

Abstract
Preequilibration of an interconverting set of isomeric allylic azides is coupled with an intramolecular Schmidt reaction to stereoselectively afford substituted lactams. The effect of substitution and a preliminary mechanistic study are reported. The synthetic potential of this method is demonstrated in the context of an enantioselective synthesis of an advanced intermediate toward pinnaic acid.
The intramolecular Schmidt reaction of alkyl azides, as exemplified in eq 1, is a useful means of converting 3-azidopropyl ketones into fused lactams.1 Although most applications of this reaction in total synthesis have R = H as defined in eq 1, the general issue of how to establish relative stereochemistry between an azide-bearing stereocenter and the rest of the molecule arises more frequently in complex synthetic projects.2 One approach is to fully establish the relevant stereochemistry of the ketone reactant, since the ring expansion step occurs with retention of configuration.1b However, this is often impractical or awkward, and we have sought alternative ways to couple the stereochemistry of the α-azido stereocenter with that adjacent to the ketone. In this paper, we present a strategy for accomplishing this goal using the kinetically controlled reaction of a rapidly equilibrating mixture of allylic azides and present an application to alkaloid synthesis. In addition, through a combination of experiments and density functional theory (DFT) calculations, we have uncovered previously unknown details about the stereochemical course of the intramolecular Schmidt reaction of cyclohexanone-containing substrates.
 |
(1) |
The rearrangement of allylic azides, which is facile at room temperature, was first discovered by Winstein and co-workers in 19603 and has since become well-known.4 Previous researchers have sought to carry out selective reactions from one allylic azide of a rapidly equilibrating pair by freezing out the rearrangement at low temperature,4a by taking advantage of significant stereochemical differences between potential substrates,4d,e or by rigging substrates so that only one isomeric azide or double bond can take part in a downstream intramolecular reaction.4g Recently, Craig and coworkers reported the dependence of product stereochemistry on the relative and interconverting configurations of a bystander azide adjacent to a reactive olefin participating in a Claisen rearrangement.5 However, we are unaware of other examples of using the Winstein rearrangement to control the stereochemistry of an emerging C–N bond.
It occurred to us that it might be possible to engage a mixture of allylic azides 1a–d in an intramolecular Schmidt reaction to selectively form 2a (Scheme 1). Our proposal was based on the following reasoning: (1) 1a–d would equilibrate faster than the Schmidt reaction would occur, (2) formation of lactams would be kinetically controlled via the preferential formation of intermediates Aeq and Aax (both arising from equatorial attack of azide onto ketone6), (3) trans alkene 1a would be unreactive and cis isomer 1b would be nearly so,7 and (4) the stereochemical outcome of the reaction would roughly reflect the relative stability of Aeq and Aax, favoring compound 2a by a ratio that would roughly reflect the A value of a vinyl group (1.49–1.68 kcal/mol8), or ca. 93:7 dr. The practical value of this procedure would lie in the ability to introduce the azide by displacement of a terminal leaving group prior to ring expansion reaction without having to pre-set the ultimately reacting secondary center.
Scheme 1.

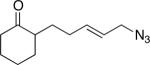
When compound 3 was subjected to Lewis acid treatment, an 83% yield of 4 was obtained, verifying that an allylic azide was a suitable partner for the intramolecular Schmidt reaction (Table 1, entry 1). In this case allylic rearrangement is redundant and only a single product is possible. Azide 5 (prepared from 2,2-dimethyl-1-phenylhex-5-en-1-one via cross-metathesis with allyl bromide and subsequent azide displacement9) rapidly reached a steady-state equilibrium of azides once formed, favoring the trans alkene 5a but containing a significant amount of rearranged isomer c bearing a secondary azide (see Scheme 1 for structures of isomer types a–d). Subjecting this mixture to the established conditions for the azido-Schmidt reaction led to lactam 6, which resulted from selective reaction of the internal azide. In this case, the overall yield of lactam exceeded the percentage of 5c in the starting azide, indicating that 1,3-allylic transposition effectively competed with the Schmidt step. Having established this, a mixture of azides 1a–d was similarly prepared and subjected to azido-Schmidt conditions. Although the reaction proceeded well, a nearly equimolar mixture of stereoisomeric lactams was obtained (entry 3), suggesting that the stereochemical hypothesis presented in Scheme 1 was, minimally, incomplete.
Table 1.
Intramolecular Schmidt reactions of allylic azides.a
| entry | allylic azide, isomer a (a:b:c:d isomer ratiob) | major product (isomer a depicted) | yield (%) (a:b ratio)b |
|---|---|---|---|
| 1 |
 3 (−) |
 4 |
83 |
| 2 |
5a (51:13:36c) |
 6 |
54 |
| 3 |
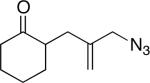
 1a (62:8:15:15) |
 2a |
68 (1.2:1) |
| 4 |
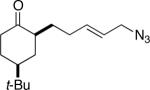 7a (67:7:13:13) |
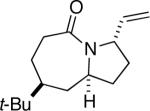 8a |
68 (3:1) |
| 5 |
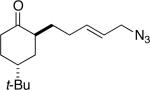 9a (63:9:14:14) |
 10a |
63 (25:1d) |
| 6 |
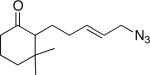 11a (67:9:12:12) |
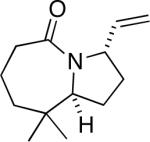 12a |
57 (>20:1) |
| 7 |
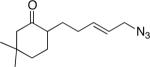 13a (63:7:15:15) |
 14a |
54 (9:1) |
| 8 |
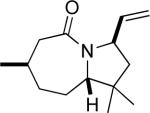
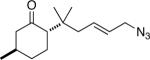 15a (90:8:1:1) |
 16a |
50e (1.6:1) |
| 9 |
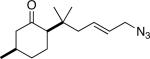 17a (94:5:0.5:0.5) |
 18a |
47e (7:1) |
| 10 |
19a (64:8:14:14) |
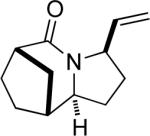 20a |
63f,g (>20:1) |
| 11 |
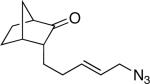 21a (63:7:15:15) |
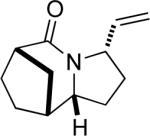 22a |
65f (15:1) |
Conditions: 1.5 equiv SnCl4, CH2Cl2, reflux.
Ratio determined by NMR of purified allylic azides and crude reaction mixtures of lactams; see Scheme 1 for structures a–d.
Only one stereoisomer containing a secondary azide is possible.
About 2% of a third lactam, assigned as a bridged isomer,10 was also observed.
Conditions: 1.5 equiv SnCl4, ClCH2CH2Cl, reflux.
Conditions: 1.5 equiv TiCl4, ClCH2CH2Cl, reflux.
Some insight into this process was obtained by making and reacting conformationally biased azide sets 7a and 9a (entries 4 and 5). The former afforded product in a slightly higher ratio than did 1a, but the reaction of axial side-chain-containing 9a led to a much higher ratio of products in favor of compound 10a. The most significant difference in this case is that the azide in the latter is obligated to react from only a single face, rendering the ratio dependent on a competition between transition structures containing an equatorial vs. axial vinyl substituent as originally proposed (Figure 1). As depicted for the reactive isomers of compounds 1, each isomeric allylic azide can react in principle by equatorial or axial attack. Equatorial attack is favored for the intramolecular additions of azide attached to a ketone substrate via oxonium ions (which forms a spirocyclic intermediate6), which is why we had initially proposed an equatorial trajectory for the addition of azide to ketone here. However, the low to modest stereoselectivities observed for 1, 7 and 15 suggest that an axial approach that also displays the vinyl group in an equatorial orientation (i.e., D in Figure 1a) might be competitive. On the other hand, constraining the azide-bearing side chain into a pseudoaxial orientation (as in compounds 9) limits the molecule to a single mode of axial azide attack onto the carbonyl. In this instance, the selectivity should be solely controlled by the expected preference for an equatorial vinyl group adjacent to the lactam nitrogen atom (Figure 1b). DFT calculations carried out on transition states emanating from intermediates A–D (Figure 1a) support this view, suggesting that 2a and 2b primarily arise from intermediates A and D, respectively.11 These experiments provide the first evidence that intramolecular Schmidt reactions of cyclohexanone-containing substrates can occur via competitive equatorial or axial azide addition to a cyclohexanone.
Figure 1.

Pathways for 1,3-allylic rearrangement/Schmidt reactions of (a) 1c and 1d (or substituted cyclohexanones bearing an equatorial side chain, like 7a) and (b) 9c and 9d. In (a), the numbers in parentheses indicate calculated energies for transition state structures from the indicated intermediate to the corresponding LA-complexed lactam product (CPCM(DCM)-B3LYP/6-31G(d,p)[SDD for Sn], relative to the lowest energy transition state structure). (c) Disfavored intermediates from isomers of 11. Proposed intermediates from (d) isomers of 13 or (e) isomers of 19.
Similarly, intermediates B and D can be disfavored by the appropriate placement of geminal dimethyl groups on C-3, where they would incur a syn-pentane interaction upon formation of a cis-fused azidohydrin intermediate (Figure 1c). Thus, azides formed upon rearrangement of 11a afford 12a and b in a >20:1 ratio (entry 6). Placement of the geminal dialkyl group elsewhere in the molecule also enhances the ratio, if less impressively, as seen in the results for compound 13 (entry 7). Here the reason for enhanced selectivity is not obvious. In this case, the axial C-5 methyl group encounters an O-metal bond following equatorial azide attack (i.e., leading to A or C) or an N(alkyl)(N2+) group for the alternative axial attack (Figure 1d). The reaction of 17, whose C-5 methyl is forced into an axial position due to the existence of a bulky C-2 side chain, further supports the role that distal substituents can have on reaction selectvity (cf. entry 9 with entry 8). Moreover, we note the very low population of internal allylic azide isomers 17c and d (< 2%), which ultimately lead to the reaction products; this provides an impressive example of the ability of the azide equilibration step in the context of intramolecular Schmidt reaction.
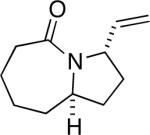
Having discerned the apparent need for a high degree of diastereofacial selectivity in affording high product ratios in the presumably chair-like examples shown so far, we examined other ring systems capable of exerting a similar bias. The norcamphor system represents a very common boat conformation for a six-membered ring and is also well known to favor exo nucleophilic attack onto a carbonyl group.12 As expected, the orientation of equatorial vinyl group was favored, probably through intermediate G, to give lactam 20a and 20b in a >20:1 ratio (Figure 1e). In comparison, a 15:1 ratio was obtained for the corresponding endo allylic azide 21 (entry 11).
We wished to demonstrate the utility of this schema for stereocontrol in the context of a synthetic approach to pinnaic acid, which was isolated by Uemura and co-workers in 1996 (Scheme 2).13,14 Specifically, we targeted an asymmetric route to lactam 28, which was an advanced intermediate in the 2004 formal synthesis of (±)-pinnaic acid by Kibayashi and coworkers.15 We proposed that a [3.2.0] bicyclic ring system ought to result in a highly diastereoselective Schmidt reaction controlled exclusively by the placement of the vinyl group in the product due to exclusive attack from the exo face.
Scheme 2.

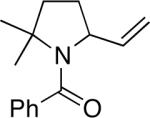
Conversion of known acid 2316 to the chiral amide 24 set up an asymmetric [2+2] cycloaddition using the protocol of Ghosez17 to afford 25 following basic hydrolysis of the intermediate iminium ions (a bridged isomer of 25 was also obtained; see SI for details). Following cross metathesis,18 NaN3 displacement gave an interconverting mixture of allylic azides 26a–d in 71% yield. The best results for the isomerization/Schmidt reaction sequence were obtained using TiCl4 treatment, which afforded a separable mixture of lactams 27a and 27b in a ca. 10:1 ratio and 68% yield. The preference for the former is presumed to result from the placement of the vinyl group in a pseudoequatorial orientation in 27a (see the ball-and-stick model shown in the scheme). Finally, hydroboration/oxidation gave Kibayashi's pinnaic acid intermediate 2815 in 83% yield.
In conclusion, we have demonstrated that it is possible to combine allylic azide rearrangement and intramolecular Schmidt reaction to stereoselectively afford substituted lactams. We are currently carrying experimental and theoretical studies to further explore the scope and applications of this combined reaction sequence.
Supplementary Material
ACKNOWLEDGMENT
We thank Michal Szostak, Thomas C. Coombs, and Erik Fenster for helpful discussions. JA acknowledges the financial support of the National Institute of General Medical Sciences (GM-049093). DJT and OG acknowledge support from NSF (CHE030089, Pittsburgh Supercomputer Center).
Footnotes
SUPPORTING INFORMATION AVAILABLE Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra of all new compounds, and details on DFT calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Aubé J, Milligan GL. J. Am. Chem. Soc. 1991;113:8965–8966. [Google Scholar]; (b) Milligan GL, Mossman CJ, Aubé J. J. Am. Chem. Soc. 1995;117:10449–10459. [Google Scholar]; (c) Lang S, Murphy JA. Chem. Soc. Rev. 2006;35:146–156. doi: 10.1039/b505080d. [DOI] [PubMed] [Google Scholar]; (d) Grecian S, Aubé J. In: Organic Azides: Syntheses and Applications. Bräse S, Banert K, editors. John Wiley and Sons; 2009. [Google Scholar]
- (2).Review: Nyfeler E, Renaud P. Chimia. 2006;60:276–284.. For some examples of total syntheses using the intramolecular Schmidt reaction see: Reddy PG, Varghese B, Baskaran S. Org. Lett. 2003;5:583–585. doi: 10.1021/ol027563v.. Wrobleski A, Sahasrabudhe K, Aubé J. J. Am. Chem. Soc. 2002;124:9974–9975. doi: 10.1021/ja027113y.. Zeng Y, Aubé J. J. Am. Chem. Soc. 2005;127:15712–15713. doi: 10.1021/ja055629m.. Ghosh P, Judd WR, Ribelin T, Aubé J. Org. Lett. 2009;11:4140–4142. doi: 10.1021/ol901645j.. Meyer AM, Katz CE, Li S, Velde DV, Aubé J. Org. Lett. 2010;12:1244–1247. doi: 10.1021/ol100113r.. Chen Z-H, Chen Z-M, Zhang Y-Q, Tu Y-Q, Zhang F-M. J. Org. Chem. 2011;76:10173–10186. doi: 10.1021/jo202042x..
- (3).Gagneux A, Winstein S, Young WG. J. Am. Chem. Soc. 1960;82:5956–5957. [Google Scholar]
- (4).For recent examples of allylic azides in synthesis, see: Klepper F, Jahn E-M, Hickmann V, Craell T. Angew. Chem., Int. Ed. 2007;46:2325–2327. doi: 10.1002/anie.200604579.. Takasu H, Tsuji Y, Sajiki H, Hitota K. Tetrahedron. 2005;61:11027–11031.. Chang Y-K, Lo H-J, Yan T-H. Org. Lett. 2009;11:4278–4281. doi: 10.1021/ol9016194.. Gagnon D, Lauzon S, Godbout C, Spino C. Org. Lett. 2005;7:4769–4771. doi: 10.1021/ol052034n.. Lauson S, Tremblay F, Gagnnon D, Godbout C, Chabot C, Mercier-Shanks C, Perreault S, DeSève H, Spino C. J. Org. Chem. 2008;73:6239–6250. doi: 10.1021/jo800817p.. Cardillo G, Fabbroni S, Gentilucci L, Perciaccante R, Piccinelli F, Tolomelli A. Org. Lett. 2005;7:533–536. doi: 10.1021/ol047815n.. Feldman AK, Colasson B, Sharpless KB, Fokin VV. J. Am. Chem. Soc. 2005;127:13444–13445. doi: 10.1021/ja050622q.. Cakmak M, Mayer P, Trauner D. Nature Chem. 2011;3:543–545. doi: 10.1038/nchem.1072..
- (5).Craig D, Harvey JW, O'Brien AG, White AJP. Org. Biomol. Chem. 2011;9:7057–7061. doi: 10.1039/c1ob05972f. [DOI] [PubMed] [Google Scholar]
- (6).Hewlett ND, Aubé J, Radkiewicz-Poutsma JL. J. Org. Chem. 2004;69:3439–3446. doi: 10.1021/jo035632t. [DOI] [PubMed] [Google Scholar]
- (7).Casadei MA, Galli C, Mandolini L. J. Am. Chem. Soc. 1984;106:1051–1056. [Google Scholar]
- (8).Eliel EL, Manoharan M. J. Org. Chem. 1981;46:1959–1962. [Google Scholar]; (b) Buchanan GW. Can. J. Chem. 1982;60:2908–2913. [Google Scholar]
- (9).Details of preparations and structure identifications may be found in the Supporting Information accompanying this paper.
- (10).Szostak M, Yao L, Aubé J. J. Org. Chem. 2010;75:1235–1243. doi: 10.1021/jo902574m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) All structures were calculated using B3LYP/6-31G(d,p)-[SDD for Sn] in DCM (CPCM with UA0 radii); see Supporting Information for details. Gutierrez O, Aubé J, Tantillo DJ. J. Org. Chem. 2012;77:640–647. doi: 10.1021/jo202338m..
- (12).Ashby EC, Laemmle JT. Chem. Rev. 1975;75:521–546. [Google Scholar]
- (13).Chou T, Kuramoto M, Otani Y, Shikano M, Yazawa K, Uemura D. Tetrahedron Lett. 1996;37:3871–3874. [Google Scholar]
- (14).Recent review: Clive DLJ, Yu M, Wang J, Yeh CSC, Kang S. Chem. Rev. 2005;105:4483–4514. doi: 10.1021/cr0406209.. Recent papers: Christie HS, Heathcock CH. Pro. Natl. Acad, Sci. USA. 2004;101:12079–12084. doi: 10.1073/pnas.0403887101.. Xu S, Arimoto H, Uemura D. Angew. Chem. Int. Ed. 2007;46:5746–5749. doi: 10.1002/anie.200701581.. Wu H, Zhang H, Zhao G. Tetrahedron. 2007;63:6454–6461..
- (15).Matsumura Y, Aoyagi S, Kibayashi C. Org. Lett. 2004;6:965–968. doi: 10.1021/ol0301431. [DOI] [PubMed] [Google Scholar]
- (16).Snider BB, Vo NH, Foxman BM. J. Org. Chem. 1993;58:7228–7237. [Google Scholar]
- (17).(a) Houge C, Frisque-Hesbain AM, Mockel A, Ghosez L, Declercq JP, Germain G, Van Meerssche M. J. Am. Chem. Soc. 1982;104:2920–2921. [Google Scholar]; (b) Depre D, Chen L-Y, Ghosez L. Tetrahedron. 2003;59:6797–6812. [Google Scholar]
- (18).Bandini M, Cozzi PG, Licciulli S, Umani-Ronchi A. Synthesis. 2004;3:409–414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.