice have <15% triads per spherule compared with >33% in age-matched WT controls (Fig. 4D). The percentage of spherules with ribbons is also noted to be lower in 711dKO mice, although not as pronounced as that in synaptic triads (∼61% control versus ∼47% 711dKO; Fig. 4E). These data indicate that the requirement of Gβ5S expression for normal OPL development is not mediated by other RGS proteins, but exclusively by RGS7 and RGS11.
Gβ5S Does Not Have Non-R7 RGS Protein Partner inRetina
With a true RGS7−/− mouse line at hand, we performed additional mating with RGS6−/− and RGS11−/− mice to generate different permutations of R7 RGS KO mice to determine the extent at which Gβ5S expression is sustained by individual R7 RGS proteins in the retina. In RGS6 and RGS7 double KO (67dKO) mice, the level of Gβ5S in the retina, presumably stabilized by RGS11, is drastically reduced to ∼5% of control (Fig. 5). Similarly, in RGS6 and RGS11 double KO (611dKO) mice, the level of Gβ5S, presumably stabilized by RGS7, is decreased to ∼35% of control (Fig. 5). In the 711dKO retina, the level of Gβ5S, presumably sustained by interaction with RGS6, is ∼60% of control (Fig. 5), demonstrating that RGS6 alone sustains more than half of Gβ5S expression in the mouse retina. To explore further whether Gβ5S may interact with and be stabilized by a non-R7 RGS protein partner, we generated RGS6, RGS7, and RGS11 tKO mice and found in their retinas that Gβ5S was undetectable, whereas the level of the photoreceptor-specific Gβ5L, stabilized by RGS9-1 (Fig. 5), remained unchanged.
FIGURE 5.
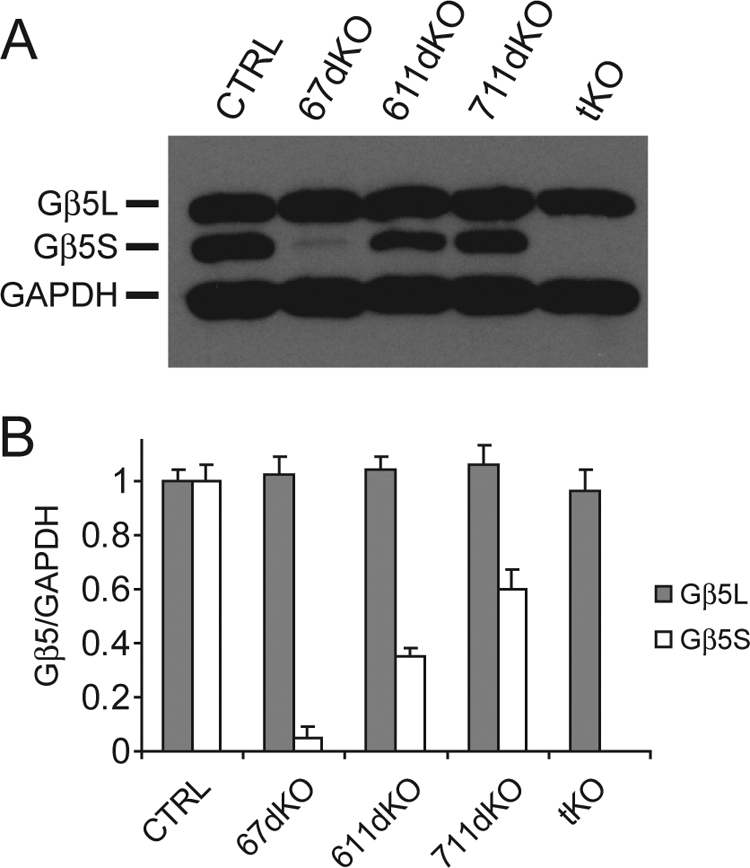
Gβ5S protein is undetectable in the RGS6, RGS7, and RGS11 tKO mouse retina. A, representative immunoblot showing the level of Gβ5L and Gβ5S from WT CTRL, 67dKO, 611dKO, 711dKO, and tKO mouse retinal extracts (15 μg) using anti-Gβ5 (CT215, 1:2,000) and anti-GAPDH (1:50,000) antibodies. B, quantification of Gβ5L and Gβ5S levels, normalized to GAPDH signal. Retinal Gβ5L protein levels do not differ across the various genotypes. However, 67dKO contains ∼5% of normal Gβ5S protein level (n = 3), 611dKO contains ∼35% of normal Gβ5S protein level (n = 3), 711dKO contains ∼60% of normal Gβ5S protein level (n = 3), and tKO has undetectable Gβ5S protein level (n = 3). Error bars are S.E.
DISCUSSION
Among the pleiotropic phenotypes of Gβ5−/− mice, the easiest one to be noticed is its smaller size (2). Because all R7 RGS proteins are down-regulated in Gβ5−/− mice (2), it was unclear whether the individual or combined loss of R7 RGS proteins is responsible for this defect. The weight of individual knock-out mice of RGS6 (14), RGS9 (21), and RGS11 (7, 8, 14) appears normal. In the hypomorphic RGS7 mutant mouse studied previously (7, 8, 14), there was no weight abnormality, either. These findings imply the combined loss of different R7 RGS proteins to be the cause. However, we found in this study a lower body weight similar to that of Gβ5−/− mice in a true RGS7-null mouse (Fig. 1D). This suggests that the loss of RGS7 in the Gβ5−/− mouse is responsible for the weight phenotype. The conclusion is further supported by the observation that 611dKO mice have normal weight profile (data not shown). Whereas some of the Gβ5−/− phenotypes have been shown to be mediated by individual loss of a specific R7 RGS protein, e.g. RGS9-1 in phototransduction (21) and perhaps RGS11 in ERG b-wave implicit time (7, 8, 22), the finding that RGS7 loss is behind this systemic defect is significant and novel. This true RGS7−/− mouse line will thus be useful to elucidate the actions of Gβ5 and R7 RGS proteins in vivo further.
ERG recordings of RGS7−/− mice revealed a delay in the b-wave implicit time that could be detected immediately after eye opening. However, the delay became less pronounced and diminished with aging of mice. RGS7 protein is detectable by Western blotting in the retina as early as P1, whereas RGS11 protein becomes detectable at around P10 (data not shown). The disappearance of the ERG b-wave phenotype in RGS7−/− mice may be the result of compensatory RGS11 expression. However, we found no significant increase of RGS11 level in adult RGS7−/− mouse retinas. This is in contrast to what was reported for RGS11−/− mice, in which Gβ5S level is unchanged but RGS7 level is significantly up-regulated (7). With the notable difference in the specific drop of Gβ5S and no change in RGS11 levels in RGS7−/− mice, this suggests that RGS6 may not be up-regulated, either. We have checked for RGS6 expression in retinal extracts of RGS7−/− mice and confirmed this speculation. Thus, overexpression of RGS6 and RGS11 does not occur when RGS7 is knocked out in the retina, suggesting that these R7 RGS proteins may have uniquely important functions in some retinal cell types. In DBCs, the failure of RGS11 to compensate may be attributable to its dependence on R9AP for expression (22, 23). Because the R9AP level determines the overall amount of RGS9-1/Gβ5L in rods (24) it may similarly limit the up-regulation of RGS11/Gβ5S level in RGS7−/− DBCs. In the 711dKO mouse retina, Gβ5S protein level is reduced to ∼60% of WT level, suggesting that the majority of retinal Gβ5S is stabilized solely by RGS6. Although the function of RGS6 has not been determined in the retina, the dramatic loss of ERG b-waves in 711dKO mice does indicate that RGS6 may only play a minor role, if any, in DBC dendritic light responses. To determine further whether Gβ5S can be stabilized in vivo in the absence of R7 RGS proteins, we generated and characterized RGS6, RGS7, and RGS11 tKO mice and found unaltered level of Gβ5L but undetectable level of Gβ5S in the retina (Fig. 5). Despite the demonstration that Gβ5 forms a complex with and function through conventional Gγ subunits (6, 13), our data eliminate the possibility in the retina and likely in the entire nervous system that Gβ5S can be stabilized by other protein partners and therefore firmly establishes the exclusivity of the long eluded in vivo obligate partnership between Gβ5 and R7 RGS proteins (2).
RGS7 and RGS11 are the two known R7 RGS proteins present in DBC dendrites (7, 8, 25). Our data that Gβ5S is undetectable and that ERG b-wave is missing in the OPL of 711dKO mice not only support this notion but also indicate that concurrent loss of RGS7 and RGS11 is responsible for compromised DBC structure in Gβ5−/− mice and that other RGS proteins previously implicated in DBC function may be dispensable. With some questions resolved, new ones emerge: how does the loss of GTPase-accelerating proteins affect DBC development and function? How general is this finding? Given that Gβ5−/− mice have many interesting CNS phenotypes (26) and that all individual R7 RGS knock-out mice are now available, these questions may be systematically examined under different physiological contexts. Unfortunately, the OPL morphological defects observed in adult 711dKO mice still preclude a clear determination of the exact role of RGS7 and RGS11 in the mGluR6/Gαo/TRPM1 signaling pathway that mediates DBC light responses. Thus, the extent to which the missing b-wave phenotype may result from deregulation of this pathway or from the developmental OPL abnormality remains to be determined unequivocally. Individual DBC responses to exogenous glutamate and/or mGluR6 antagonist may provide further insight to this particular question (27, 28).
Whereas the roles of transducin and RGS9-1 in phototransduction are well established (29–31), we have little understanding of how similar but distinct set of G protein-related molecules located a mere synapse away in the DBC subserve vertebrate vision. Future investigations will greatly benefit from having new mouse models, such as a conditional Gβ5 knock-out mouse and/or a mouse line allowing inducible expression of Gαo with prolonged activity in DBCs at different stages of retinal development.
Acknowledgments
Confocal and electron microscopy was performed at the Virginia Commonwealth University Department of Anatomy and Neurobiology Microscopy Facility, supported in part with funding from NINDS/National Institutes of Health Center Core Grant 5P30NS047463-02.
This work was supported, in whole or in part, by National Institutes of Health Grants EY013811 (to C.-K. C.), MH094626 (to A. R. M.), and GM075033 (to R. A. F.).
- RGS
- regulators of G protein signaling
- DEP
- Dishevelled/Egl-10/Pleckstrin homology domain
- DHEX
- DEP helical extension domain
- GGL
- G-protein gamma subunit like domain
- DBC
- depolarizing bipolar cell
- dKO
- double knock-out
- DTA
- diphtheria toxin cassette
- ERG
- electroretinogram
- MCS
- multiple cloning site
- NEO
- neomycin phosphotransferase cassette
- OPL
- outer plexiform layer
- P
- postnatal day
- tKO
- triple knock-out.
REFERENCES
- 1. Ross E. M., Wilkie T. M. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827 [DOI] [PubMed] [Google Scholar]
- 2. Chen C. K., Eversole-Cire P., Zhang H., Mancino V., Chen Y. J., He W., Wensel T. G., Simon M. I. (2003) Instability of GGL domain-containing RGS proteins in mice lacking the G protein β-subunit Gβ5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheever M. L., Snyder J. T., Gershburg S., Siderovski D. P., Harden T. K., Sondek J. (2008) Crystal structure of the multifunctional Gβ5-RGS9 complex. Nat. Struct. Mol. Biol. 15, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snow B. E., Krumins A. M., Brothers G. M., Lee S. F., Wall M. A., Chung S., Mangion J., Arya S., Gilman A. G., Siderovski D. P. (1998) A G protein γ subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gβ5 subunits. Proc. Natl. Acad. Sci. U.S.A. 95, 13307–13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rao A., Dallman R., Henderson S., Chen C. K. (2007) Gβ5 is required for normal light responses and morphology of retinal ON-bipolar cells. J. Neurosci. 27, 14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watson A. J., Aragay A. M., Slepak V. Z., Simon M. I. (1996) A novel form of the G protein β subunit Gβ5 is specifically expressed in the vertebrate retina. J. Biol. Chem. 271, 28154–28160 [DOI] [PubMed] [Google Scholar]
- 7. Chen F. S., Shim H., Morhardt D., Dallman R., Krahn E., McWhinney L., Rao A., Gold S. J., Chen C. K. (2010) Functional redundancy of R7 RGS proteins in ON-bipolar cell dendrites. Invest. Ophthalmol. Vis. Sci. 51, 686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mojumder D. K., Qian Y., Wensel T. G. (2009) Two R7 regulator of G-protein signaling proteins shape retinal bipolar cell signaling. J. Neurosci. 29, 7753–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J., Jeffrey B. G., Morgans C. W., Burke N. S., Haley T. L., Duvoisin R. M., Brown R. L. (2010) RGS7 and -11 complexes accelerate the ON-bipolar cell light response. Invest. Ophthalmol. Vis. Sci. 51, 1121–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morgans C. W., Zhang J., Jeffrey B. G., Nelson S. M., Burke N. S., Duvoisin R. M., Brown R. L. (2009) TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. U.S.A. 106, 19174–19178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watson A. J., Katz A., Simon M. I. (1994) A fifth member of the mammalian G-protein β-subunit family: expression in brain and activation of the β2 isotype of phospholipase C. J. Biol. Chem. 269, 22150–22156 [PubMed] [Google Scholar]
- 12. Yoshikawa D. M., Hatwar M., Smrcka A. V. (2000) G protein β5 subunit interactions with α subunits and effectors. Biochemistry 39, 11340–11347 [DOI] [PubMed] [Google Scholar]
- 13. Yost E. A., Mervine S. M., Sabo J. L., Hynes T. R., Berlot C. H. (2007) Live cell analysis of G protein β5 complex formation, function, and targeting. Mol. Pharmacol. 72, 812–825 [DOI] [PubMed] [Google Scholar]
- 14. Yang J., Huang J., Maity B., Gao Z., Lorca R. A., Gudmundsson H., Li J., Stewart A., Swaminathan P. D., Ibeawuchi S. R., Shepherd A., Chen C. K., Kutschke W., Mohler P. J., Mohapatra D. P., Anderson M. E., Fisher R. A. (2010) RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gregg R. G., Mukhopadhyay S., Candille S. I., Ball S. L., Pardue M. T., McCall M. A., Peachey N. S. (2003) Identification of the gene and the mutation responsible for the mouse nob phenotype. Invest. Ophthalmol. Vis. Sci. 44, 378–384 [DOI] [PubMed] [Google Scholar]
- 16. Li S., Chen D., Sauvé Y., McCandless J., Chen Y. J., Chen C. K. (2005) Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis 41, 73–80 [DOI] [PubMed] [Google Scholar]
- 17. Li Q., Muma N. A. (2009) Program 715.26/C16. 2009 Neuroscience Meeting Planner, October 17–21, 2009, Society for Neuroscience, Chicago, IL [Google Scholar]
- 18. Pardue M. T., McCall M. A., LaVail M. M., Gregg R. G., Peachey N. S. (1998) A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 39, 2443–2449 [PubMed] [Google Scholar]
- 19. Koike C., Obara T., Uriu Y., Numata T., Sanuki R., Miyata K., Koyasu T., Ueno S., Funabiki K., Tani A., Ueda H., Kondo M., Mori Y., Tachibana M., Furukawa T. (2010) TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc. Natl. Acad. Sci. U.S.A. 107, 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pearring J. N., Bojang P., Jr., Shen Y., Koike C., Furukawa T., Nawy S., Gregg R. G. (2011) A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J. Neurosci. 31, 10060–10066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen C. K., Burns M. E., He W., Wensel T. G., Baylor D. A., Simon M. I. (2000) Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature 403, 557–560 [DOI] [PubMed] [Google Scholar]
- 22. Cao Y., Masuho I., Okawa H., Xie K., Asami J., Kammermeier P. J., Maddox D. M., Furukawa T., Inoue T., Sampath A. P., Martemyanov K. A. (2009) Retina-specific GTPase accelerator RGS11/Gβ5S/R9AP is a constitutive heterotrimer selectively targeted to mGluR6 in ON-bipolar neurons. J. Neurosci. 29, 9301–9313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song J. H., Song H., Wensel T. G., Sokolov M., Martemyanov K. A. (2007) Localization and differential interaction of R7 RGS proteins with their membrane anchors R7BP and R9AP in neurons of vertebrate retina. Mol. Cell. Neurosci. 35, 311–319 [DOI] [PubMed] [Google Scholar]
- 24. Krispel C. M., Chen D., Melling N., Chen Y. J., Martemyanov K. A., Quillinan N., Arshavsky V. Y., Wensel T. G., Chen C. K., Burns M. E. (2006) RGS expression rate-limits recovery of rod photoresponses. Neuron 51, 409–416 [DOI] [PubMed] [Google Scholar]
- 25. Morgans C. W., Weiwei Liu, Wensel T. G., Brown R. L., Perez-Leon J. A., Bearnot B., Duvoisin R. M. (2007) Gβ5-RGS complexes co-localize with mGluR6 in retinal ON-bipolar cells. Eur. J. Neurosci. 26, 2899–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J. H., Pandey M., Seigneur E. M., Panicker L. M., Koo L., Schwartz O. M., Chen W., Chen C. K., Simonds W. F. (2011) Knockout of G protein β5 impairs brain development and causes multiple neurologic abnormalities in mice. J. Neurochem. 119, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nawy S. (1999) The metabotropic receptor mGluR6 may signal through Go, but not phosphodiesterase, in retinal bipolar cells. J. Neurosci. 19, 2938–2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Snellman J., Nawy S. (2004) cGMP-dependent kinase regulates response sensitivity of the mouse on bipolar cell. J. Neurosci. 24, 6621–6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen C. K. (2005) The vertebrate phototransduction cascade: amplification and termination mechanisms. Rev. Physiol. Biochem. Pharmacol. 154, 101–121 [DOI] [PubMed] [Google Scholar]
- 30. Fu Y., Yau K. W. (2007) Phototransduction in mouse rods and cones. Pflugers Arch. 454, 805–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo D. G., Xue T., Yau K. W. (2008) How vision begins: an odyssey. Proc. Natl. Acad. Sci. U.S.A. 105, 9855–9862 [DOI] [PMC free article] [PubMed] [Google Scholar]