

Abstract
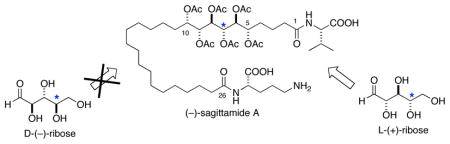
The absolute stereostructure of sagittamide A (1), a O-hexacetyl long-chain hexahydroxy-α, ω-dicarboxylic acid, was assigned using a progressive-convergent approach that integrates three powerful regimens for stereochemical analysis of acyclic natural products: J-based analysis, 13C NMR universal database comparisons and exciton coupling circular dichroism.
The structure of (–)-sagittamide A (1)1–an unprecedented polyacetoxy, long-chain α,ω-dicarboxylic acid isolated from a tropical didemnid tunicate–was solved by application of conventional 2D NMR spectroscopic methods, however, only partial stereochemistry could assigned. Although configurations of the terminal amino acids (L-ornithine and L-valine) were determined readily by conventional methods, the contiguous 5,6,7,8,9,10-hexol hexaacetate in 1 represented a significantly more complex NMR problem, in part, because of isolated stereohexad C5–C10 flanked by CH2 groups2 and equivocal interpretations of J coupling.
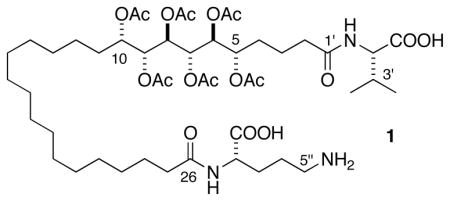
We now report the complete stereostructure of 1 using a progressive-convergent approach that integrates three powerful regimens for stereochemical analysis of natural products: use of Murata’s J-based analysis (3JHH and 2,3JHC),3 application of Kishi’s Universal Database4 (pairwise-comparison of 13C NMR chemical shifts with stereo-defined models) and highly sensitive exciton coupling circular dichroism (ECCD).5 The integrated approach rapidly converges upon a unique stereochemical assignment for 1 with internal validation.
A basis set of 3JHH and 2,3JCH values were obtained by 2D heteronuclear 2D NMR experiments of 1 (COSY and HSQMBC, respectively, see Supporting Information) and used to predict an all anti-relative configuration for C6–C9 for 1. Consequently, the number of remaining possible diastereomers of 1 was reduced from N=32 to 4. A synthetic route to six model compounds, representing permutations of the six stereocenters C5–C10 congruent with those proposed for 1, was conceived and executed starting with D-xylose (see Supporting Information).6 In order to address an equivocal C8 3JCH value in 1, a parallel set of models 2–9 was also prepared from D-ribose as described below (Scheme 1).
Scheme 1.

a) In°, allyl bromide, H2O; b) TrCl, pyridine, reflux 53% (2 steps); c) CSA, acetone, CH3C(OCH3)2CH3 58%, 10:11 dr 2:1); d) SiO2-HPLC 1:19:EtOAc hexanes; e) H2, 1 atm, Pd/C, CF3CH2OH, 35–69%; f) i. (COCl)2, DMSO, CH2Cl2, −78 °C ii. Et3N; g) i. 15, DME, NaHMDS, −78 °C, ii. aldehyde, 25%, dr 3:1 (2 steps); h) i. 14, THF, NaHMDS, −78 °C, ii. aldehyde, dr>19:1,16% 2 steps; i) OsO4, NMO, acetone, H2O; dr 1.7:1, 93%; j) K3Fe(CN)6, K2OsO4, K2CO3 (DHQ)2PHAL, t-BuOH, H2O, CH3SO2NH2, dr 3.8:1, 86%; k) 2% TMS-Cl, MeOH, ii. CH2N2, ether/MeOH, iii. Ac2O, pyridine 6h: 22% 3 (3 steps), 48%; 2 (3 steps), 44%, 6 (4 steps), 26%, 7 (4 steps).
Indium-promoted Barbier reaction of D-ribose with allyl bromide gave a 2:1 mixture of epimeric homoallylic alcohols7 10 and 11 after protection. Each acetonide was deprotected and hydrogenated (Pd/C, CF3CH2OH, 1 atm H2)8 followed by Swern oxidation to the corresponding C9 aldehydes and homologation using two stereocomplementary methods (Z-selective Wittig olefination using phosphonium salt 14 and E-selective Julia-Kocienski olefination with tetrazole 159) to give 12 and 13.
Stereoselective OsO4 dihydroxylation10 of 12 gave diols 16 and 17. In this manner, E- and Z-olefins were converted to diol diastereomers and purified by HPLC, prior to deprotection to the hexaols. Peracetylation of each hexaol furnished the eight C7–C9 ribo-model compounds 2–9 and six xylo-models (Supporting information). The correct relative configuration of 1 emerged from 13C NMR comparisons with the model compounds (Figure 1).4
Figure 1.

13C NMR (125MHz, d6-DMSO, T=298 K) Δδ values (δC 1– δC model) of ribo-model compounds 2–9
The 1H and 13C NMR spectra of each peracetate model were carefully assigned from COSY and HMBC spectra. Pairwise comparisons of the differences of the 13C chemical shifts (Δδ) for C4–C11 in model compounds and 1 clearly showed an excellent match for the C8 epimer 6 obtained from D-ribose, but an mismatch for the corresponding xylo-C8 epimer (e.g. C8: Δδ = +0.05 and −3.93 ppm, respectively, see Supporting Information). A valuable object lesson is revealed here that promotes a progressive-convergent approach to stereochemistry. Although anomalous 3JCH’s in 1 predicted an erroneous xylo-configuration during J-based analysis,11 this was readily rectified in the progressive 13C Δδ analysis allowing reassignment of C8 configuration to that of 6.
The absolute stereochemistry of 1 was secured by transformation of the natural product, and hexaol diastereomers corresponding to 6 and 7, to the per-benzoate ester derivatives, 18, 19 and 20, respectively,12 and comparison of their corresponding CD spectra (Figure 2). Since the fingerprint Cotton effects observed in the CD spectra of 18 and 19 were equal in magnitude but opposite in sign, the absolute configuration of 1 corresponds to ent-19 and is related to L-ribose.12 Thus, the complete configuration of sagittamide A (1) is depicted as (5S,6S,7S,8R,9R,10S).
Figure 2.

CD spectra of sagittamide A derivative 18 (—), together with models 19 (…) and 20 (-– -), (CH3CN, c=10 μM).
In summary, we have deployed an integrated approach to solve the configuration of sagittamide A (1). The power of this triple-combination of methodologies lies in judicious interpretation of homonuclear 3J and heteronuclear 2,3J to provide partial stereochemical information which is then used to inform correct choices for synthesis of model compounds to be used in the next stage: 13C NMR comparative analysis.
A significant advantage is gained by a requirement for only a limited sub-set of stereo-model compounds without the necessity for synthesis of all 64 possible permutations. The progressive-convergent approach succeeds where other singular methods based on NMR may become irreducibly complex13 or rendered equivocal by second-order effects that militate against reliable stereochemical assignments.
Supplementary Material
Acknowledgments
We thank J. DeRopp (UC Davis) for assistance with NMR measurements and R. New (UC Riverside) and Y.X. Su (UCSD) for MS data. This work was supported by the NIH (CA85602).
Footnotes
Supporting Information Available: Preparation of ribo- and xylo-model model compounds, and their stereochemical assignments, Δδ’s of xylo-models, 1H, 13C NMR and MS spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lievens SC, Molinski TF. Org Lett. 2005;7(11):2281–2284. doi: 10.1021/ol050717x. [DOI] [PubMed] [Google Scholar]
- 2.All attempts to convert 1 to a C1-O5 δ-lactone (potentially useful for stereochemical assignment) using acid catalysis were unsuccessful
- 3.Murata M, Nakamura H, Tachibana K. J Org Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kobayashi Y, Hayashi N, Kishi Y. Org Lett. 2002;4:411–414. doi: 10.1021/ol0171160. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi Y, Tan CH, Kishi Y. J Am Chem Soc. 2001;123:2076–2078. doi: 10.1021/ja004154q. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi Y, Lee J, Tezuka K, Kishi Y. Org Lett. 1999;1:2177–2180. doi: 10.1021/ol9903786. [DOI] [PubMed] [Google Scholar]
- 5.(a) Vazquez JT, Wiesler WT, Nakanishi K. J Am Chem Soc. 1987;109:5586–5592. [Google Scholar]; (b) Zhou P, Berova N, Nakanishi K, Knani M, Rohmer M. J Am Chem Soc. 1991;113:4040–4042. [Google Scholar]; (c) Harada N, Nakanishi K. Circular Dichroic Spectroscopy: Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley: 1983. [Google Scholar]
- 6.The carbons numbered C7, C8 and C9 in 1 map to C4, C3 and C2 of ribose or xylose, respectively. Thus, the stereochemical descriptors ‘xylo-‘ and ‘ribo-‘ in the context of this work refer to C7–C9 of 1.
- 7.The configuration of the major isomer was assigned by analogy with the well-known 1,2-syn-stereopreference for In°-promoted allylation of aldohexoses [ Kim E, Gordon DM, Schmid W, Whitesides GM. J Chem Org. 1993;58:5500–5507.Kobayashi S, Nagayama S. J Org Chem. 1996;61:2256–2257.] and subsequent conversion to the acetonides 10 and 11.
- 8.Deprotection of 10 and 11 to the corresponding primary alcohols was rapidly effected when CF3CH2OH was used as solvent for hydrogenolysis. No reaction was observed in ethanol, even after several days at 3 atm H2.
- 9.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998;(1):26–28.Both 14 and 15 were prepared from δ-valerolactone in three and four steps, respectively (see Supporting Information).
- 10.Diastereomeric assignments of 5,6-diols were based on the expectation of anti-selectivity of OsO4 addition to allylic alcohols and confirmed by the outcomes from double-diastereoselection using the Sharpless asymmetric dihydroxylation (Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–547.) and observed pseudo-C2 symmetry in the 1H and 13C NMR spectra of 2 and 8. See Supporting Information.
- 11.This observation suggests caution in using J-based methodology and over-reliance on the underlying assumption of all-staggered conformations and the accuracy of J’s measured in strongly coupled contiguous polyols that may not be amenable to first-order spin analysis.
- 12.The lactam-mono methyl ester that formed spontaneously upon treatment of 1 (CH2N2, MeOH-ether, ref. 1) and the hexaols corresponding to 6 and 7 were each converted (excess BzCl, pyridine, 40 °C) to hexabenzoates 17, 18, and 19, respectively, after HPLC purification. Benzoylation at higher temperatures (60–90 °C) lead to significant formation of tetrabenzoyloxy-tetrahydrofuran.
- 13.The similarity of CD spectra of diastereomeric 19 and 20 reflect the dominance of the C7–C10 configuration on the Cotton effects.
- 14.Rychnovsky SD, Rogers B, Yang G. J Org Chem. 1993;58:3511–3515. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.